The Biocompatibility of Pharmaceutical Packaging Systems and Their Materials of Construction
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- 1. INTRODUCTION
- 2. SCOPE
- 3. OVERVIEW OF BIOCOMPATIBILITY EVALUATION
- 4. RISK-BASED APPROACH TO BIOCOMPATIBILITY EVALUATION
- 5. BIOLOGICAL REACTIVITY TEST CONSIDERATIONS
- 5.1 Test Article Selection and Sample Preparation Considerations
- 5.2 In Vitro Test Selection
- 5.3 Illustrative Examples Cytotoxicity Reactive Grades
- 5.4 Factors to Consider When Investigating a Biological Reactivity Test Failure
- 5.5 Risk-Based Approach to Investigating a Failed Biological Reactivity Test
- 5.6 Role of Chemical Assessment in Assessing Biological Reactivity
- 6. OVERALL BIOCOMPATIBILITY EVALUATION
- GLOSSARY
- APPENDIX
- REFERENCES
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
DOWNLOAD PDF HERE
1 1. INTRODUCTION
The purpose of this chapter is to provide guidance for biocompatibility evaluation of polymeric materials of construction and polymeric components for pharmaceutical packaging systems. Background information and principles for the application of Biological Reactivity Tests, In Vitro 〈87〉 and Biological Reactivity Tests, In Vivo 〈88〉 are provided. Additionally, principles for the toxicological evaluation of chemicals found during the application of Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems 〈1663〉 or Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 〈1664〉 are discussed.
Biological reactivity test results and/or chemical safety assessments (CSA) may be used to evaluate the biological impact of elastomers as described in Elastomeric Components in Injectable Pharmaceutical Product Packaging/Delivery Systems 〈381〉 and plastic materials as described in Plastic Materials of Construction 〈661.1〉 and Plastic Packaging Systems for Pharmaceutical Use 〈661.2〉. Utilization of the approaches described herein will enable an overall determination of biocompatibility for polymeric materials of construction and polymeric components for pharmaceutical packaging systems.
2 2. SCOPE
This chapter presents a framework for the biocompatibility evaluation of polymeric materials of construction, and polymeric components for pharmaceutical packaging systems as defined in Packaging and Storage Requirements 〈659〉. The scope of this chapter encompasses packaging for drugs, and for combination products regulated by the US Food and Drug Administration (FDA)'s Center for Drug Evaluation and Research (CDER). For medical devices and combination products regulated as medical devices (e.g., delivery devices) and for packaging that is considered a device-constituent part, users should refer to additional device-specific FDA Guidances for Industry (1). It is a regulatory expectation that such packaging systems are composed of materials and components that are considered safe for use with the dosage form and the route of administration (2). This chapter outlines a risk-based approach to biocompatibility evaluation, which includes the use of chemical characterization of materials of construction and/or packaging components.
3 3. OVERVIEW OF BIOCOMPATIBILITY EVALUATION
Biocompatibility evaluation is typically performed on packaging systems for a specific intended use. Chemical constituents of packaging systems (e.g., additives and processing aids) are potentially bioavailable and therefore may produce a biological effect. Information on a component’s material of construction is necessary but may not be sufficient for a complete evaluation. The component’s manufacturing and assembly process should also be known and understood, including processing aids, fabrication by molding or extrusion (e.g., parameters of time, temperature, and pressure), and conditions used (e.g., solvents, temperature, radiation type) in post-manufacturing processes, such as cleaning, passivation, curing, surface treatment, coating, and sterilization.
Prior knowledge may contribute toward establishing a baseline level of biocompatibility assurance for a component in a pharmaceutical packaging system, including confirmation that the materials of construction meet food contact and/or medical grade regulations and USP–NF compendial physicochemical and biological reactivity requirements. Chemical safety assessment and/or biological reactivity studies on finished components or systems can be used to further evaluate biocompatibility for a specific packaging system associated with a particular dosage form and route of administration.
