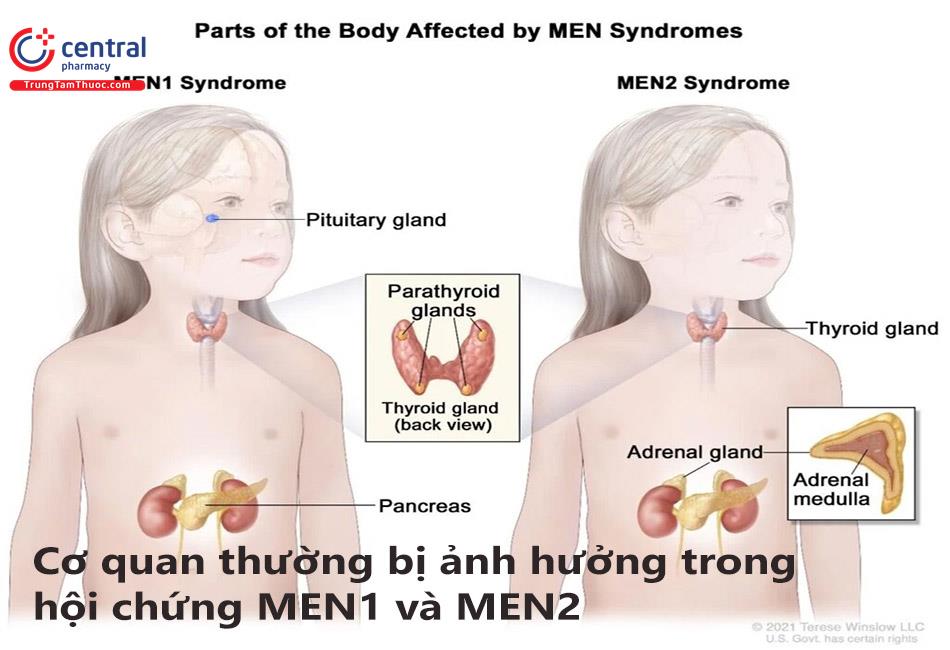
Tổng quan hội chứng bệnh lý u tân sinh đa tuyến nội tiết
1 ĐẠI CƯƠNG
Hội chứng u tân sinh đa tuyến nội tiết (Multiple endocrine neoplasia - MEN) là những rối loạn tân sinh đơn lẻ hoặc di truyền của nhiều hơn một tuyến nội tiết. Nói chung, có hai hội chứng riêng biệt: MEN1 và MEN2 với đặc tính di truyền trội và có bệnh trên toàn bộ đối tượng nhưng biểu hiện rất thay đổi. MEN2 có ba dưới nhóm: MEN2A, ung thư tuyến giáp thể tủy có tính chất gia đình và MEN2B.
MEN1 có nguyên nhân là do mất chức năng hoặc bất hoạt gen ức chế khối u, trong khi MEN2 có nguyên nhân là tăng chức năng hoặc hoạt hóa tiền sinh gen ung thu (proto-oncogene).
1.1 Định nghĩa
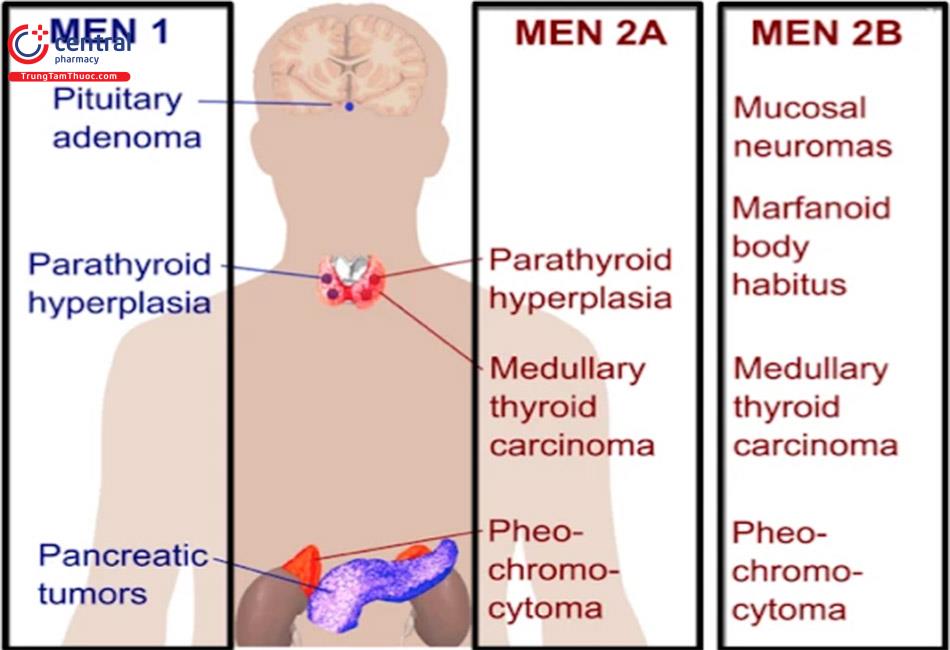
MEN1 được định nghĩa là sự có mặt của ít nhất hai trong ba loại u chính: u cận giáp, u tuyến yên và u tụy-ruột. MEN1 được coi là có tính chất gia đình nếu có một bệnh nhân đầu tiên cộng với một người thân có ít nhất một trong ba loại u chính của MEN1.
MEN2 được phân chia thành 3 hội chứng: MEN2A, ung thư tuyến giáp thể tủy có tính chất gia đình (FMTC), MEN2B.
- MEN2A: gồm ung thư tuyến giáp thể tủy, u tuỷ thượng thận (pheochromocytoma) và quá sản tuyến cận giáp.
- Ung thư tuyến giáp thể tủy có tính chất gia đình là một biến thể của MEN2A, trong đó có khuynh hướng mạnh phát triển thành ung thư tuyến giáp thể tủy mà không phải là u tuỷ thượng thận hay cường cận giáp.
- MEN2B gồm ung thư tuyến giáp thể tủy, pheochromocytoma và u hạch thần kinh của ruột và niêm mạc miệng (ngoài ra còn có hình dáng kiểu Marfan, dày sợi thần kinh giác mạc tủy).
==> Bạn đọc có thể tham khảo thêm: Ung Thư Tuyến Giáp: Sự Tăng Sinh Bất Thường Các Tế Bào Tuyến Giáp
1.2 Dịch tễ học
MEN1 được di truyền theo tính trội qua nhiễm sắc thể thường với tần suất mới mắc từ 2 - 20 ca trên 100.000 người trong cộng đồng (xem Bảng 37.1) [1].
- 1% đến 18% ở những bệnh nhân cường cận giáp nguyên phát.
- 16% đến 38% ở những bệnh nhân u tiết gastrin.
- Dưới 3% ở những bệnh nhân u tuyến yên (xem Bảng 37.2) [1,2].
MEN2 là một hội chứng hiếm gặp, di truyền theo tính trội qua nhiễm sắc thể thường với tần suất mới mắc ước tính từ 1-10 ca trên 100.000 người trong cộng đồng. Cho đến nay đã phát hiện được 500 - 1000 dòng họ gia đình trên toàn thế giới, trong đó MEN2A chiếm 80%, ung thư tuyến giáp thể tủy có tính chất gia đình chiếm 15% và MEN2B chiếm 5% các trường hợp [3].
1.3 Bệnh căn
1.3.1 Hội chứng u tân sinh đa tuyến nội tiết typ 1 (MEN)
Gen đột biến gây MEN1 (xem Bảng 37.1) [1] là một gen ức chế u, nằm trên cánh dài của nhiễm sắc thể 11 (11q13) và mã hóa cho một protein nhân có 610 Amino acid được đặt tên là menin. Chức năng đầy đủ của menin còn chưa được biết rõ, mặc dù một số nghiên cứu gợi ý protein này có thể có vai trò điều hòa phiên mã. Có 10% các đột biến ở MEN1 là đột biến mới và hơn 400 loại đột biến tế bào mầm khác nhau đã được phân lập.
Không có mối liên quan giữa kiểu gen và kiểu hình ở bệnh nhân MEN1, gây khó khăn cho việc sàng lọc di truyền và điều trị can thiệp từ gốc.
