Tổng quan hội chứng đa tuyến nội tiết tự miễn (APS)
1 NGUYÊN LÝ CHUNG

Bệnh lý đa tuyến nội tiết tự miễn là một nhóm các rối loạn không đồng nhất dẫn đến phá hủy hoặc rối loạn chức năng của nhiều tuyến nội tiết và có thể liên quan đến cả các mô khác. Cơ chế bệnh sinh có thể qua trung gian miễn dịch, xâm nhập, phá hủy tế bào do các khiếm khuyết di truyền khác nhau hoặc cơ chế kết hợp.
Hội chứng đa tuyến nội tiết tự miễn (APS) là dạng phổ biến nhất của bệnh lý đa tuyến nội tiết, có đặc trưng bởi mất khả năng dung nạp miễn dịch với các tự kháng nguyên dẫn đến tổn thương tự miễn các tuyến nội tiết và các mô khác.
Thomas Addison là người đầu tiên mô tả các đặc điểm lâm sàng và bệnh học của suy tuyến thượng thận ở các bệnh nhân có thiếu máu ác tính năm 1849 [1]. Sau đó, Schmidt mô tả sự thâm nhiễm bạch cầu lympho tại tuyến giáp và tuyến thượng thận khi mổ tử thi hai bệnh nhân tử vong do suy thượng thận cấp (hội chứng Schmidt) [2].
Bệnh lý đa tuyến nội tiết tự miễn (trước đây gọi là hội chứng đa tuyến nội tiết tự miễn) có thể được phân chia thành các typ 1 (APS1) và typ 2 - 4 (APS2, APS3, APS4). Đặc điểm các typ được trình bày trong Bảng 39.1.
2 ĐIỀU TRỊ
Hiện nay không có điều trị nào là an toàn và hiệu quả nhắm vào cơ chế bệnh sinh tự miễn của APSs. Các bệnh nhân APS cần được theo dõi chặt và các thầy thuốc lâm sàng nên duy trì mức cảnh giác cao với sự xuất hiện thêm bệnh lý tự miễn ở những bệnh nhân này.
Điều trị các bệnh lý thành phần trong hội chứng này được thảo luận trong các chương khác và quản lý mỗi bệnh về cơ bản là giống nhau đối với bệnh nhân APS. Có một số điểm cần lưu ý đặc biệt là:
- Các bệnh nhân APS1 và nhiễm nấm miệng Candida mạn tính nên được điều trị tích cực bằng thuốc chống nấm (Fluconazole) và theo dõi chặt vì có tăng nguy cơ ung thư miệng. Ngoài ra, vệ sinh miệng sạch và kiêng hút thuốc, uống rượu sẽ giúp ngăn ngừa bệnh nấm Candida.
- Điều trị hormon giáp thay thế ở bệnh nhân suy giáp có thể thúc đẩy cơn suy thượng thận cấp đe dọa tính mạng nếu có mắc cả suy thượng thận. Nên đánh giá chức năng tuyến thượng thận bằng các test động ở những bệnh nhân có nhiều bệnh lý nội tiết tự miễn trước khi bắt đầu điều trị levothyroxin.
- Các cơn hạ đường máu và giảm nhu cầu insulin ở các bệnh nhân đái tháo đường typ 1 có thể là một trong những dấu hiệu sớm nhất của suy thượng thận.
- Thiếu máu ác tính được điều trị bằng uống Vitamin B12 liều cao hoặc tiêm bắp Cyanocobalamin.
- Bệnh Celiac thường đáp ứng với chế độ ăn không có gluten; tuy nhiên, có thể cần bổ sung các vitamin và chất khoáng nếu giảm hấp thu nhiều kéo dài.
3 KIỂM TRA VÀ THEO DÕI
Phải hết sức cảnh giác mỗi khi có một bệnh tự miễn được chẩn đoán để ngăn ngừa bệnh tật và tử vong do các bệnh đi kèm khác.
Việc theo dõi các trẻ bị nhiễm nấm Candida mạn tính là cực kỳ quan trọng. Trẻ bị nhiễm nấm Candida mạn tính cần được sàng lọc và xem xét có bị APS1 không.
Các bệnh nhân nên được theo dõi bởi các bác sĩ chuyên khoa, đặc biệt là bởi một chuyên gia nội tiết.
Những bệnh nhân này cần được hỗ trợ tốt về mặt tinh thần và xã hội.
Các bệnh nhân nên được giáo dục về các triệu chứng của các bệnh mới tiềm tàng nặng trong nhóm.