The drug product manufacturer is responsible for gathering the information described above and managing the biocompatibility evaluation process. Because of the proprietary nature of specific component, material, and process details, it may be necessary for a drug product manufacturer to establish a collaborative relationship with a unique component or material supplier to share information under conditions of confidentiality as is appropriate. Pharmaceutical products in early development require baseline information, while products in later stages of development require product-specific information. Material and component selection may involve performing selected tests from 〈87〉 and/or 〈88〉, while material qualification may involve CSA along with additional biological reactivity tests. Products that are associated with routes of administration for which there is a high degree of concern require more information than those for which there is a low degree of concern (see 〈1664〉, Table 1). Ideally, the biocompatibility evaluation process begins at the time of material and/or component selection.
Biocompatibility evaluation could include CSA of leachables and/or potential leachables (extractables) as well as biological reactivity testing. CSA begins with chemical characterization through baseline physicochemical testing according to 〈381〉, Plastic Packaging Systems and Their Materials of Construction 〈661〉, 〈661.1〉, and 〈661.2〉. Depending on the route of administration and the dosage form, extractables and leachables may be identified and quantitated as described in 〈1663〉 and 〈1664〉. CSA of leachable compounds may include analyses of structure–activity relationship (SAR) or quantitative structure–activity relationship (QSAR) (3), evaluation of the scientific literature, and review of information from proprietary sources. Biological reactivity of chemical mixtures that leach or are extracted from components is assessed through in vitro or in vivo assays as described in 〈87〉 and 〈88〉.
The overall biocompatibility evaluation is drug product-specific and should be a collaborative effort involving experts including the packaging/material engineer, drug product development scientist, chemical analyst, and toxicologist (4). Materials of construction and components that comprise a finished packaging system are deemed biocompatible when their interaction with a living system or a specific tissue is not expected to cause an adverse biological effect.
3.1 3.1 Pharmaceutical Grade Polymeric Packaging Materials
Classifying polymeric materials as Classes I–VI in 〈88〉 was based historically on a series of in vivo biological reactivity tests in which the classes were differentiated by the number and type of solvents used for extraction and then in vivo biological reactivity tests performed.
Thus, Class I designation was obtained by injecting into test animals an extract from the test material derived using a single solvent, whereas Class VI designation was obtained by injecting an extract derived from four different solvents plus an implantation test. Over time, a Class VI designation has become the predominant standard for evaluating and describing polymeric materials, including plastic materials of construction, plastic and elastomeric components, and any other organic polymeric components used in primary packaging or delivery systems for pharmaceuticals or biopharmaceuticals. Therefore, the distinction into six classes no longer serves a current purpose.
The term "Pharmaceutical Grade Polymeric Packaging Materials" replaces the Classification of Plastics Classes I - VI and is designed to facilitate communication among suppliers, users, and manufacturers of polymeric materials by summarizing the tests to be performed for prospective polymeric packaging materials. Establishing this designation is based on several steps (Figure 1), beginning with a chemical risk assessment of supplier information and prior knowledge, as well as responses to an in vitro test in 〈87〉 for cytotoxicity [Minimum Essential Medium (MEM) Elution, Direct Contact or Neutral Red Uptake (NRU) test] plus the in-vitro Reconstructed Skin Epidermis (RhE) Test for irritation. Obtaining a passing result for all in vitro tests permits application of the term "Pharmaceutical Grade Polymeric Packaging Materials". Failure of any of the in vitro tests may be investigated by chemical characterization (see 〈1663〉) with toxicological assessment of extractable chemical entities, followed by performance of the 〈88〉, 3. Systemic Injection Test for which extracts, materials, and routes of administration are specified. The choice of extracting solutions is representative of the vehicles in preparations with which the polymers are likely to be in contact. This test is not required for polymeric packaging materials and systems for oral or topical use. [Note - Elastomeric components for packaging materials and systems that meet the requirements of 〈87〉 are not required to undergo 〈88〉 testing.]