Nhiều hội chứng khối u nội tiết và các gen liên quan mới được phát hiện bao gồm thể MEN4 khác biệt, có biểu hiện lâm sàng giống MEN1 nhưng nguyên nhân là do đột biến CDKN1B [4].
| Bảng 37.1. Các đặc điểm chung của hội chứng MEN1 và MEN2 | ||
| MEN1 | MEN2 | |
|
Tần suất mắc mới Di truyền Gen Gen sản xuất |
2 - 20/100.000 người Di truyền trội Gen MEN1 Menin (protein nhân)
|
1 - 10/100.000 người Di truyền trội Gen RET RET (protein gắnkết tyrosine kinase xuyên màng) |
|
Vị trí có đột biến
Chức năng Typ đột biến trong u Liên quan kiểu gen - kiểu hình Xét nghiệm di truyền hướng dẫn can thiệp để dự phòng và điều trị tiệt căn ung thư.
|
Nhiễm sắc thể 11 (11q13) Gen ức chế khối u Bất hoạt Không Không |
Nhiễm sắc thể 10 (10q11-2) Gen gây tiền sinh ung thư Hoạt hoá Có Có |
| Theo Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658-5671. | ||
1.3.2 Hội chứng u tân sinh đa tuyến nội tiết typ 2 (MEN2)
Hầu như tất cả các bệnh nhân hội chứng MEN2 đều sẽ xuất hiện ung thư tuyến giáp thể tủy, xuất phát từ tế bào C có nguồn gốc từ mào thần kinh hơn là từ tế bào nang tuyến giáp. Xấp xỉ 25% các bệnh nhân ung thư tuyến giáp thể tủy có một trong các biến thể của MEN2. Pheochromocytoma là u thường gặp thứ hai trong MEN2, xuất hiện ở 50% số bệnh nhân.
Gen đột biến gây MEN2 là gen sinh tiền ung thư RET (REarranged during Transfection), nằm trên nhiễm sắc thể 10 (10q11-2), chứa 21 exons và mã hóa cho một thụ thể tyrosine kinase gắn màng.
Có mối liên quan chặt chẽ giữa kiểu gen và kiểu hình ở MEN2. Từ 80% đến 98% các trường hợp MEN2A và ung thư tuyến giáp thể tủy có tính chất gia đình có liên quan đến đột biến ở exon 10 hoặc exon 11 dẫn đến hiện tượng nhị trùng hóa đồng nhất không phụ thuộc gắn kết của thụ thể gây hoạt hóa thể tạng và truyền tín hiệu xuôi dòng theo con đường kinase MAP (protein hoạt hóa phân bào).
Ở đa số các gen gây MEN2A, một trong năm gốc cysteine (C609, C611, C618, C620, C634) thuộc phần ngoại bào của gen RET bị đột biến.
Hơn 95% các trường hợp MEN2B có biểu hiện một đột biến M918T ở exon 16 và 2 - 3% có đột biến codon 883 ở exon 15 dẫn phosphoryl hóa và thay đổi tính đặc hiệu của cơ chất [3].
Có thể dự đoán được bệnh cảnh lâm sàng của MEN2 dựa trên loại đột biến nào đang thúc đẩy diễn biến bệnh. Ví dụ, 50% những người MEN2A mà có đột biến ở vị trí C634 sẽ xuất hiện cả cường cận giáp và pheochromocytoma thêm vào ung thư tuyến giáp thể tủy so với chỉ 4% ở những người có đột biến C609 [3]. Ở các bệnh nhân MEN2B, ung thư tuyến giáp thể tủy sẽ ít ác tính hơn nếu có đột biến A883F so với đột biến M918T [3]. Do có tương quan chặt chẽ giữa kiểu gen với kiểu hình nên cả sàng lọc di truyền và điều trị can thiệp khỏi bệnh đều dễ dàng hơn.
| Bảng 37.2. Các biểu hiện lâm sàng của MEN1, MEN2A và MEN2B | ||
| MEN1 | MEN2A | MEN2B |
|
Cường cận giáp (90 - 95%)
U tuyến yên (15 - 90%)
Các u khác
|
Ung thư tuyến giáp thể tủy (< 100%) Pheochromocytoma (xấp xỉ 50%) Cường cận giáp (< 30%) Khác
|
Ung thư tuyến giáp thể tủy (< 100%) Pheochromocytoma (< 50%) Khác
|
|
GH: hormon tăng trưởng; VIP: peptid hoạt tính trong ruột Theo Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Metab 2001;86:5658-5671; Lakhani VT, You YN, Wells SA. The multiple endocrine neoplasia syndromes. Ann Rev Med 2007;58:253-265. |
||
CHẨN ĐOÁN
Biểu hiện lâm sàng
1.3.3 MEN1
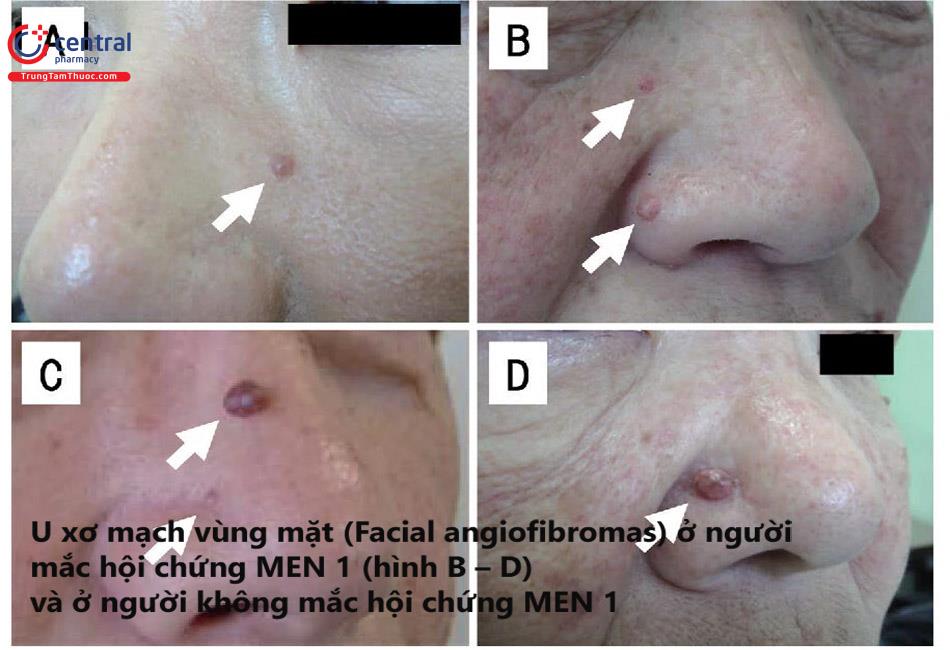
Về mặt lâm sàng, một bệnh nhân cường cận giáp nguyên phát và có kèm theo hoặc u tuyến yên hoặc là u tế bào tiểu đảo tụy thì được coi là có MEN1.
Cường cận giáp nguyên phát là biểu hiện phổ biến nhất và sớm nhất (95% các bệnh nhân ở tuổi 50). Cường cận giáp trong MEN1 so với cường cận giáp tản phát, thường xuất hiện ở độ tuổi 20 - 25 (so với 55 - 60), với tỷ lệ nam/nữ là tương đương (so với tỷ lệ 1 nam/3 nữ) và có quá sản không đồng đều cả bốn tuyến (so với adenoma một tuyến). Mặc dù đa số các bệnh nhân không có triệu chứng, một số có thể có các biểu hiện không điển hình của Canxi máu' data-type-link='internal' target='_blank'>tăng canxi máu (đái nhiều, đau cơ, mệt mỏi và sỏi thận). Xem Bảng 37.2 [1,2].
Các u tụy ruột là loại u thường gặp thứ hai (gặp ở 30 - 80% những người bị MEN1). Các u này có thể tiết (có chức năng) hoặc không tiết (không có chức năng). Các triệu chứng của tăng quá mức hormon thường xảy ra ở tuổi 40 nhưng có thể phát hiện được các khối u không triệu chứng ở những người mang gen trẻ hơn bằng các xét nghiệm sinh hóa và hình ảnh học. Khối u có kích thước to hơn có liên quan trực tiếp với các biểu hiện nặng hơn, phân độ bệnh học theo WHO tồi hơn và khả năng di căn cao hơn [5].
U tiết Gastrin là u tụy ruột phổ biến nhất (gặp ở 50% các bệnh nhân MEN1). Nên cân nhắc MEN1 ở những bệnh nhân được chẩn đoán ban đầu là u tiết gastrin. Các khối u làm tăng nồng độ gastrin trong máu với tăng sản xuất acid dịch vị (hội chứng Zollinger - Ellison). Những bệnh nhân này có thể biểu hiện bằng loét dạ dày, tiêu chảy, suy kiệt và đau bụng, thường có nhiều chỗ và có nguy cơ trở thành ác tính. Hơn một nửa các u tiết gastrin trong MEN1 đã có di căn tới hạch trước khi được chẩn đoán. Những khối u này là nguyên nhân chính gây tử vong và tàn tật ở bệnh nhân MEN. Nó thường nằm ở tá tràng (>85%) và có thể đi kèm với các khối u tụy không tiết [6].