Các bệnh nhân nên được cung cấp một bản in hướng dẫn về bệnh hoặc được cho đeo một vòng tay cảnh báo y tế trong trường hợp cấp cứu.
| Bảng 39.1. Hội chứng đa tuyến tự miễn (APS) | ||
| Hội chứng đa tuyến nội tiết tự miễn typ 1 | Hội chứng đa tuyến nội tiết tự miễn typ 2 - 4 | |
| Tỷ lệ mắc bệnh | Hiếm gặp | Thường gặp |
| Khởi phát | Trẻ sơ sinh/trẻ nhỏ | Trẻ lớn/trẻ trưởng thành |
| Di truyền | Đột biến AIRE (chromosome21), di truyền lặn | Đa gen, kết hợp với HLA |
| Giới tính | Nam = nữ | Nữ > Nam |
| Kiểu hình phổ biến | Nhiễm nấm Candida da-niêm mạc Suy cận giáp Suy thượng thận Loạn dưỡng móng Thiểu sản men răng | Suy thượng thận tháo đường typ 1 Viêm tuyến giáp |
| Các bệnh đi kèm | Suy sinh dục Hói đầu Bạch biến Bệnh Celiac Đái tháo đường typ 1 Thiếu máu ác tính Viêm tuyến giáp Viêm gan mạn hoạt động | Suy sinh dục Hói đầu Bạch biến Thiếu máu ác tính Bệnh nhược CƠ Bệnh Celiac Viêm khớp dạng thấp Hội chứng Sjögren |
APS, hội chứng đa tuyến tự miễn; AIRE, gen điều hòa tự miễn, nhiễm sắc thể 21; HLA, kháng nguyên bạch cầu người. | ||
4 HỘI CHỨNG ĐA TUYỂN NỘI TIẾT TỰ MIỄN TYP 1
4.1 Nguyên lý chung
4.1.1 Định nghĩa
Hội chứng đa tuyến nội tiết tự miễn typ 1 (APS1) hay còn gọi là bệnh lý đa tuyến nội tiết tự miễn - nhiễm nấm Candida - loạn dưỡng ngoại bì (APECED), là một rối loạn tự miễn di truyền lặn đơn gen hiếm gặp bao gồm tam chứng kinh điển là nhiễm nấm Candida da-niêm mạc mạn tính, suy cận giáp và suy thượng thận (bệnh Addison). APECED xuất hiện ở trẻ nhỏ và là hậu quả của bệnh tự miễn đặc hiệu với mô, dẫn đến mất chức năng ở nhiều cơ quan.
Ngoài các biểu hiện lâm sàng chính, bệnh nhân APS1 có thể có suy sinh dục nguyên phát, đái tháo đường typ 1, bệnh tuyến giáp tự miễn, viêm tuyến yên thâm nhiễm lympho bào, viêm teo dạ dày mạn tính với thiếu máu ác tính, bệnh Celiac, hói đầu, bạch biến và viêm gan tự miễn. đái tháo đường typ 1 và bệnh tuyến giáp tự miễn ít gặp hơn so với các typ APS khác.
4.1.2 Dịch tễ học
APS1 là tình trạng bệnh hiếm gặp, phổ biến nhất ở người Sardinians (1/14.000), người Phần Lan (1/25.000) và người Do Thái gốc Iran (1/9000) [3].
4.1.3 Bệnh căn
APS1 có nguyên nhân là do đột biến gen AIRE (21.422.3) [4], nó mã hóa cho protein AIRE, một yếu tố sao chép được biểu lộ trên các tế bào biểu mô tủy của tuyến ức. Protein AIRE làm trung gian cho sự biểu lộ các kháng nguyên mô ngoại biên, một chức năng cần thiết để loại bỏ các tế bào T tự phản ứng và thiết lập tình trạng tự dung nạp. Đột biến gen này được cho là sẽ dẫn đến chọn lọc ngược không hoàn toàn (incomplete negative selection) và thoát các tế bào T tự phản ứng ra ngoại biên, từ đó gây khởi phát quá trình phá hủy các mô của cơ thể [5]. Cho đến nay, đã có xấp xỉ 60 kiểu đột biến gen AIRE được báo cáo [6].
Đột biến gen AIRE dẫn đến tăng sinh các tế bào T tự phản ứng và hình thành các tự kháng thể kèm phá hủy các cơ quan bị ảnh hưởng [78].
4.2 Chẩn đoán
4.2.1 Biểu hiện lâm sàng
Khai thác tiền sử và bệnh sử
- Khai thác tiền sử bao gồm tiền sử gia đình hoàn chỉnh và kỹ lưỡng là mấu chốt để đánh giá và điều trị đúng các bệnh nhân có hội chứng đã tuyến nội tiết tự miễn.