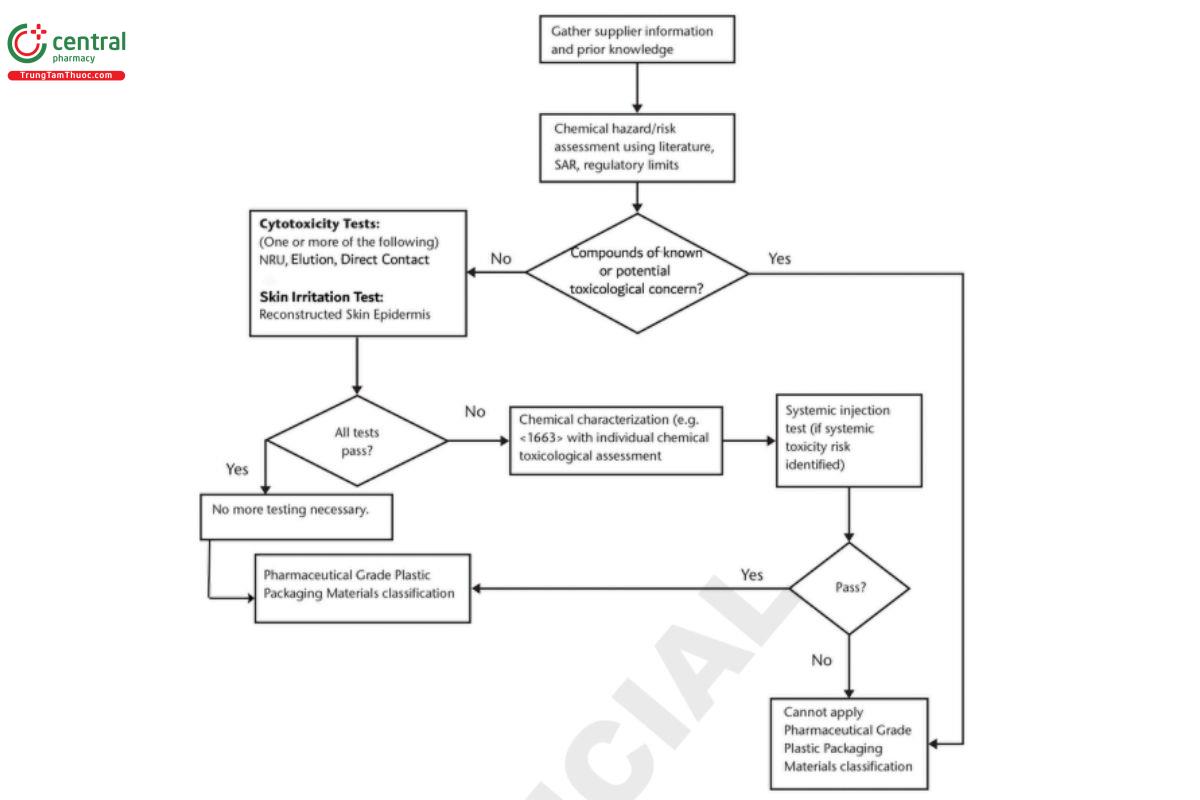
3.2 3.2 Current Regulatory Expectations for Biocompatibility
The goal of biocompatibility evaluation is to assess endpoints appropriate for a dosage form and route of administration. An endpoint, such as acute systemic toxicity, is not necessarily a test but rather an evaluation of the biological effect. In some cases, a particular biological reactivity test(s) may be used and/or a CSA performed. Both approaches take into consideration the risks associated with the route of administration and potential dosage form interaction with the packaging system.
4 4. RISK-BASED APPROACH TO BIOCOMPATIBILITY EVALUATION
In recent years there has been an increasing emphasis on applying a risk-based approach to drug product development (5,11). For combination products, where the packaging system is considered a device-constituent part, additional device-specific FDA guidance should be followed (1). Chapters 〈661〉, 〈661.1〉, 〈87〉, 〈88〉, 〈381〉, and 〈661.2〉 on polymeric packaging materials address a risk-based approach, and Evaluation of Plastic Packaging Systems for Pharmaceutical Use and Their Materials of Construction 〈1661〉 addresses both risk assessment and risk management. This section outlines an approach to biocompatibility evaluation for packaging systems. An overarching, general process for biocompatibility evaluation is shown in Figure 2.