U tiết Insulin là u tụy phổ biến thứ hai (gặp ở 10% các bệnh nhân MEN1). Phần lớn các u tiết insulin có nguồn gốc tự phát vì chỉ dưới 5% các bệnh nhân u tiết insulin có hội chứng MEN1. Các bệnh nhân thường có biểu hiện hạ đường máu lúc đói. Các xét nghiệm lúc hạ đường máu thấy tăng nồng độ insulin, C-peptide và proinsulin không thích hợp gợi ý khả năng cao là do u tiết insulin. Các khối u thường rất nhỏ nên khó phát hiện bằng chụp CT hay chụp MRI nhưng có thể phát hiện được khi làm siêu âm tụy trong khi mổ. (Xem thêm chi tiết ở Chương 38).
U tuyến yên. Adenoma thùy trước tuyến yên là u xuất hiện đầu tiên ở 10 - 25% các trường hợp MEN1. Hai phần ba là microadenomas, thường là u có tiết, chủ yếu là tiết prolactin, gây ra các triệu chứng của quá thừa prolactin (vô kinh và tiết sữa ở phụ nữ, bất lực ở nam). Gần một phần tư các u này có tiết GH, gây bệnh to đầu chi và một tỷ lệ nhỏ hơn có tiết ACTH gây bệnh Cushing. Biểu hiện, chẩn đoán và điều trị tương tự như các u tuyến yên tản phát (xem chương 5).
Các u Carcinoid xuất hiện ở dưới 3% các bệnh nhân MEN1. Gần như toàn bộ các u carcinoid trong MEN1 có nguồn gốc từ các mô phát sinh từ ruột trước của phôi thai. Carcinoid tuyến ức gặp chủ yếu ở nam giới, có thể không có triệu chứng cho tới giai đoạn muộn và có xu hướng nặng hơn các u tản phát. Ngược lại các u carcinoid phế quản gặp chủ yếu ở nữ giới, có thể tiết ACTH và có thể gây hội chứng Cushing. Các u carcinoid ở dạ dày với các tế bào giống tế bào ưa chrom ruột có thể được phát hiện tình cờ khi soi dạ dày cho những bệnh nhân MEN1 có u tiết gastrin. Hội chứng carcinoid thường không xảy ra trừ khi khối u di căn đến gan (xem Chương 38).
Các khối u khác. Các bệnh nhân MEN1 có thể có biểu hiện với đa khối u mỡ, u xơ mạch ở mặt và u tế bào Collagen. Các khối u vỏ thượng thận, cả tiết và không tiết hormon, xảy ra ở 5 - 40% những bệnh nhân MEN1.
Lưu ý, tăng cortisol máu có thể phụ thuộc ACTH (u tuyến yên hoặc u tiết ACTH ngoại lai) hoặc không phụ thuộc ACTH (adenoma tuyến thượng thận). Mặc dù về mặt thống kê thì đa số các trường hợp là u tuyến yên nhưng vẫn cần chẩn đoán phân biệt các nguyên nhân khác nhau dựa vào các xét nghiệm sinh hóa (xem Chương 15).
1.3.4 MEN2
Các biểu hiện của MEN2 phụ thuộc nhiều vào các phân nhóm (xem Bảng 37.2) [1,2]. Tuy nhiên đặc điểm chung cơ bản ở hầu hết các bệnh nhân MEN2 là sự xuất hiện của ung thư tuyến giáp thể tủy, nó là nguyên nhân phổ biến nhất gây tử vong và tàn phế ở các bệnh nhân hội chứng MEN2.
Ung thư tuyến giáp thể tủy là biểu hiện lâm sàng đầu tiên ở các gia đình MEN2 vì khả năng xâm lấn sớm hơn và cao hơn. Giai đoạn trước của ung thư tuyến giáp thể tủy là quá sản tế bào C, với kết quả là tiết Calcitonin và CEA (carcinoembryonic antigen), có vai trò như là một chi dấu khối u trong máu rất giá trị. Quá sản tế bào C sẽ tiến triển thành ung thư tuyến giáp thể tủy vi thể, tiếp đó là ung thư tại chỗ (thường là nhiều ổ) và thậm chí là di căn (phổ biến là đến hạch, phổi, gan và xương).
Ung thư tuyến giáp thể tủy thường biểu hiện như là nhân giáp và/hoặc tăng calcitonin máu.
Ung thư tuyến giáp thể tủy thường ác tính hơn trong MEN2B, đa số xuất hiện trước 5 tuổi và có thể sớm ngay ở tuổi thơ ấu.
Ung thư tuyến giáp thể tủy là biểu hiện duy nhất của ung thư tuyến giáp thể tủy có tính chất gia đình và có diễn tiến lâm sàng thầm lặng. Ung thư tuyến giáp thể tủy có tính gia đình xuất hiện muộn, với lứa tuổi hay gặp nhất là 40 - 50. Nó có xu hướng ít ác tính hơn các phân nhóm khác của MEN2. Các tiêu chuẩn của dòng họ có ung thư tuyến giáp thể tủy có tính chất gia đình gồm có hơn 10 người trong dòng họ là bị ung thư tuyến giáp thể tủy, nhiều người bị bệnh hoặc nhiều thành viên trong gia đình tuổi trên 50 bị tác động và khai thác tiền sử đầy đủ để loại trừ pheochromocytoma hoặc cường cận giáp. Một số bệnh nhân MEN2A có thể chỉ biểu hiện bằng ung thư tuyến giáp thể tủy nên bị chẩn đoán nhầm là ung thư tuyến giáp thể tủy có tính chất gia đình, hậu quả là bỏ sót chẩn đoán pheochromocytoma.
U tuỷ thượng thận (pheochromocytoma). So với các trường hợp tản phát, pheochromocytoma trong MEN2 gần như luôn là lành tính, hai bên, chỉ khu trú trong tuyến thượng thận và xuất hiện sớm trong đời. Nếu không được chẩn đoán nó có thể biểu hiện như là cơn tăng huyết áp kịch phát trong khi phẫu thuật ung thư tuyến giáp thể tủy sớm ở trẻ nhỏ. Biểu hiện lâm sàng, chẩn đoán và điều trị bệnh giống như những những trường hợp tản phát (xem Chương 16).
Cường cận giáp nguyên phát gặp ở khoảng 1/3 số bệnh nhân MEN2A nhưng không gặp ở MEN2B. Nó thường do quá sản bốn tuyến không đối xứng nhưng không nặng bằng MEN1. Biểu hiện lâm sàng, chẩn đoán, điều trị tương tự như những bệnh nhân MEN1 và những trường hợp tản phát (xem chương 23). Điều quan trọng cần làm là đánh giá các tuyến cận giáp trong khi mổ cắt tuyến giáp ở các bệnh nhân MEN2A vì nó có thể to mặc dù nồng độ canxi trước mổ là bình thường.
U hạch thần kinh xảy ra ở 95% các bệnh nhân MEN2B, có thể biểu hiện ở môi, mí mắt, lưỡi, khiến cho những bệnh nhân này có kiểu hình đặc trưng có thể nhận ra ngay lúc sinh. U hạch thần kinh ở ruột có thể xảy ra sớm ở trẻ sơ sinh gây rối loạn nhu động dạ dày ruột. Một nghiên cứu báo cáo 90% các bệnh nhân có rối loạn nhu động đại tràng, thường gây táo bón mạn tính ngay từ lúc sinh [8].
Các đặc điểm khác của MEN2A/ung thư tuyến giáp thể tủy có tính chất gia đình: Bệnh amyloidois liken của da là tổn thương phối hợp phổ biến nhất với đột biến RET ở codon 634. Bệnh biểu hiện bằng ngứa rất nhiều, thường ở phần lưng trên và đôi khi rất khó điều trị [9]. MEN2A còn có thể có bệnh Hirschprung. Một nghiên cứu loạt ca bệnh thấy 50% số bệnh nhân có đột biến RET ở vị trí C620 có bệnh Hirschprung [10].
Các đặc điểm khác trong MEN2B: Các bệnh nhân MEN2B cũng có biểu hiện hình dáng marfan đặc trưng nhưng không có trật đồng tử hay bệnh động mạch chủ [11].