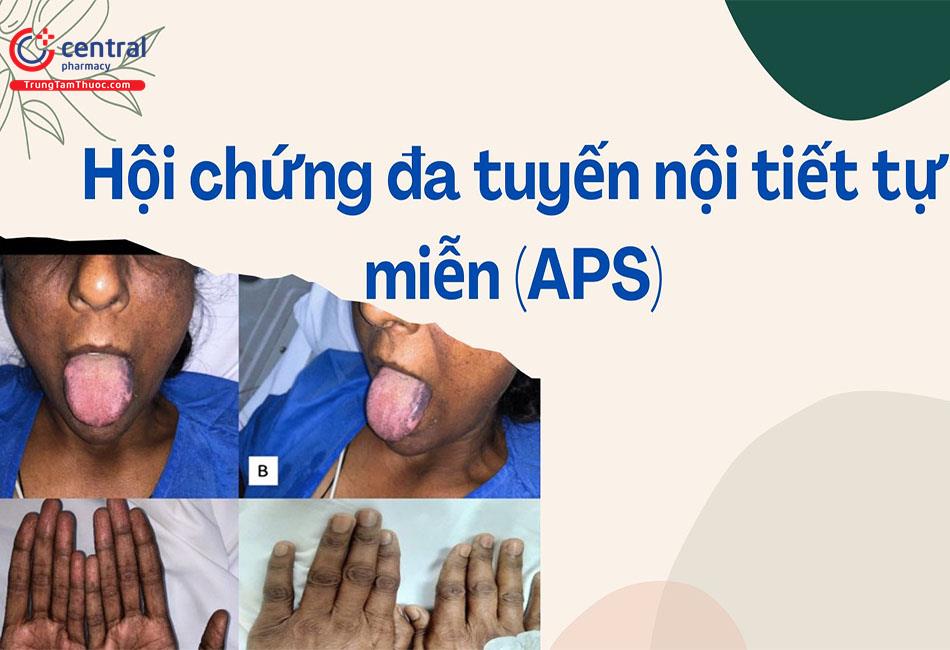
- Nhiễm nấm Candida albicans da và niêm mạc mạn tính thường xảy ra như là biểu hiện chính phổ biến nhất của APS1, thường xuất hiện ở tuổi ấu thơ. Nó có ở 97% các bệnh nhân lúc 30 tuổi [6].
- Các triệu chứng lâm sàng của nhiễm nấm Candida miệng gồm đau, đỏ, loét miệng và các mảng xám-trắng trong miệng. Viêm thực quản gây nuốt đau và đau sau xương ức. Các tổn thương ở ruột gây đau bụng, đầy bụng và tiêu chảy. Nhiễm trùng ngoài da biểu hiện bằng phát ban, nấm móng mạn tính và viêm xuất tiết ở bộ phận sinh dục. Đã có trường hợp chuyển thành ung thư tế bào vảy khi nhiễm nấm Candida miệng và thực quản không được điều trị tốt [9].
- Suy cận giáp là bệnh thành phần chính phổ biến thứ hai của APS1 với các triệu chứng của hạ Canxi máu bao gồm chuột rút cơ, cơn tetany, loạn cảm quanh miệng và co giật, cũng như tắc nghẽn đường thở trong trường hợp nặng. Suy cận giáp thường biểu hiện trước khi trẻ 10 tuổi. Suy thượng thận thường là bệnh thành phần chính xuất hiện cuối cùng trong hội chứng với các triệu chứng như mệt mỏi, thèm muối, sút cân, xạm da và niêm mạc và tụt huyết áp. Tuổi trung bình khi được chẩn đoán suy thượng thận là 15 tuổi.
- Ở một số bệnh nhân, các bệnh thành phần này có thể có các tiền triệu như tiêu chảy mạn tính, viêm giác mạc, phát ban chu kỳ kèm theo sốt, táo bón nặng, viêm gan tự miễn, rụng tóc hoặc bạch biến [9].
Khám thực thể

- Các bằng chứng về bệnh nhiễm nấm Candida ở niêm mạc, thiểu sản men răng và loạn dưỡng móng ở bệnh nhân APS1.
- Các dấu hiệu Chvostek và Trousseau, máy cơ và chuột rút gợi ý suy cận giáp.
- Tụt huyết áp tư thế, mệt, hạ natri máu và tăng Kali máu là các dấu hiệu của suy thượng thận. Suy thượng thận nếu không được chẩn đoán trong thời gian dài có thể dẫn đến hội chứng Nelson, đặc trưng bởi tăng cao ACTH và xạm da, đặc biệt là xạm các nếp gấp lòng bàn tay.
4.2.2 Tiêu chuẩn chẩn đoán
APS1: Các tiêu chuẩn chẩn đoán kinh điển bao gồm sự xuất hiện của ít nhất hai trong số các bệnh chính: nhiễm nấm Candida niêm mạc mạn tính, suy cận giáp và suy thượng thận nguyên phát. Khẳng định chẩn đoán bằng các xét nghiệm di truyền được khuyến cáo.
5 HỘI CHỨNG ĐA TUYẾN NỘI TIẾT TỰ MIỄN TYP 2 - 4
5.1 Nguyên lý chung
5.1.1 Định nghĩa

APS2, phổ biến hơn APS1, có đặc trưng bởi sự cùng xuất hiện của suy thượng thận tự miễn với bệnh tuyến giáp tự miễn và/hoặc đái tháo đường typ 1. Các bệnh thành phần có thể không được chẩn đoán cùng lúc, hậu quả là hội chứng có thể không biểu hiện đầy đủ cho đến tuổi trưởng thành. Trước đây, sự có mặt của cả suy thượng thận tự miễn và viêm tuyến giáp tự miễn được gọi là hội chứng Schmidt. Sự kết hợp của đái tháo đường typ 1 với suy thượng thận tự miễn và/hoặc viêm tuyến giáp tự miễn được gọi là hội chứng Carpenter [10].
Các bệnh nhân mắc rối loạn tự miễn đặc hiệu cơ quan phổ biến hơn như đái tháo đường typ 1 và bệnh tuyến giáp tự miễn thường có các bệnh nội tiết đồng thời mà không có bệnh Addison. Do đó, một số chuyên gia đề xuất phân loại chúng thành các typ APS 3 và 4 [11].