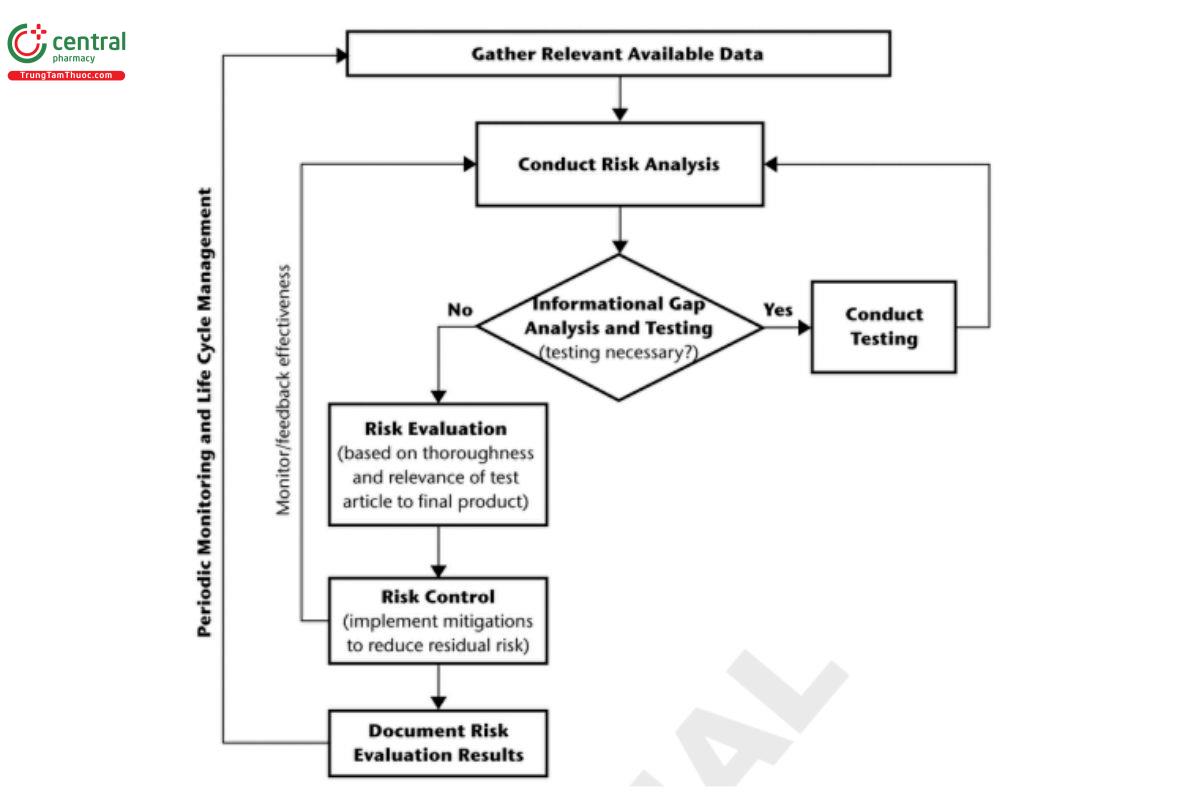
Gather Relevant Available Data: For the purpose of classifying the risk, the materials of construction and components that are most likely to contribute leachables or potentially elicit biological reactivity should be identified. Typically, these would be the materials or components that are in contact with the patient or drug. Data may include information that can be gathered externally (e.g., from the material or component supplier) or internally from historical evaluations such as:
- Composition (e.g., base polymer, additives, color additives)
- Compliance statements [e.g., USP pharmaceutical grade polymeric packaging materials; food contact and safety; transmissible spongiform encephalopathies (TSEs) and bovine spongiform encephalopathy (BSE); phthalates; and Registration, Evaluation, Authorization and Restriction of Chemicals (REACH)] (6)
- Test with relevant USP chapters (e.g., 〈87〉, 〈88〉, 〈381〉, 〈661〉, 〈661.1〉, and 〈661.2〉)
For components constructed from each material, the following information may include:
Chemical additives (e.g., processing aids, coatings, surface treatments)
Processing steps (e.g., washing, siliconization, sterilization)
- Physicochemical test compliance (e.g., 〈381〉, 〈661〉, 〈661.1〉, 〈661.2〉)
- Biological reactivity compliance (〈87〉, 〈88〉)
- Extraction study results (e.g., volatiles, semi-volatiles, non-volatiles, elemental analysis)
- Presence of compounds with specific safety concerns (e.g., phthalates, polynuclear aromatics, N-nitrosamines, 2-mercaptobenzothiazole, azoxy compounds, endocrine disruptors)
- Leachables study results (internal use for pharmaceutical manufacturer)
Conduct Risk Analysis: Risk analysis for materials of construction used in patient- or drug-contacting components of a packaging system depends on the intended product use. Drug formulations can significantly impact the leachable outcome and related bioreactivity of materials of construction. A cross-functional team, including design engineers, materials specialists, drug product development scientists, manufacturing representatives, toxicology experts, clinical experts, and regulatory and quality representatives, may provide the appropriate expertise to analyze the risk associated with the materials.