1.4 Tiêu chuẩn chẩn đoán
Chẩn đoán MEN1 nếu bệnh nhân có hai trong ba khối u chính liên đến MEN1: U cận giáp, u tuyến yên và u tụy ruột. Chẩn đoán MEN1 có tính chất gia đình khi có ít nhất một trường hợp MEN1 cộng với một người thân trực hệ có một trong ba u.
Chẩn đoán MEN2 ở một bệnh nhân có tiền sử cá nhân hoặc gia đình bị ung thư tuyến giáp thể tủy và có đột biến mầm gen RET. Đây là ví dụ điển hình về một bệnh trong đó xét nghiệm di truyền cho phép chẩn đoán sớm và can thiệp ngoại khoa dự phòng hiệu quả. Ở những
Ӧ bệnh nhân có u tuyến giáp nghi ngờ, chọc hút tế bào bằng kim nhỏ (FNA) có thể xác lập chẩn đoán ung thư tuyến giáp thể tủy.
Chẩn đoán MEN2A ở một bệnh nhân có tiền sử cá nhân hoặc gia đình bị ung thư tuyến giáp thể tủy, pheochromocytoma và cường cận giáp nguyên phát kèm theo đột biến gen RET. Có thể chẩn đoán lâm sàng MEN2A nếu có ít nhất 2 đặc điểm lâm sàng của MEN2A trong ca bệnh đầu tiên hoặc ở hai thế hệ, ngay cả khi không có di truyền trội có tính gia đình trên nhiễm sắc thể thường hoặc đột biến gen RET. Khi có đột biến gen RET và không có bất kỳ đặc điểm lâm sàng nào, bệnh nhân vẫn có nguy cơ xuất hiện đặc điểm lâm sàng của MEN2A nên tuân theo các biện pháp quản lý y tế phù hợp.
Ung thư tuyến giáp thể tủy có tính chất gia đình là một biến thể lâm sàng của MEN2A. Để chứng minh ung thư tuyến giáp thể tủy có tính chất gia đình, cần xác định không có pheochromocytoma hoặc cường cận giáp nguyên phát trong ít nhất hai thế hệ thuộc gia đình đó hoặc có đột biến RET được phát hiện chỉ ở họ hàng người ung thư tuyến giáp thể tủy có tính chất gia đình.
MEN2B được chẩn đoán khi một bệnh nhân có tiền sử cá nhân hoặc gia đình mắc ung thư tuyến giáp thể tủy và có đột biến gen RET ở vị trí 918 hoặc 883. Ca bệnh hoặc các thành viên trong gia đình có thể có các biểu hiện lâm sàng của ung thư tuyến giáp thể tủy, pheochromocytoma, các bệnh đi kèm với MEN2B khác như hình dạng marfan, u hạch thần kinh và u hạch thần kinh ở ruột.
1.5 Chẩn đoán cận lâm sàng
1.5.1 Cận lâm sàng
MEN1
Xét nghiệm chẩn đoán giống như những trường hợp tản phát. Các xét nghiệm nhằm tìm bằng chứng của cường cận giáp (PTH, canxi toàn phần và ion hóa), u tuyến yên (prolactin, ACTH, IGF-1, alpha subunit, TSH) và các u tụy ruột.
Xét nghiệm DNA để chẩn đoán và sàng lọc MEN1 vẫn còn tranh cãi, tuy nhiên các hướng dẫn thực hành hiện tại khuyến cáo thực hiện xét nghiệm di truyền MEN1 cho những bệnh nhân nghi ngờ cao trên lâm sàng và nếu dương tính thì thực hiện cho những người thân trực hệ của bệnh nhân [12,13].
MEN2
Không yêu cầu các xét nghiệm sinh hóa để chẩn đoán MEN2 nhưng nó có thể hữu ích cho chẩn đoán và theo dõi bệnh cường cận giáp và pheochromocytoma. Các xét nghiệm hữu ích gồm calcitonin, CEA, ca (PTH nếu nghi cường cận giáp), metanephrin và normetanephrin tự do trong máu và metanephrin và normetanephrin/nước tiểu 24 giờ.
Khi phát hiện ca bệnh đầu tiên (bệnh nhân bất kỳ bị ung thư tuyến giáp thể tủy), người đó nên được gửi đi tư vấn di truyền. Nội dung tư vấn nên bao gồm nhưng không chỉ giới hạn, về phạm vi và mức độ nghiêm trọng của bệnh, tác động tiềm tàng đến khả năng bảo hiểm, mặc cảm của người sống sót, khả năng không nên có con, trách nhiệm của bệnh nhân hoặc của người giám hộ có thẩm quyền trong việc thông báo cho các thành viên trong gia đình về xét nghiệm và lựa chọn xét nghiệm chẩn đoán trước sinh hoặc trước khi cấy phôi nếu bệnh nhân đang trong độ tuổi sinh đẻ.
Phần lớn các Labo sàng lọc năm codon đột biến phổ biến nhất ở các exons 10 (609, 611, 618, 620) và 11 (634) cho MEN2A và codons 918 và 883 cho MEN2B. Nếu phân tích ban đầu là âm tính, thì các exon còn lại sẽ được giải trình tự.
Để tìm labo có thể đáp ứng các nhu cầu chuyên biệt cho bệnh nhân của bạn, hãy vào trang web http://www.ncbi.nlm.nih.gov/gtr. Nếu kết quả xét nghiệm của bệnh nhân dương tính, chỉ nên mở rộng xét nghiệm di truyền phân tử RET đến người thân trực hệ của họ (bố mẹ và con cái). Nếu bố hoặc mẹ có kết quả dương tính, tất cả các thành viên trong gia đình đều có nguy cơ và nên được xét nghiệm tìm đột biến gen.
Các chỉ định chính của xét nghiệm di truyền phân tử gồm:
- Khẳng định chẩn đoán MEN2A, ung thư tuyến giáp thể tủy có tính gia đình và MEN2B.
- Sàng lọc các thành viên trong gia đình có nguy cơ trước khi có triệu chứng.
- Phát hiện các đột biến mầm để phân biệt ung thư tuyến giáp thể tủy tản phát và có tính gia đình.
Nếu một người có xét nghiệm âm tính với đột biến RET, người đó hầu như không có nguy cơ mắc hội chứng MEN2 (âm tính giả từ 2 - 5%).
Khả năng có đột biến RET mới hoặc bất thường cao là rất thấp. Mặc dù việc chẩn đoán người mang gen bệnh dựa hoàn toàn vào phân tích đột biến RET nhưng xét nghiệm calcitonin vẫn được sử dụng trong những trường hợp không thể xác định được người mang gen MEN2 bằng xét nghiệm ADN hoặc không phát hiện được đột biến RET.
Xét nghiệm gen RET không thay thế được các xét nghiệm sinh hóa cần thiết để phát hiện pheochromocytoma hoặc cường cận giáp ở bệnh nhân MEN2. Ngoài ra, xét nghiệm gen RET trước khi xuất hiện các triệu chứng không thể xác định được các đột biến tự phát chưa xảy ra.
1.5.2 Hình ảnh học
MEN1
Nên làm siêu âm vùng cổ cho bệnh nhân cường cận giáp nguyên phát. Xét nghiệm có độ nhạy từ 72 - 89% trong phát hiện các adenoma đơn độc.
Xạ hình bằng 99mTc-sestamibi. Độ nhạy tương đương với siêu âm, từ 68 - 95%, trong phát hiện các adenoma đơn độc. Một ưu điểm của xạ hình là nó có thể phát hiện được các tuyến lạc chỗ nằm ngoài vùng cổ. Do đó, một số người thích kết hợp các phương pháp để đánh giá trước mổ, nó được chứng minh dự báo chính xác các adenoma hơn các phương pháp riêng rẽ.
Nên chụp cộng hưởng từ tuyến yên khi các xét nghiệm sinh hóa gợi ý có u tuyến yên. Cộng hưởng từ là phương pháp nhạy và đặc hiệu nhất trong chẩn đoán các tổn thương ở tuyến yên.
Xạ hình 111-Indium-penetreotide (octreoscan) gắn với thụ thể Somatostatin và chụp SPECT (single-photon emission computed tomography) có độ nhạy cao hơn tất cả các phương pháp chẩn đoán hình ảnh khác trong việc xác định vị trí các khối u tụy ruột và đặc biệt hữu ích trong việc phát hiện các di căn xương và gan. Các tổn thương dương tính với chụp PET-FDG (Fluorodeoxyglucose-positron emission tomography) tiến triển nặng hơn và có thể ảnh hưởng đến quyết định phẫu thuật hay chỉ theo dõi các khối u thần kinh nội tiết ở tụy [14]. Chụp PET với Ga Dotatate là kỹ thuật mới có khả năng phát hiện các khối u thần kinh nội tiết ở tụy tương đương nhưng có ưu điểm là phát hiện được cả các tổn thương ở tuyến yên, tuyến cận giáp và tuyến thượng thận [15].