APS3 biểu hiện giống như nhóm bệnh của APS2 nhưng không có bất kỳ bệnh vỏ thượng thận nào.
APS4 bao gồm sự kết hợp không điển hình của hai hoặc nhiều hơn bệnh tự miễn đặc hiệu với cơ quan nhưng không xếp được vào bất kỳ typ nào nêu trên, ví dụ như sự biểu hiện của APS2 với nhiễm nấm Candida da - niêm mạc.
Giống như trong APS1, không hiếm gặp bệnh tự miễn của các cơ quan khác, có thể là suy sinh dục nguyên phát, đái tháo đường typ 1, viêm tuyến giáp mạn, viêm tuyến yên thâm nhiễm lympho bào, viêm teo dạ dày với thiếu máu ác tính, bệnh Celiac, rụng tóc, bạch biến và viêm gan tự miễn.
5.1.2 Dịch tễ học
Tỷ lệ mắc APS2 ước tính trong dân số chung là 1,4 - 2/100.000 [11]. Hội chứng này xảy ra ở xấp xỉ một nửa số ca mắc bệnh Adddison tự miễn [12]. Bệnh gặp ở nữ nhiều gấp ba lần nam. Suy thượng thận là rối loạn nội tiết khởi phát ở một nửa số trường hợp [2].
APS3 và 4 là phổ biến nhất, với tỷ lệ lên tới 1% quần thể.
5.1.3 Bệnh căn
Chùm bệnh mắc APS2 có tính chất gia đình và các bệnh lý thành phần gợi ý mạnh yếu tố di truyền. Phân tích phả hệ của hầu hết các gia đình có người mắc APS2 cho thấy sự di truyền đa gen. Mối liên quan của các allele HLA trên nhiễm sắc thể 6p21 với các bệnh tự miễn đã được biết từ vài thập kỷ. Các halotyp HLA II DR3 và DR4 có liên quan chặt chẽ với các bệnh lý thành phần của hội chứng này. Nguy cơ cao nhất xuất hiện cả bệnh Addison và đái tháo đường typ 1 liên quan với kiểu gen dị hợp tử HLA-DR4-DQ8/HLA-DR3-DQ2 [7]. Các gen không phải HLA bao gồm kháng nguyên tế bào lympho T gây độc tế bào 4 (CTLA4) và tương tác với môi trường cũng đã được đề xuất trong bệnh sinh APS2 [8].
5.1.4 Các yếu tố nguy cơ
Ngoài yếu tố di truyền, các yếu tố môi trường cũng có liên quan với APS2. Điều trị interferon-a có liên quan với sự xuất hiện tự kháng thể kháng 21- hydroxylase, tự kháng thể kháng tiểu đảo tụy và tự kháng thể tuyến giáp cùng với các bệnh tự miễn tương ứng [7].
Có thai thường phối hợp với giảm chức năng miễn dịch, tuy nhiên viêm tuyến giáp sau đẻ được thấy ở gần 1/3 các bệnh nhân đái tháo đường typ 1.
Prolamins, hay các protein giàu proline và glutamine có khả năng kháng với các peptidase đường ruột, được coi là tác nhân gây bệnh Celiac. Gliadin (từ lúa mì) và các protein liên quan trong lúa mạch, lúa mạch đen, ngô và một số yến mạch có ảnh hưởng đến cơ chế bệnh sinh bệnh Celiac.
Giả thuyết vệ sinh cho rằng sự suy giảm các bệnh truyền nhiễm, đặc biệt là nhiễm giun sán, góp phần làm tăng tỷ lệ mắc các bệnh dị ứng, hen và bệnh tự miễn [3].
5.2 Chẩn đoán
5.2.1 Biểu hiện lâm sàng
Khai thác bệnh sử và tiền sử
- Tiền sử gia đình và thông tin về tăng nguy cơ mắc các rối loạn nội tiết khác là chìa khóa để chẩn đoán hội chứng đa bệnh lý nội tiết
- Một nửa các trường hợp APS 2 có biểu hiện khởi đầu là bệnh lý thượng thận. Các bệnh thành phần khác có thể xuất hiện trước khi chẩn đoán suy thượng thận hoặc xuất hiện sau vài năm đến vài chục năm.
- Tiền sử đái nhiều và uống nhiều gợi ý bệnh đái tháo đường typ 1 trong khi tăng/giảm cân, sợ nóng/lạnh và táo bón/đại tiện nhiều lần gợi ý bệnh tuyến giáp tự miễn.
- Vô kinh và nóng bừng mặt ở phụ nữ trẻ nhắc nhở phải kiểm tra suy buồng trứng, trong khi giảm ham muốn và giảm chức năng tình dục gợi ý suy tinh hoàn ở nam giới. Trong cả 2 trường hợp, xét nghiệm sẽ thấy LH và FSH tăng, còn estrogen và testosteron thấp. Do không phát hiện được các kháng thể đặc hiệu với tuyến sinh dục nên những chẩn đoán này dựa vào thay đổi sinh lý ở những người nhạy cảm.