For example, there may be materials of known biocompatibility concern (e.g., irritation) or chemicals of known safety concern (e.g.,mutagens, sensitizers) or chemicals subject to regulated limits (e.g., phthalates) present in the composition, manufacturing process, or assembly process. An additional safety concern is that a chemical reaction with the formulation could result in reaction products with toxicity concerns. One approach might be to utilize this information to correlate each material or component with a unique risk rating. Such a risk rating may be numerical, for example, one that is calculated in a failure mode and effect analysis (FMEA) as the product of numerical scores for probability and severity of a particular harm that may result from a specified hazard. Alternatively, a risk rating may be a qualitative assignment (e.g., low, moderate, high). Any values assigned are based on a ranking system developed prior to a risk analysis.
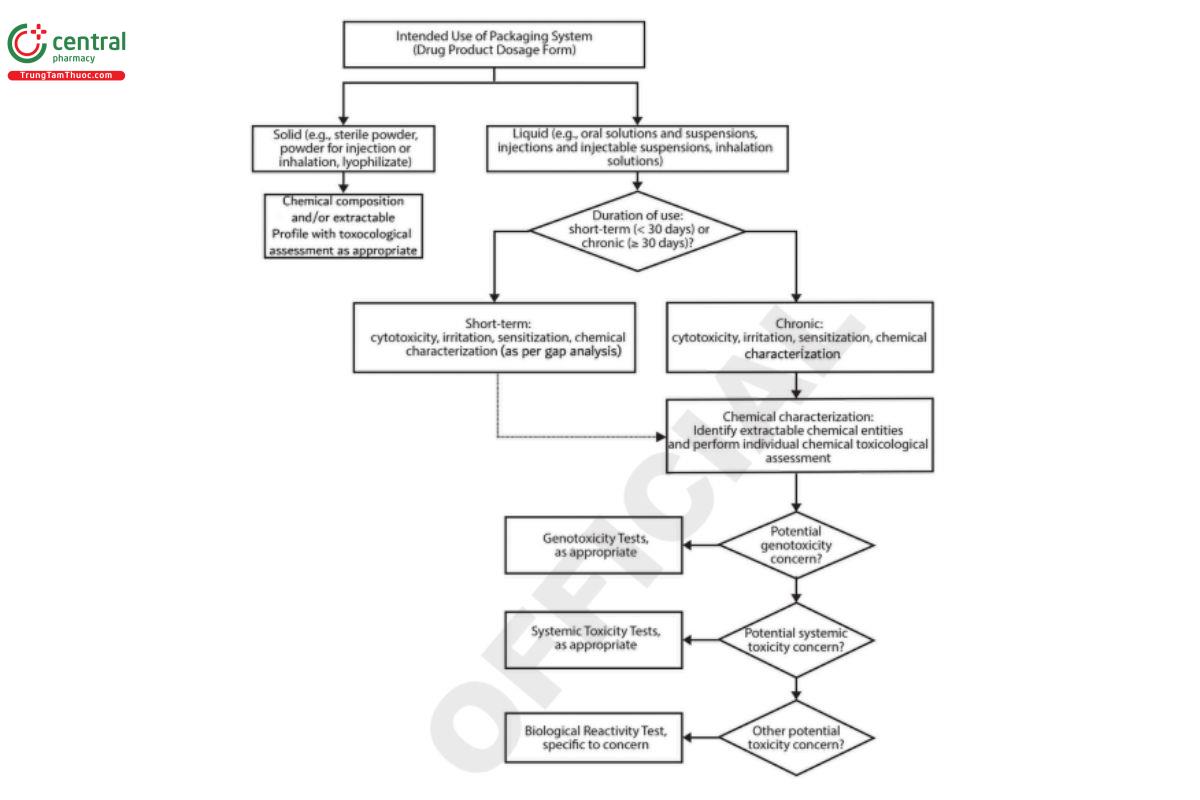
Informational Gap Analysis and Testing: Materials of construction and components that are considered a high risk may require additional studies or need to be replaced. Gaps in the available information are identified, and a strategy is developed to address the gaps. There may be additional chemical composition, manufacturing process information, or in vitro or in vivo test results (e.g., prior knowledge) that could be used to reduce the risk rating. Alternatively, it may be appropriate to perform an extractables assessment to understand potential leachables and their impact on various endpoints. Other product-specific endpoints include: genotoxicity, systemic toxicity, carcinogenicity, and reproductive toxicity. To determine which tests may be appropriate, the flow chart in Figure 3 may be used. If the intended use is unknown, a conservative approach that considers all potential intended uses may be appropriate.