Siêu âm nội soi đặc biệt có giá trị trong phát hiện các khối u nội tiết nhỏ ở tụy.
MEN2
Siêu âm vùng cổ phải do chuyên gia siêu âm giàu kinh nghiệm thực hiện để kiểm tra các vùng trung thất trên và vùng cổ trung tâm và vùng cổ bên ở các bệnh nhân MEN2 hoặc người nghi ngờ ung thư tuyến giáp thể tủy.
Chụp CT ngực, CT cổ, chụp CT gan đa đầu dò 3 pha có tiêm thuốc cản quang hoặc chụp MRI có thuốc cản quang được chỉ định khi nghi ngờ có ung thư tuyến giáp thể tủy di căn.
Cần chụp CT hoặc MRI ổ bụng để xác định vị trí pheochromocytomas, nó thường nằm trong tuyến thượng thận ở bệnh nhân MEN2. Xem thêm chi tiết ở Chương 16.
1.5.3 Thủ thuật chẩn đoán
MENI
Chẩn đoán và theo dõi các bệnh lý thành phần trong MEN1 dựa chủ yếu vào các xét nghiệm sinh hóa và hình ảnh.
Sinh thiết qua nội soi có thể hữu ích trong chẩn đoán một số khối u tụy ruột. Sinh thiết da có thể hữu ích trong chẩn đoán các khối u da đi kèm với MEN1, như là u xơ mạch và u collagen.
MEN2
Chọc tế bào kim nhỏ là an toàn cho chẩn đoán ung thư tuyến giáp thể tủy và các hạch lympho nghi ngờ di căn. Xét nghiệm calcitonin trong dịch chọc hút kim nhỏ từ các ổ nghi tái phát hay các hạch nghi di căn có thể có độ nhạy và độ đặc hiệu cao hơn.
Sinh thiết da có thể hữu ích trong chẩn đoán các u hạch thần kinh trong MEN2B và bệnh amyloid liken da trong MEN2A.
Sinh thiết trực tràng hoặc sinh thiết ruột qua nội soi có thể hữu ích trong chẩn đoán các u hạch thần kinh.
2 ĐIỀU TRỊ
MEN1
Không có chỉ định điều trị cho đến khi bệnh có các biểu hiện lâm sàng hoặc sinh hóa. Tuy nhiên, một bệnh nhân đã biết có đột biến MEN1 nên được theo dõi chặt bằng chứng các khối u đặc trưng liên quan với hội chứng này.
Cường cận giáp
- Nguyên tắc và phương thức điều trị cường cận giáp trong MEN1 tương tự như cường cận giáp tản phát. Xem chi tiết ở Chương 23.
- Cắt tuyến cận giáp xâm lấn tối thiểu không được khuyến cáo, vì cường cận giáp ở bệnh nhân MEN1 luôn có sự quá sản của cả 4 tuyến, do đó phương pháp phẫu thuật phổ biến nhất là cắt 4 tuyến cận giáp kèm cấy ghép tự thân hoặc cắt 3,5 tuyến cận giáp [13].
- Giảm nồng độ PTH trong mổ > 50% so mức ban đầu chứng tỏ đã cắt bỏ đủ mô cận giáp. Tỷ lệ tái phát khá cao. Một nghiên cứu chùm ca bệnh theo dõi trong hơn 20 năm, thấy các bệnh nhân được cắt bỏ ít hơn 3 tuyến cận giáp có tỷ lệ tái phát cao (55%) trong khi cắt toàn bộ tuyến cận giáp kèm ghép tự thân có tỷ lệ cao bị suy cận giáp sau mổ cần phải điều trị thay thế (50%) [16].
- Thuốc Calcimimetic (cinacalcet) có vai trò trong điều trị tăng canxi máu dai dẳng hoặc tái phát sau mổ ở các bệnh nhân MEN1 [17].
Các u thần kinh nội tiết tụy: Điều trị nội khoa và phẫu thuật được thảo luận riêng dưới đây. Các công nghệ mới đang đem đến nhiều phương thực điều trị hơn. Liệu pháp phóng xạ hạt nhân thụ thể peptide cải thiện sống sót không tiến triển và tỷ lệ sống sót chung ở các bệnh nhân u thần kinh nội tiết tụy có tiết và không tiết hormon [18].
U tiết Gastrin (hội chứng Zollinger - Ellison)
- Thuốc ức chế bơm proton là điều trị được lựa chọn để kiểm soát hiệu quả tình trạng tăng gastrin máu nhưng nó được dùng với liều cao gấp đôi thông thường (ví dụ omeprazol uống 40 mg/ngày, pantoprazol uống 80 mg/ngày).
- Vai trò của phẫu thuật trong điều trị u tiết gastrin trong MEN1 vẫn còn tranh cãi. Cả hướng dẫn của NANETS và ENETS khuyến cáo hướng điều trị bảo tồn đối với bệnh nhân MEN1 có các u nhỏ mà các triệu chứng đã được kiểm soát [19,20]. Các bệnh nhân có u tiết gastrin nhỏ (< 2 cm) ở tụy và tá tràng không phẫu thuật có tỷ lệ sống thêm 15 năm từ 90 - 100% [6]. Tuy nhiên, ở những bệnh nhân MEN1 đã có di căn, tỷ lệ khỏi bệnh gần như bằng 0 với tỷ lệ sống thêm 10 năm ở bệnh nhân đã có di căn gan là từ 15 - 25% [4]. Do đó, phẫu thuật thường được dành cho những bệnh nhân: (a) kháng trị hoặc không dung nạp với điều trị nội khoa; (b) có u tiết gastrin > 2 cm; (c) có nguy cơ di căn cao (tiền sử gia đình) và (d) không có di căn gan.
- Các bệnh nhân vẫn còn u tiết gastrin hoặc u tái phát sau phẫu thuật có thể phẫu thuật lại hoặc điều trị nội khoa với 5-FU, Octreotide hoặc interferon.
- Với những bệnh nhân có di căn gan, phẫu thuật làm giảm khối u tối đa và/hoặc nút hóa chất động mạch gan có thể được chỉ định. Cũng có thể cân nhắc hóa trị liệu với streptozotocin và Doxorubicin cho những bệnh nhân u tiết gastrin đã có di căn. Tuy nhiên, phương pháp phù hợp nên là theo dõi do nhiều bệnh nhân ít có di căn và hiệu quả tương đối thấp của các thuốc này.
U tiết Insulin
- Phẫu thuật là điều trị được lựa chọn cho u tiết insulin và thường là chữa khỏi bệnh. Với các u tế bào tiểu đảo khác, phẫu thuật vẫn được chỉ định đầu tiên vì điều trị nội khoa đơn thuần không có hiệu quả.
- U tái phát được điều trị triệu chứng bằng các thuốc như octreotide.
Các u có tiết khác, u tiết VIP, u tiết glucagon và u tiết somatostatin.
- Những u này hiếm gặp nhưng có nguy cơ ác tính cao. Từ 30 - 50% các bệnh nhân đã có di căn khi có biểu hiện lâm sàng.
- Điều trị nội khoa đầu tay thường là đồng phân somatostatin tác dụng kéo dài. Có thể cần cho thêm a-Interferon để kiểm soát các triệu chứng [20].
- Phẫu thuật làm giảm tối đa khối u được chỉ định cho những trường hợp kháng trị nhưng còn tranh cãi vì không có các nghiên cứu đủ mạnh để đánh giá hiệu quả của phẫu thuật lên thời gian sống thêm hoặc cải thiện triệu chứng [20].
- Những bệnh nhân đã có di căn gan có thể được chỉ định đốt sóng cao tần, đốt lạnh, nút mạch hoặc nút hóa chất động mạch gan [18-20].
Các khối u tá- tụy (pancreaticcoduodenal tumors) không tiết
- Phần lớn các chuyên gia chỉ định phẫu thuật để điều trị (khỏi) hoặc dự phòng u chuyển thành ác tính và di căn nếu u > 2 cm [21]. Phổ biến là cắt khối u đầu tụy và đồng thời cắt (80%) đuôi và gần toàn bộ thân tụy. Tuy nhiên, có rất ít dữ liệu về thời điểm phẫu thuật.