- Da mất sắc tố loang lổ, rụng tóc, phát ban sẩn, thiếu máu không rõ nguyên nhân, tiêu chảy kéo dài hay vàng da là những biểu hiện điển hình của các bệnh tự miễn thứ phát đi kèm bao gồm bạch biến, rụng tóc từng mảng/toàn thân, thiếu máu ác tính, bệnh Celiac hoặc viêm gan tự miễn.
- Sự xuất hiện thêm các rối loạn nội tiết có thể làm phức tạp hơn bệnh cảnh lâm sàng của bệnh đã có từ trước. Ví dụ, tụt huyết áp có triệu chứng hoặc hạ đường máu dẫn đến phải giảm liều insulin ở bệnh nhân đái tháo đường typ 1 có thể là biểu hiện của suy thượng thận. Ở một bệnh nhân suy giáp tự miễn và APS, việc tăng liều hormon giáp để duy trì bình giáp có thể là hậu quả của giảm hấp thu thuốc do hiện bệnh Celiac.
Khám thực thể
- Các dấu hiệu của suy thượng thận nguyên phát bao gồm tụt huyết áp, hạ natri máu và xạm da (xem phần Hội chứng đa tuyến nội tiết tự miễn typ 1).
- Các bệnh nhân đái tháo đường typ 1 sẽ có các dấu hiệu của tăng đường máu, thường xuất hiện rầm rộ hơn ở các bệnh nhân trẻ nhưng âm thầm hơn ở các bệnh nhân lớn tuổi (xem Chương 29).
- Giảm phản xạ gân gót và phù quanh hốc mắt là dấu hiệu của suy giáp, còn lồi mắt, co cơ mi và run nhanh nhỏ đầu chi gợi ý cường giáp (xem Chương 8 và Chương 9).
- Thiếu máu ác tính có thể biểu hiện bằng thiếu máu hoặc triệu chứng của bệnh lý thần kinh ngoại biên. Nếu không được chẩn đoán, thiếu máu ác tính có thể gây thiếu máu nặng và các triệu chứng thần kinh bao gồm dấu hiệu tiểu não và chứng thất điều.
- Bạch biến và hói đầu là những bệnh lý ít gặp trong hội chứng đa tuyến nội tiết tự miễn. Bạch biến trong hội chứng này có tính chất tiến triển, phân bố không thành từng mảng và thường xuất hiện tại các vị trí bị cọ sát và chấn thương [14]. Rụng tóc từng mảng biểu hiện bằng hói đầu từng mảng không liên tục và có thể tiến triển thành rụng tóc toàn bộ trên da đầu (hói toàn đầu) hoặc rụng toàn bộ lông trên cơ thể (hói toàn thể) [15].
- Tổn thương dạng sẩn ngứa trên bề mặt da gợi ý viêm da dạng mụn nước, nó có liên quan đến bệnh Celiac.
5.2.2 Tiêu chuẩn chẩn đoán
APS2: Chẩn đoán cần sự hiện diện của bệnh Addison tự miễn với bệnh tuyến giáp tự miễn và/hoặc đái tháo đường typ 1. Suy thượng thận tự miễn là thành phần chính yếu của hội chứng này.
APS3 - 4: Chẩn đoán dựa trên sự hiện diện của nhiều bệnh lý tự miễn trên cùng một người, xuất hiện theo bất kỳ trình tự nào và ở bất kỳ lứa tuổi nào.
5.2.3 Chẩn đoán phân biệt
Một số hội chứng di truyền và mắc phải bao gồm suy hoặc rối loạn chức năng đa tuyến nội tiết. Các hội chứng đa bệnh lý nội tiết này được đề cập trong Bảng 39.2 [16-23].
Các hội chứng khác liên quan đến rối loạn chức năng tự miễn tuyến, cụ thể được ghi nhận xảy ra cùng với bệnh Lupus ban đỏ hệ thống, viêm khớp dạng thấp và các bệnh mạch máu Collagen khác. Nó bao gồm kháng insulin typ B, giảm tiểu cầu miễn dịch, giảm bạch cầu tự miễn dịch và thiếu máu tan máu.