Risk Evaluation: The complete information package, including any additional testing performed in this step, is used to complete the risk evaluation. The final risk evaluation depends on the intended use of the pharmaceutical product. When the chemical safety assessment indicates that there may be an exposure risk, controls may be recommended that include raw material and component monitoring, process monitoring, or change to a process, raw material, or component. These can only be determined for a specific set of materials of construction or components used in a particular packaging system.
Risk Control: The recommended controls generated from the risk evaluation may need to be implemented as a control strategy to mitigate chemical exposure risk. Examples of such controls are composition control by upstream supplier(s) or changes in a process step to eliminate a specific chemical. The impact of risk controls is evaluated to ensure that the risk is mitigated appropriately. Modifications to controls may be necessary with life cycle management.
Document Risk Evaluation Results: To be effective, the risk evaluation should be documented. This enables future reference to historical information throughout the life cycle of the product. The details of the raw materials, components, and packaging system should be captured to allow traceability. The information package and biocompatibility evaluation complete the documentation.
Periodic Monitoring and Life Cycle Management: The risk evaluation should be reviewed periodically as is appropriate (e.g., on an annual basis). There may be events that trigger an ad hoc re-evaluation such as:
Process change:
- Sterilization
- Surface treatment
- Fabrication
- Assembly and handling
Material of construction or component change:
- Availability
- Vendor
- Production facility
- Formulation
- Storage conditions
- Intended use
If there are any changes to the product profile or information package, these should be documented and accompanied by an impact assessment. Depending on assessment of the anticipated impact, additional activities may be required (e.g., specific chemical safety assessments, additional testing, corrective or preventive actions, alternate controls, or regulatory notifications or submissions).
5 5. BIOLOGICAL REACTIVITY TEST CONSIDERATIONS
5.1 5.1 Test Article Selection and Sample Preparation Considerations
When it has been determined that testing should be performed, it is essential that appropriate test articles are selected and handled properly. Biological reactivity test results are meant to be representative of the material of construction or component. The selection considerations may include:
- Product development stage [e.g., prototype, first article from operational qualification (OQ) of tool, good manufacturing practices (GMP), production]
- Inherent material or component variability (e.g., cross-linking, catalyst residuals)
- Processing variability (e.g., no controls, well-established controls)
- Lot size and number of lots available
- Representative of the components used in commercial products
For any test article, it is expected that there be a traceability from the source point of raw materials, through the manufacturing process and control, to all the in-process testing and release test results.
Sample Handling
Appropriate handling practices are critical for test articles that are subjected to extraction. It is important that samples are collected and handled in such a way as not to introduce any additional chemical substances at trace levels. Sampling procedures should prevent direct human contact with the materials and components [e.g., by use of gloves (powder free) or sampling tools (cleaned with isopropanol and allowed to air dry)]. Samples should be packaged in containers or bags that are normally used at the manufacturer for such purposes (e.g., food grade, free of particulates and compounds such as antistatic agents or other surface treatment that may transfer to and compromise or contaminate the sample) and sealed in a clean area. The use of clean dry air to remove particulates or to dry samples should be avoided due to trace levels of oil emanating from the compressor; however, an appropriate alternative such as high purity nitrogen (e.g., from a cylinder or Dewar ask) may be used for this purpose. Samples should be stored at the temperature and relative humidity recommended by the manufacturer. Shipment conditions should ensure protection from extended exposure to moisture, freezing, and excessive heat, and, where necessary, from light as defined in 〈659〉, as these conditions could change the chemical profile of the test article.