Các khối u tân sinh trong lồng ngực
- Các khối u carcinoid phế quản và tuyến ức hiếm gặp với tỷ lệ mắc ước tính ở các bệnh nhân MEN1 lần lượt là 4,9% và 2,0% [22]. U carcinoid tuyến ức hầu như chỉ xảy ra ở nam giới và có tiên lượng tồi [22]. Cắt tuyến ức qua vùng cổ, được khuyến cáo cùng lúc với mổ cắt tuyến cận giáp, để làm giảm tỷ lệ tử vong và tàn phế [13].
U tuyến yên và các u khác: điều trị tương tự những bệnh nhân mắc các u này tản phát. Sau điều trị phẫu thuật hoặc điều trị nội khoa, các bệnh nhân nên được theo dõi về nguy cơ u còn tồn tại hoặc tái phát.
MEN2
Ung thư tuyến giáp thể tủy
- Cắt toàn bộ tuyến giáp kèm theo nạo vét hạch vùng là điều trị được ưu tiên cho ung thư tuyến giáp thể tủy. Nếu chẩn đoán bệnh đã tiến triển nặng thì phẫu thuật cắt bỏ rộng có thể là phương pháp thích hợp cùng với cân nhắc xạ trị ngoài hoặc điều trị thuốc ức chế tyrosine kinase [9].
- Nếu biết có đột biến gen của ung thư tuyến giáp thể tủy từ trước khi bệnh xuất hiện, thời điểm phẫu thuật cắt tuyến giáp dự phòng, thường ở trẻ em, phụ thuộc vào codon đột biến RET. Có nhiều khuyến cáo đã được công bố. Dưới đây là hướng dẫn của Hội Tuyến giáp Hoa Kỳ đã được sửa đổi năm 2016 (xem Bảng 37.3) [9].
- Trường hợp đã có biểu hiện rõ trên lâm sàng, các xét nghiệm trước mổ nên bao gồm calcitonin, CEA và canxi (đã điều chỉnh theo Albumin máu và ion hóa) và xét nghiệm đột biến gen tiền ung thư RET. Hướng điều trị phẫu thuật và hóa trị liệu tiềm năng phụ thuộc vào mức độ lan rộng của bệnh trên thăm dò hình ảnh trước mổ (xem Bảng 37.4) [9].
- Nếu calcitonin trước mổ < 20, rất ít khả năng có di căn hạch [9].
- Với những bệnh nhân đã có di căn hạch vùng được phát hiện trên lâm sàng hoặc bằng siêu âm hoặc calcitonin>500 pg/dL, thì có thể cân nhắc làm thêm các chẩn đoán hình ảnh trước mổ như chụp CT ngực, CT cổ có tiêm thuốc cản quang, chụp CT gắn đa đầu dò 3 pha có tiêm thuốc cản quang hoặc chụp MRI có tiêm thuốc cản quang. Chụp cộng hưởng từ cắt ngang (Axial MRI) và xạ hình xương có thể được sử dụng để phát hiện di căn xương [23].
- Sau khi cắt tuyến giáp, các bệnh nhân được theo dõi bằng cách xét nghiệm calcitonin liên tục, vì calcitonin thường là chỉ dấu đầu tiên của ung thư tuyến giáp thể tủy còn tồn tại hoặc tái phát. Bệnh tại chỗ có thể được phẫu thuật cắt bỏ, còn di căn đã lan rộng thì rất khó chữa vì hóa trị liệu và xạ trị thường quy kém hiệu quả, tuy nhiên có 2 thuốc ức chế tyrosine kinase đã được chấp thuận cho điều trị ung thư tuyến giáp thể tủy tại chỗ giai đoạn muộn hoặc đã có di căn (Vandetanib và cabozantanib), ngoài ra một số thuốc khác đang được nghiên cứu [9].
Pheochromocytoma: điều trị pheochromocytoma trong MEN2 tương tự như các trường hợp tản phát (xem Chương 16). Nếu pheochromocytoma được phát hiện cùng lúc với ung thư tuyến giáp thể tủy, thì nên thực hiện cắt tuyến thượng thận nội soi trước khi cắt tuyến giáp với điều trị thuốc chẹn giao cảm phù hợp để tránh cơn tăng huyết áp trong mổ.
Cường cận giáp:
- Những người có nguy cơ bị cường cận giáp (đột biến ở các codon 609, 611, 618, 620, 630 634, 804, 891) nên được sàng lọc hằng năm bắt đầu từ tuổi 15 bằng các xét nghiệm canxi ion hóa (hoặc canxi toàn phần và albumin) và hormon cận giáp toàn phần (intact PTH).
- Điều trị cường cận giáp bằng cắt tuyến cận giáp gần toàn bộ hoặc toàn bộ cùng với ghép tự thân. Nếu phát hiện quá sản tuyến cận giáp khi mổ tuyến giáp, thì nên coi như là cường cận giáp ngay cả khi không có bằng chứng sinh hóa [9].
| Bảng 37.3. Khuyến cáo khi nào nên thực hiện cắt tuyến giáp | |||
|
Nguy cơ cao nhất (ATA-HST) |
Nguy cơ cao (ATA-H) | Nguy cơ trung bình (ATA-MOD) | |
| Khuyến cáo phẫu thuật | Cắt tuyến giáp < 1 tuổi, có thể nạo vét hạch trung tâm | Cắt tuyến giáp < 5 tuổi | Cắt tuyến giáp trước khi calcitonin tăng và khi nhân tuyến giáp < 5 mm |
| Đột biến gen | M918T (MEN2b) | C634, A883F | Tất cả các đột biến khác |
| Tuổi bắt đầu phải sàng lọc pheochromocytoma | 11 tuổi | 11 tuổi | 16 tuổi |
| Phỏng theo Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25:567-610. | |||
| Bảng 37.4. Hướng điều trị phẫu thuật của ung thư tuyến giáp thể tuỷ | |||
| Chẩn đoán hình ảnh phát hiện bệnh tại chỗ | Chẩn đoán hình ảnh phát hiện đã có di căn hạch | Chẩn đoán hình ảnh phát hiện di căn xa | |
|
Cắt tuyến giáp Nạo vét hạch cổ trung tâm |
Có | Có | Có |
| Nạo vét hạch cổ cùng bên | Không | Có | Phẫu thuật làm giảm khối u tối đa |
| Nạo vét hạch cổ đối bên | Không | (Nếu calcitonin > 200) | Phẫu thuật làm giảm khối u tối đa |
| Thuốc ức chế tyrosine kinase | Không | (Nếu tiến triển nhanh) | (Nếu tiến triển nhanh) |
| Phỏng theo Wells SA Jr, Asa SL, Dralle H, et al. Revised American Thyroid Association guidelines for the management of medullary thyroid carcinoma. Thyroid 2015;25:567-610. | |||
2.1 Giáo dục bệnh nhân
Tư vấn cho các bệnh nhân và gia đình là một phần quan trọng của điều trị bệnh. Nên gửi họ đi tư vấn di truyền. Các website hữu ích gồm http:// ghr.nlm.nih.gov/condition/multiple-endocrine-neoplasia và http://www. amend.org.uk/.
3 KIỂM TRA VÀ THEO DÕI
3.1 MEN1
Khi phát hiện ca bệnh MEN1 đầu tiên nên cân nhắc xét nghiệm và sàng lọc di truyền nhưng không bắt buộc cho tất cả các thành viên trong gia đình. Tuổi bắt đầu cần sàng lọc vẫn còn tranh cãi. Phân tích trực tiếp DNA tìm đột biến trong gen gây bệnh MEN1 xác định những bệnh nhân có nhận một allele đột biến và có khả năng xuất hiện MEN1.
Khi phát hiện một người có nguy cơ cao bị MEN1 (xét nghiệm gen dương tính hoặc tiền sử gia đình), thì nên sàng lọc sinh hóa định kỳ để phát hiện các triệu chứng liên quan đến dư thừa hormon liên quan với các u đặc trưng trong MEN1. Tuy nhiên, điều trị dự phòng không có tác dụng ở các bệnh nhân MEN1. Các hướng dẫn đồng thuận hiện tại đề xuất sàng lọc các u ở người có xét nghiệm gen khẳng định đột biến MEN1 hoặc chẩn đoán MEN1 [24,25], xem Bảng 37.5 [13].
3.2 MEN2
Ung thư tuyến giáp thể tủy
- Tất cả các bệnh nhân được cho là ung thư tuyến giáp thể tủy tản phát nên được xét nghiệm đột biến gen RET vì 40 - 50% các trường hợp giả định là tản phát được chứng minh là có đột biến RET .