| Bảng 39.2. Hội chứng đa bệnh nội tiết | ||
| Hội chứng đa bệnh nội tiết | Nguyên nhân | Đặc điểm lâm sàng |
| Bất thường nhiễm sắc thể (hội chứng Down và hội chứng Turner) | Không rõ | Bệnh tuyến giáp tự miễn, đái tháo đường typ 1, bệnh Celiac, viêm gan tự miễn, rụng tóc từng mảng, bạch biến. |
| Hội chứng DiGeorge, hội chứng velocardiofacial OMIM 188400 | Mất đoạn nhiễm sắc thể rất nhỏ 22q11.2 | Biến dạng sọ mặt, hầu họng và tim, chậm phát triển trí tuệ. Nội tiết: suy cận giáp, không có tuyến giáp và hoặc bệnh tuyến giáp tự miễn hoặc cường giáp, đái tháo đường typ 1, xuất huyết giảm tiểu cầu tự miễn (ITP), thiếu GH. |
| Bệnh nhiễm thiết huyết tố di truyền [16] | Đột biến gen HFE | Tam chứng cổ điển gồm gan to, đái tháo đường typ 1và xạm da cùng với suy tim xung huyết. Các bệnh nội tiết khác: suy tuyến yên, suy sinh dục, suy giáp. |
| Bệnh Hirata, còn gọi là hội chứng insulin tự miễn (IAS) [17] | HLA phối hợp với HLA DR4 + do thuốc | Hạ đường máu do cường insulin tự miễn. Phối hợp với bệnh basedow, đặc biệt là khi điều trị bằng methimazol. |
| IPEX (rối loạn điều hào miễn dịch, bệnh lý đa tuyến nội tiết, bệnh ruột, liên quan đến nhiễm sắc thể X) [18] OMIM 304930 | Đột biến gen FoxP3 dẫn đến bệnh tự miễn | Tam chứng lâm sàng gồm bệnh ruột (tiêu chảy nặng), bệnh nội tiết (đái tháo đường typ 1 hoặc viêm tuyến giáp) và viêm da. Thường xuất hiện ở trẻ sơ sinh Các bệnh khác: hói toàn thể, thiếu máu tan máu tự miễn, giảm tiểu cầu, giảm bạch cầu, viêm thận kẽ, viêm gan tự miễn |
| Hội chứng Kearns - Sayre [19] | Đứt đoạn DNA ty lạp thể | Tam chứng gồm đau vùng mắt ngoài tiến triển, bệnh võng mạc hắc tố không điển hình và rối loạn dẫn truyền trong tim. Xuất hiện trước khi 20 tuổi. Bệnh nội tiết: lùn, suy sinh dục, đái tháo đường, bệnh tuyến giáp, cường aldosteron, suy cận giáp. |
| Hội chứng McCune - Albright [20] OMIM 174800 | Kích hoạt đột biển ở tiểu đơn vị anpha của thụ thể gắn với protein G | Đặc trưng bởi tam chứng gồm các chấm cà phê sữa trên da, loạn sản dạng sợi đa sinh (Polyostotic fibrous dysplasia) và rối loạn chức năng nhiều tuyến nội tiết. Bệnh nội tiết: dậy thì sớm, to đầu chi, u tuyến yên tiết Prolactin, cường giáp, hội chứng Cushing, nhuyễn xương giảm phosphat máu, nhiễm độc testosteron |
| Hội chứng POEMS [21] | Không rõ; gammopathy thể đơn dòng gợi ý rối loạn tế bào huyết tương, tăng cytokine và VEGF khá phổ biến. | Bệnh lý đa dây thần kinh, bệnh lý các tạng, bệnh lý nội tiết, M protein (rối loạn tăng sinh huyết tương đơn dòng), thay đổi về da (POEMS). Khác: tổn thương xơ cứng xương, bệnh Castleman, phù gai thị, phù, tràn dịch màng phổi/cổ trướng. Bệnh nội tiết: suy sinh dục, đái tháo đường typ 2, suy giáp, suy thượng thận, tăng PTH. |
| Hội chứng kháng insulin typ B [22] | Kháng thể kháng thy the insulin (không có sản phẩm thương mại) | Tăng đường máu, cường androgen, Lupus ban đỏ hệ thống, viêm tuyến giáp hashimoto, xơ gan ứ mật nguyên phát, hạ Glucose máu (hiếm gặp). |
| Hội chứng Wolfram [23] OMIM 222300 | Đột biến gen WFS1 | Đái tháo nhạt, đái tháo đường, teo gai thị và điếc (DIDMOAD). Khác: mất thính lực, bệnh bàng quang thần kinh, thất điều, loạn vận ngôn, sa sút trí tuệ bệnh tâm thần, các rối loạn chức năng nội tiết khác (suy sinh dục, suy giáp và chậm lớn) xảy ra trong hội chứng tiến triển này. |
| GH = hormon tăng trưởng; HLA = Kháng nguyên bạch cầu người; ITP = Xuất huyết giảm tiểu cầu miễn dịch; OMIM = Số phân loại bộ gen; VEGF: (vascular endothelial growth factor) = Yếu tố tăng trưởng nội mô mạch máu. | ||
| Bảng 39.3. Các tự kháng thể đặc hiệu cơ quan (organ-specific autoantibodies) | ||
| Bệnh tự miễn | Kháng thể đi kèm | Chẩn đoán cận lâm sàng |
| Đái tháo đường typ 1 | Kháng thể kháng glutamic acid decarboxylase (GAD65), kháng thể kháng protein tyrosine phosphatase (IA-2A & IA-2B)*, tự kháng thể kháng insulin (IAA), kháng thể kháng chất vận chuyển Kẽm 8 (ZnT8). | Đường máu lúc đói, nghiệm pháp dung nạp glucose đường uống, HbA1c |