The quantity of test articles needed for extract preparation depends on the type of test and is typically determined from the required surface area for the test. It is calculated from the surface area per volume of extraction medium required per recommendations in 〈88〉 and in collaboration with the laboratory performing the test. After the extract is prepared, it should be stored in a non-reactive, sealed container, protected from light and at a temperature appropriate to maintain the integrity of the sample. Extracts for biological reactivity should typically be used on the day that the sample preparation is complete, or per specific test recommendations.
5.2 5.2 In Vitro Test Selection
If a biological reactivity test is determined to be the only way to evaluate a biocompatibility endpoint, there are several considerations that should be made in the selection of the test. The use of an in vitro test (if available) is recommended over the use of an in vivo test in alignment with the "three Rs" approach of replacement, reduction, and refinement to the use of live animals in testing (7), where the method provides information consistent with that obtained with an in vivo method. Any additional considerations in selecting a test other than those represented in the published recommendations of the regulatory authorities should be provided.
For cytotoxicity tests, mammalian cells are grown to near confluence and then exposed directly, or indirectly via their extracts, to test and control samples. There are advantages and disadvantages to each type of test as summarized in Table 1.
Table 1. Cytotoxicity Test Selection
| Test | Test Description | Advantages | Disadvantages |
|---|---|---|---|
| Elution | Sample extracted in cell media for at least 24 h | Examines cell morphologically as a direct determination of cell cytotoxicityAppropriate for dose-response evaluations | Semi-quantitative |
| Direct contact | Tests cell cytotoxicity of sample in direct contact with cell monolayers | Able to test small quantities of samples (too small for use in the elution test)Able to test a single surface of a sample (useful for coated and laminated materials) | Semi-quantitativeMorphological toxicity changes to cells not evaluated; zones of cell lysis are measuredHeavier samples or pressure when placing samples on cells can cause physical damage to cells under the sample, which can be misinterpreted as a reactivity grade of 1 or 2 |
| Neutral red uptake | Tests cell viability in the presence of sample extracts | QuantitativeVery sensitiveAppropriate for dose-response evaluations | Indirect measurement of cell toxicity by dye uptake in viable cellsColored extracts and precipitates can interfere with the assay |
These tests can be semi-quantitative, using a grading system to score cell reactivity under a microscope, such as in the elution and direct tests; or quantitative as in the NRU test. A semi-quantitative test has the advantage of direct visual observation and evaluation of cellular toxicity. Typically, the elution test, a more rigorous semi-quantitative test, is performed using cell media extracts of the test and control samples. Direct cytotoxicity tests are useful if only small quantities of samples are available, or if only one surface needs to be evaluated.
The NRU test provides a sensitive quantitative dose-response evaluation of both cell integrity and growth inhibition. This test is designed for the evaluation of extracts of materials of construction and components. The NRU test is based on the ability of viable, uninjured cells to incorporate and bind neutral red (NR), a supravital dye. NR readily diffuses through the plasma membrane and concentrates in lysosomes.
Cytotoxicity is expressed as a concentration dependent reduction of the uptake of NR after substance exposure to the cells, thus providing a sensitive, integrated signal of both cell integrity and growth inhibition.
The skin irritation test utilizes reconstructed skin epidermis RhE to replace an in vivo intracutaneous test. The advantages and disadvantages of this test are summarized in Table 2.
Table 2. Reconstructed Skin Epidermis (RhE) Test
| Test | Test Description | Advantages | Disadvantages |
|---|---|---|---|
| In vitro RhE Test | Tests cell viability in RhE tissue constructs incubated with sample extracts, using the MTT cytotoxicity test | Quantitative; replaces in vivo test for detection of potential irritants | Not applicable to evaluate irritation by direct contact of solid materials with body; may not be appropriate for some contact (ocular, mucosal) |
a 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide.
The in vitro RhE test for irritation utilizes cultured multi-layered human epidermis. This method was developed and validated for neat chemicals to detect skin irritation potential and standardized as OECD Method 439 (8) and was later adapted and validated for extracts of materials used in medical devices (9–10). Sample extracts are incubated with RhE constructs for up to 24 h at 37°, rinsed, dried, then viability assessed by the quantitative MTT (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) cytotoxicity test to determine cell viability.