- Với những bệnh nhân có đột biến RET được chọn chờ phẫu thuật cắt tuyến giáp dự phòng nên tiến hành xét nghiệm calcitonin và siêu âm cổ mỗi 6-12 tháng.
- Với những bệnh nhân đã đạt tiêu chí khỏi bệnh hoàn toàn về sinh hóa, theo dõi lâu dài về xét nghiệm sinh hóa nồng độ calcitonin hằng năm được khuyến cáo.
- Với những bệnh nhân có nồng độ calcitonin nền sau mổ tăng nên tiến hành xét nghiệm nồng độ calcitonin nền và CEA khoảng mỗi 6 tháng để xác định thời gian tăng gấp đôi. Việc xét nghiệm theo dõi các dấu ấn khối u này và khám thực thể nên thực hiện ở một phần tư thời gian tăng gấp đôi ngắn nhất hoặc hằng năm, tùy theo thời điểm nào ngắn hơn. Thời điểm thực hiện theo dõi hình ảnh giải phẫu có thể dựa trên tính ổn định của các xét nghiệm này, có hay không có triệu chứng và vị trí di căn đã biết hoặc vị trí nhiều khả năng có di căn đến.
- Những bệnh nhân ở trong một gia đình thỏa mãn các tiêu chuẩn lâm sàng của MEN2A hoặc 2B hoặc ung thư tuyến giáp thể tủy có tính chất gia đình nhưng không phát hiện có đột biến gen RET, thì nên được sàng lọc định kỳ ung thư tuyến giáp thể tủy (siêu âm cổ, xét nghiệm calcitonin), cường cận giáp nguyên phát (canxi hiệu chỉnh theo albumin hoặc canxi ion hóa) và pheochromocytoma (metanephrines và normetanephrines tự do trong máu hoặc metanephrines và normetanephrines niệu/24 giờ) theo kiểu hình của gia đình. Nên sàng lọc liên tục mỗi 1 – 3 năm, ít nhất cho đến tuổi 50 hoặc 20 năm quá tuổi được chẩn đoán ban đầu lớn nhất trong gia đình, tùy theo tuổi nào lớn nhất.
Pheochromocytoma
- Sàng lọc pheochromocytoma ở tất cả các bệnh nhân MEN2 bằng xét nghiệm hằng năm metanephrin phân đoạn trong máu và/hoặc nước tiểu. Tuổi bắt đầu sàng lọc cũng phụ thuộc vào codon đột biến cụ thể. Sàng lọc bắt đầu giữa tuổi 10 - 15 ở các gia đình có nguy cơ đột biến cao (codon 918, 634 và 883) và sau tuổi 16 ở các gia đình có đột biến ở các codon nguy cơ thấp hơn. Nếu xét nghiệm sinh hóa bất thường thì tiếp đó sẽ chụp CT hoặc MRI để xác định vị trí khối u.
- Những phụ nữ có đột biến RET đi kèm MEN2 mà có thai hoặc dự kiến có thai nên được sàng lọc pheochromocytoma về sinh hóa.
- Các bệnh nhân pheochromocytoma nên được xét nghiệm sinh hóa sau mổ từ 4 - 6 tuần, rồi hằng năm nếu huyết áp được kiểm soát. Nếu cắt cả 2 tuyến thượng thận, khoảng cách sàng lọc có thể được kéo dài ra vì rất hiếm gặp các pheochromocytoma ngoài thượng thận trong hội chứng MEN.
Cường cận giáp: Xem phần MEN1 nêu trên.
| Bảng 37.5. Đề xuất sàng lọc các u ở người có MEN1 | ||||
| Xét nghiệm sinh hóa | Tần suất | Chẩn đoán hình ảnh | Tần suất | |
| Cường cận giáp | Alb, Ca toàn phần hoặc ion hóa, PTH. | Hằng quý | Không | |
| U tụy ruột |
Gastrin, glucagon, VIP, pancreatic polypeptide, chromogranin A, glucose và insulin lúc đói.
|
Hằng quý |
CT, MRI ổ bụng hoặc siêu âm nội soi |
Không rõ ràng |
| U tuyến yên | Prolactin, IGF-1; xét nghiệm thêm khác nếu chụp phát hiện u. | Hằng quý. | MRI tuyến yên theo protocol | Mỗi 3 - 5 năm/ lần |
| U tuyến ức, phế quản phổi và dạ dày | Không | CT hoặc MRI từ ngực Nội soi và sinh thiết. | Mỗi 1-2 năm/lần Mỗi 3 năm/lần | |
| U thượng thận | Nghiệm pháp ức chế 1 mg dexamethason qua đêm, renin: aldosteron cho các u > 1 cm. | CT hoặc MRI ngực | Mỗi 3 năm/lần | |
| Phỏng theo Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97:2990-3011. | ||||
4 KẾT CỤC/ TIÊN LƯỢNG
4.1 MEN1
Không có nhiều tài liệu về tỷ lệ tử vong ở bệnh nhân MEN1. Ung thư tụy và carcinoid ác tính của tuyến ức là những nguyên nhân chính gây tử vong liên quan đến bệnh ở MEN1[26]. Báo cáo của viện chúng tôi cho thấy gần một nửa các bệnh nhân (46%) chết do các nguyên nhân liên quan đến MEN1 ở tuổi trung bình là 50. Một báo cáo từ Mayo Clinic cho biết 28% bệnh nhân chết do các nguyên nhân có liên quan đến MEN1, phổ biến nhất là ung thư tiểu đảo (tụy) di căn (58.8%). Tỷ lệ sống thêm 20 năm của các bệnh nhân MEN1 là 64%, so với 81% của nhóm chứng cùng tuổi, cùng giới [27].
Rất ít bệnh nhân tử vong vì cường cận giáp. Ung thư tuyến cận giáp rất hiếm gặp nhưng cũng đã được báo cáo [28]. Rắc rối chủ yếu của cường cận giáp là tái phát sau mổ. Tỷ lệ tái phát sau mổ cắt tuyến cận giáp toàn bộ hoặc gần toàn bộ trong vòng 10 năm ở bệnh nhân MEN1 cao đến 55% [29].
U tiết insulin ác tính là nguyên nhân chính gây tử vong liên quan đến bệnh. Tỷ lệ ác tính của u tiết insulin trong MEN1 cao hơn so với u tiết insulin tản phát. Lúc được chẩn đoán, đã có hơn 50% các bệnh nhân có di căn.
U tiết gastrin trong MEN1 cũng có tỷ lệ ác tính cao hơn. Tỷ lệ điều trị khỏi bằng phẫu thuật là rất thấp (0 - 10%). Đa số (50 - 70%) các bệnh nhân có u>2 cm trên phim chụp đã có di căn hạch. Tuy nhiên, những bệnh nhân này có tỷ lệ sống thêm lâu dài không phẫu thuật là rất tốt, ngay cả khi đã có di căn thì tỷ lệ sống thêm 15 năm vẫn là 52% [30].
Các khối u tụy không tiết hiện là lý do phổ biến nhất yêu cầu phải phẫu thuật [31]. Các khối u không tiết chiếm từ 35 - 55% tổng số các khối u tụy nội tiết và biểu hiện chủ yếu ở độ tuổi từ 40 đến 60. Xấp xỉ hai phần ba các khối u tụy nội tiết không tiết là ác tính.
4.2 MEN2
Về lâu dài, tất cả các bệnh nhân đều xuất hiện ung thư tuyến giáp thể tủy, nó là nguyên nhân chính gây tử vong liên quan đến MEN2. Theo hệ thống phân loại TNM trước đây, tỷ lệ sống thêm 10 năm của bệnh nhân giai đoạn I, II, III và IV lần lượt là 100%, 93%, 71% và 21% [32]. Các nghiên cứu mới gợi ý rằng đáp ứng ban đầu với phẫu thuật (calcitonin vẫn tăng hoặc có bằng chứng còn sót tổ chức bệnh) có thể cung cấp thêm thông tin cho phân giai đoạn TNM để dự đoán tỷ lệ tử vong [33-35].
Ung thư tuyến giáp thể tủy ở bệnh nhân MEN2B ác tính hơn ung thư tuyến giáp thể tủy trong MEN2A hay ung thư tuyến giáp thể tủy có tính chất gia đình và phẫu thuật chỉ có khả năng chữa khỏi trước khi bệnh có biểu hiện lâm sàng [9].