| Hội chứng Insulin tự miễn (bệnh Hirata) | Tự kháng thể Insulin (IAA), đơn dòng hoặc đa dòng. | Tìm bằng chứng hạ đường máu (Đường máu lúc đói hoặc sau ăn), nồng độ insulin và C-peptide, các kháng thể |
| Viêm tuyến giáp | Kháng thể kháng Peroxidase tuyến giáp (Anti-TPO), TSI (thyroid-stimulating immunoglobulin) và kháng thể kháng thyroglobulin. | TSH, T4 tự do và T3 toàn phần |
| Suy thượng thận nguyên phát | Kháng thể kháng 21-hydroxylase (ACA), kháng 17a - hydroxylase Ab, kháng P450scc Ab (SCA). | Nghiệm pháp kích thích ACTH |
| Viêm dạ dày tự miễn, thiếu máu ác tính | Kháng thể kháng tế bào diềm. Kháng thể kháng yếu tố nội (intrinsic factor). | Vitamin B12, xét nghiệm kháng thể, gastrin |
| Bệnh Celiac | Kháng thể kháng tTG, kháng thể kháng endomysial IgA, kháng thể kháng gliadin. | Xét nghiệm kháng thể (cùng với IgA huyết thanh), sinh thiết ruột non |
| Suy cận giáp | Kháng thể kháng thụ thể nhạy cảm canxi. | PTH, canxi và phosphat máu, Canxi niệu/24 giờ |
| Viêm gan tự miễn | ANA, kháng thể kháng cơ trơn, kháng thể kháng ty lạp thể, kháng thể kháng LKM1, kháng thể kháng LC1. | Chức năng gan, xét nghiệm các kháng thể, sinh thiết gan |
| Suy sinh dục | Kháng thể kháng 17a-hydroxylase, enzyme P450 phân cắt chuỗi nhánh, 3B hydroxysteroid dehydrogenase, kháng thể kháng tinh trùng. | FSH, LH, testosteron, Estradiol, progesteron |
| Bạch biến | Kháng thể kháng tyrosinase. | Khám đèn Wood |
| *Kháng thể kháng protein xuyên màng thuộc gia đình protein tyrosine phosphatase còn được gọi là kháng nguyên phối hợp với insulinoma (insulinoma-associated: IA-2A và IA-2B), IA-2A cũng còn được gọi là ICA512. ACTH, adrenocorticotrophic hormone: Hormon hướng vỏ thượng thận; ANA, antinuclear antibody: Kháng thể kháng nhân; FSH, follicle-stimulating hormone: Hormon kích thích tạo nang buồng trứng; LC1, liver cytosol type 1; LH, luteinizing hormone: Hormon kích thích tạo hoàng thể; LKM1, liver kidney microsome type 1; PTH, parathyroid hormone: Hormon cân giáp; TSH, thyroid-stimulating hormone: Hormon kích thích tuyến giáp; tTG, tissue transglutaminase. | ||
5.2.4 Chẩn đoán cận lâm sàng
Nhiều rối loạn tự miễn kiến tạo nên APS có các giai đoạn tiền căn kéo dài, trong thời gian đó có sự xuất hiện các tự kháng thể đặc hiệu mô trong huyết thanh. Lượng tự kháng thể hiện diện trong một cá nhân nhất định đóng vai trò dự báo khả năng xuất hiện bệnh vì nguy cơ mắc một bệnh cụ thể có xu hướng tăng lên khi số lượng và số loại tự kháng thể nhắm vào mô đó tăng lên. Ví dụ, nguy cơ tiến triển thành đái tháo đường typ 1 trong 5 năm ở những người thân trực hệ của người bệnh là > 50% nếu có nhiều loại tự kháng thể kháng tế bào B.
Các phương pháp xét nghiệm chính để chẩn đoán APS là các xét nghiệm tự kháng thể trong huyết thanh kháng các tuyến và các mô liên quan và đánh giá chức năng các cơ quan đích và tiết hormon.
Các tự kháng thể trung hòa IgG chống lại interferon typ 1, bao gồm các phân nhóm interferon-a và interferon-0, gần như xác định chẩn đoán APS1 khi biểu hiện lâm sàng không đáp ứng tiêu chuẩn chẩn đoán và khi đã loại trừ u tuyến ức và bệnh nhược cơ. Hiện đã có các xét nghiệm di truyền tìm đột biết gen AIRE. Các Labor tốt có thể phát hiện được đột bién gen AIRE ở > 95% các trường hợp [6].
Kiểm tra sự hiện diện các tự kháng thể của tuyến thượng thận (21-hydroxylase), tuyến giáp (peroxidase và thyroglobulin), tế bào tiểu đảo tụy (insulin, glutamic acid decarboxylase và IA-2A), tế bào diềm (H'/K*-ATPase) (Bảng 39.3) có thể hỗ trợ khẳng định nghi ngờ lâm sàng của bệnh tự miễn mô hoặc đánh giá nguy cơ bị các rối loạn nội tiết trong tương lai, mặc dù xét nghiệm huyết thanh không thay thế được thăm khám lâm sàng cẩn thận các bệnh theo cơ quan.
Nên đánh giá chức năng các tuyến nội tiết bằng các xét nghiệm hormon phù hợp: tuyến thượng thận (nghiệm pháp kích thích ACTH, điện giải đồ máu, aldosterone và renin), tuyến giáp (TSH và FT4), tiểu đảo tụy (đường máu lúc đói và nghiệm pháp dung nạp glucose), tuyến cận giáp (canxi ion hóa và PTH intact) và tuyến sinh dục (estrogen hoặc Testosterone, FSH và LH).