Pheochromocytoma xuất hiện ở xấp xỉ 40 - 50% các bệnh nhân MEN2A và có lẽ tỷ lệ ở bệnh nhân MEN2B cũng tương đương. Rất hiếm gặp các trường hợp ác tính. Sự xuất hiện pheochromocytoma dường như không làm tồi thêm tiên lượng chung ở các bệnh nhân MEN2, đặc biệt khi có đột biến C634 [9].
Cường cận giáp nguyên phát xảy ra ở 10 – 25% các bệnh nhân MEN2A và gần như luôn là cường đa tuyến. Tại các trung tâm phẫu thuật tuyến cận giáp lớn, tỷ lệ tái phát sau cắt thành công gần toàn bộ các tuyến cận giáp là rất thấp.
5 TÀI LIỆU THAM KHẢO
1. Brandi ML, Gagel RF, Angeli A, et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2. J Clin Endocrinol Me- tab 2001;86(12):5658-5671.
2. Lakhani VT, You YN, Wells SA. The multiple endocrine neopla- sia syndromes. Ann Rev Med 2007;58:253-265.
3. Wells SA, Pacini F, Robinson BG, Santoro M. Multiple endocrine neoplasia type 2 and familial medullary thyroid carcinoma: An update. J Clin Endocrinol Me tab 2013;98(8):3149–3164.
4. Pellegata NS. MENX and MEN4. Clinics (Sao Paulo) 2012;67 (Suppl 1), 13-18.
5. Conemans EB, Brosens LAA, Raicu-Ionita GM, et al. Prognos- tic value of WHO grade in pan- creatic neuro-endocrine tumors in multiple endocrine neoplasia type 1: Results from the Dutch- MEN1 Study Group. Pancreatolo- gy 2017;17(5):766–772.
6. Ito T, Igarashi H, Jensen RT. Zollinger-Ellison syndrome: Recent advances and controversies. Curr Opin Gastroenterol 2013; 29 (6): 650-661.
7. Wohllk N, Schweizer H, Erlic Z, et al. Multiple endocrine neoplasia type 2. Best Pract Res Clin Endo- crinol Metab 2010;24(3):371-387.
8. American Thyroid Association Guidelines Task Force, Kloos RT, Eng C, et al. Medullary thyroid cancer: Management guidelines of the American Thyroid Associa- tion. Thyroid 2009;19(6):565-612.
9. Wells SA, Asa SL, Dralle H, et al. Revised American Thyroid Asso- ciation guidelines for the manage- ment of medullary thyroid carcino- ma. Thyroid 2015; 25(6):567-610.
10. Butter A, Gagné J, Al-Jazaeri A, Emran MA, Deal C, St-Vil D. Prophylactic thyroidectomy in pediatric carriers of multiple endo- crine neoplasia type 2A or familial medullary thyroid carcinoma: Mutation in C620 is associated with Hirschsprung's disease. J Pediatr Surg 2007;42(1):203-206.
11. Eng C, Clayton D, Schuffenecker I, et al. The relationship between specific RET proto-oncogene mutations and disease phenotype in multiple endocrine neoplasia type 2. International RET mutation consortium analysis. JAMA 1996; 276 (19):1575-1579.
12. Newey PJ, Thakker RV. Role of multiple endocrine neoplasia type 1 mutational analysis in clinical practice. Endocr Pract 2011;17 (Suppl 3):8-17.
13. Thakker RV, Newey PJ, Walls GV, et al. Clinical practice guidelines for multiple endocrine neoplasia type 1 (MEN1). J Clin Endocrinol Metab 2012;97(9):2990–3011.
14. Kornaczewski Jackson ER, Poin- ton OP, Bohmer R, Burgess JR. Utility of FDG-PET imaging for risk stratification of pancreatic neuroendocrine tumors in MEN1. J Clin Endocrinol Metab 2017; 102(6):1926-1933.
15. Lastoria S, Marciello F, Faggiano A, et al. Role of (68)Ga-DO- TATATE PET/CT in patients with multiple endocrine neo- plasia type 1 (MEN1). Endo- crine 2016;52(3):488–494.
16. Fyrsten E, Norlen O, Hessman O, Stalberg P, Hellman P. Long-term surveillance of treated hyperpara- thyroidism for multiple endocrine neoplasia type 1: Recurrence or hypoparathyroidism? World J Surg 2016; 40(3): 615–621.
17. Moyes VJ, Monson JP, Chew SL, Akker SA. Clinical use of Cinacalcet in MEN1 hyper- parathyroidism. Int J Endocri- nol 2010;2010:906163.
18. Katona BW, Roccaro GA, Sou- len MC, et al. Efficacy of peptide receptor radionuclide therapy in a United States-based cohort of metastatic neuroendocrine tu- mor patients: Single-institution retrospective analysis. Pancre- as 2017;46(9):1121–1126.
19. Falconi M, Eriksson B, Kaltsas G, et al. ENETS consensus guidelines update for the management of patients with functional pancre- atic neuro-endocrine tumors and non-functional pancreatic neuroen- docrine tumors. Neuroendocrinolo- gy 2016; 103(2):153–171.
20. Kulke MH, Anthony LB, Bush- nell DL, et al. NANETS treatment guidelines: Well-differentiated neuro-endocrine tumors of the stomach and pancreas. Pancre- as 2010;39(6):735-752.
21. Nell S, Verkooijen HM, Pieterman CRC, et al. Management of MEN1 related nonfunctioning pancre- atic NETS: A shifting paradigm: Results from the dutchMEN1 study group. Ann Surg 2018; 267(6):1155-1160.
22. Singh Ospina N, Thompson GB, Nichols F, Cassivi SD, Young, WF. Thymic and bronchial carcinoid tumors in multiple endocrine neo- plasia type 1: The Mayo Clinic Ex- perience from 1977 to 2013. Horm Cancer 2015;6(5-6):247-253.
23. Machens A, Dralle H. Biomark- er-based risk stratification for previously untreated medullary thyroid cancer. J Clin Endocrinol Metab 2010;95(6), 2655–2663.
24. Falchetti A. Genetic screening for multiple endocrine neoplasia syn- drome type 1 (MEN-1): When and how. F1000 Med Rep 2010;2:14.
25. Waldmann J, Fendrich V, Habbe N, et al. Screening of patients with multiple endocrine neopla- sia type 1 (MEN-1): A critical analysis of its value. World J Surg 2009;33(6):1208–1218.
26. Goudet P, Murat A, Binquet C, et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs En- docrines) cohort study among 758 patients. World J Surg 2010;34(2): 249-255.
27. Dean PG, van Heerden JA, Far- ley DR, et al. Are patients with multiple endocrine neoplasia type I prone to premature death? World J Surg 2000; 24(11):1437-1441.
28. Shih RY, Fackler S, Maturo S, True MW, Brennan J, Wells D. Parathyroid carcinoma in multiple endocrine neoplasia type 1 with a classic germline mutation. Endocr Pract 2009;15(6):567–572.
29. Tonelli F, Marcucci T, Giudi- ci F, Falchetti A, Brandi ML. Surgical approach in hereditary hyper-parathyroidism. Endocr J 2009;56(7):827-841.
30. Norton JA. Surgical treatment and prognosis of gastrinoma. Best Pract Res Clin Gastroenterol 2005; 19(5):799–805.
31. Lairmore TC, Chen VY, DeBene- detti MK, Gillanders WE, Norton JA, Doherty GM. Duodenopan- creatic resections in patients with multiple endocrine neoplasia type 1. Ann Surg 2000;231(6):909-918.
32. Cupisti K, Wolf A, Raffel A, et al. Long-term clinical and biochemi- cal follow-up in medullary thyroid carcinoma: A single institution's experience over 20 years. Ann Surg 2007;246(5):815–821.
33. Kwon H, Kim WG, Jeon MJ, et al. Dynamic risk stratification for medullary thyroid cancer according to the response to initial therapy. Endocrine 2016;53(1):174-181.
34. Lindsey S, Ganly I, Palmer F, Tuttle RM. Response to initial therapy predicts clinical outcomes in medullary thyroid cancer. Thy- roid 2015;25(2): 242-249.
35. Yang JH, Lindsey SC, Camacho CP, et al. Integration of a postop- erative calcitonin measurement into an anatomical staging system improves initial risk stratification in medullary thyroid cancer. Clin Endocrinol (Oxf) 2015;83(6): 938-942.
36. Thomas J.Braranski, MD, PhD; Janet B.McGill, MD, MA, FACE; Julie M.Silverstein, MD và các tác giả khác tham gia biên soạn, Khoa nội tiết chuyển hóa và nghiên cứu