Ở một bệnh nhân thiếu máu, nồng độ vitamin B, trong huyết thanh thấp gợi ý thiếu máu ác tính, nó có thể cần được đánh giá thêm bằng xét nghiệm kháng thể kháng tế bào diềm, kháng thể kháng yếu tố nội và nồng độ gastrin máu lúc đói.
Những bệnh nhân có thiếu Sắt mà không có tiền sử mất máu nên được kiểm tra bệnh Celiac bằng xét nghiệm tự kháng thể IgA transglutaminase mô và nồng độ tổng IgA. Nội soi kèm sinh thiết ruột non là cần thiết để khẳng định bệnh Celiac.
6 TÀI LIỆU THAM KHẢO
1. Addison T. Anemia: Disease of the suprarenal capsules. Lond Med Gaz 1849;12:535-546.
2. Schatz DA, Winter WE. Autoim- mune polyglandular syndrome. II: Clinical syndrome and treatment. Endocrinol Metab Clin North Am 2002;31:339–352.
3. Betterle C, Greggio NA, Volpato M. Clinical review 93: Autoim- mune polyglandular syndrome type 1. J Clin Endocrinol Me- tab 1998;83:1049-1055.
4. The Finnish-German APECED Con- sortium. An autoimmune disease, APECED, caused by mutations in a novel gene featuring two PHD-type zince-finger domains. Nat Gen- et 1997;17:399–403.
5. Waterfield M, Anderson MS. Clues to immune tolerance: The mono- genic autoimmune syndromes. Ann NY Acad Sci 2010;1214:138-155.
6. Husebye ES, Perheentupa J, Rautemaa R, Kämpe O. Clinical manifestation and management of patients with autoimmune poly- endocrine syndrome type I. J Intern Med 2009;265:514–529.
7. Eisenbarth GS, Gottlieb PA. Autoimmune polyendocrine syn- dromes. N Engl J Med 2004; 350: 2068-2079.
8. Michels AW, Gottlieb PA. Au- toimmune polyglandular syn- dromes. Nat Rev Endocrinol 2010; 6:270–277.
9. Perheentupa J. Autoimmune poly- endocrinopathy-candidiasis-ecto- dermal dystrophy. J Clin Endocri- nol Metab 2006;91:2843-2850.
10. Kriegel MA, Lohmann T, Gabler C, Blank N, Kalden JR, Lorenz HM. Defective function suppressor of human CD4+CD25+regulatory T cells in autoimmune polyglan- dular syndrome type II. J Exp Med 2004;199:1285–1291.
11. Betterle C, Dal Pra C, Mantero F, Zanchetta R. Autoimmune adre- nal insufficiency and autoimmune polyendocrine syndromes: Au- toantibodies, autoantigens, and their applicability in diagnosis and disease prediction. Endocr Rev 2002;23:327-364.
12. Falorni A, Laureti S, Santeusanio F. Autoantibodies in autoimmune polyendocrine syndrome type II. Endocrinol Metab Clin North Am 2002;31: 369–389.
13. Gale EA. A missing link in the hygiene hypothesis? Diabetolo- gia 2002;45:588–594.
14. Taieb A, Picardo M. Vitiligo. N Engl J Med 2009; 360:160–169.
15. Shapiro J. Hair loss in women. N Engl J Med 2007; 357:1620-1630.
16. Utzschneider KM, Kowdley KV. Hereditary hemochromatosis and diabetes mellitus: implications for clinical practice. Nat Rev Endocri- nol 2010;6:26-33.
17. Uchigata Y, Eguchi Y, Takaya- ma-Hasumi S, Omori Y. Insulin autoimmune syndrome (Hirata disease): Clinical features and epi- demiology in Japan. Diabetes Res Clin Pract 1994;22:89-94.
18. Torgerson TR, Ochs HD. Immune dysregulation, polyendocrinopathy, enteropathy, X-linked: Forkhead box protein 3 mutations and lack of regulatory T cells. J Allergy Clin Immunol 2007; 120:744-750.
19. Harvey JN, Barnett D. Endocrine dysfunction in Kearns-Sayre syndrome. Clin Endocrinol (Oxf) 1992;37:97-103.
20. Dumitrescu CE, Collins MT. Mc- Cune-Albright syndrome. Orphanet J Rare Dis 2000;3:12.
21. Gandhi GY, Basu R, Dispen- zieri A, Basu A, Montori VM, Brennan MD. Endocrinopathy in POEMS syndrome: the Mayo clinic experience. Mayo Clin Proc 2007;82:836-842.
22. Arioglu E, Andewelt A, Diabo C, Bell M, Taylor SI, Gorden P. Clinical course of the syndrome of autoantibodies to the insulin receptor (type b insulin resistance): A 28 year perspective. Medicine (Baltimore) 2002;81:87-100.
23. Rohayem J, Ehlers C, Wiedemann B, et al. Diabetes and neurode- generation in Wolfram syndrome: A multicenter study of pheno- type and genotype. Diabetes Care 2011;34:1503-1510.
24. Thomas J.Braranski, MD, PhD, Janet B.McGill, MD, MA, FACE, Julie M.Silverstein, MD và các tác giả khác tham gia biên soạn, Khoa nội tiết chuyển hóa và nghiên cứu