The MTT test is based on the quantitation of cellular reduction of MTT to a formazan salt, resulting in a purple color and measured absorbance with a spectrophotometer (570 or 630 nm) as a measure of metabolic activity/viability. Cell viability is expressed as a percentage of negative control, and cell viability ≤50% indicates an irritant; cell viability >50% indicates a non-irritant.
For genotoxicity tests, no single test can detect all relevant genotoxic agents. Therefore, the usual approach is to conduct a battery of genetic toxicity tests. There are advantages and disadvantages to each type of test as summarized in Table 3.
Table 3. Genotoxicity Test Selection
| Test | Advantage | Disadvantage |
|---|---|---|
| Bacteria reverse mutation | Detects point mutations produced by the majority of genotoxic carcinogens | Cannot detect clastogens or aneugens |
| In vitro mammalian cell chromosomal aberration | Detects structural and numerical chromosomal aberrations | Cannot detect point mutations or aneugens |
| In vitro mammalian cell micronucleus test | Detects clastogenic effects and aneugens | Cannot detect point mutations |
| In vitro mammalian cell gene mutation test using L5178Y or TK6 cells | Detects both point mutations and clastogenic effects | Cannot detect aneugens or identify types of clastogenic damage |
The maximum test concentration will depend on cytotoxicity of the sample extract. If the extract is cytotoxic, it should be diluted to a noncytotoxic level before use in the genotoxicity test. Otherwise, the sample extract should be evaluated using the maximum test concentrations recommended for each test (see 〈87〉).
Bacterial reverse mutation assays have been shown to detect relevant genetic changes produced by most of the genotoxic carcinogens detected by rodent assays. Certain classes of genotoxin (e.g., alkyl halides) are not detected.
The potential of samples to produce DNA damage in bacterial systems might not be relevant to their likely effects in eukaryotic cells, and, therefore, testing in mammalian cell test systems is also performed unless otherwise justified. Several mammalian cell systems are in use:
- Systems that detect gross chromosomal damage (in vitro tests for structural and numerical chromosomal aberrations)
- Systems that detect micronuclei in the cytoplasm of interphase cells (in vitro tests for aneugens and clastogens)
- A system that detects gene mutations and clastogenic effects [mouse lymphoma thymidine kinase (TK) assay with both colony number and size determination]
Results from these tests have a relatively high level of congruence for compounds that are regarded as genotoxic but yield negative results in the bacterial reverse mutation assay. The chromosomal aberration test, the in vitro micronucleus test, and the in vitro mammalian cell gene mutation test using L5178Y or TK6 cells are currently considered equally acceptable when used with the bacterial reverse mutation assay in a standard battery for genotoxicity testing. Therefore, the genetic toxicity battery should include a test for gene mutations in bacteria (bacterial reverse mutation test) and any one of the following tests (also see 〈87〉):
- In vitro mammalian cell gene mutation test using L5178Y or TK6 cells
- In vitro mammalian cell micronucleus test
- In vitro mammalian cell chromosomal aberration test
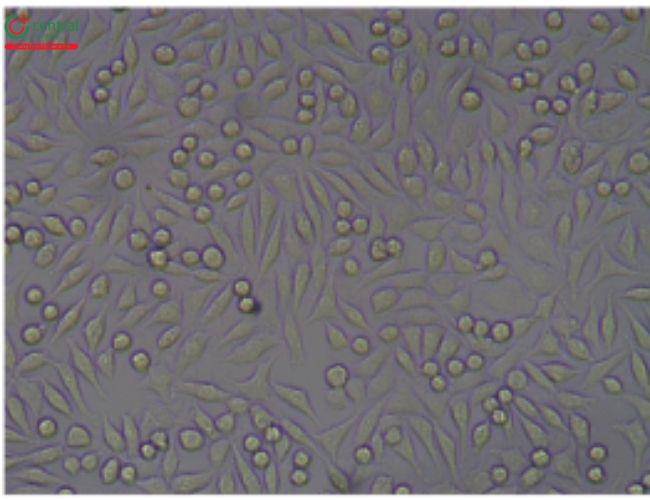
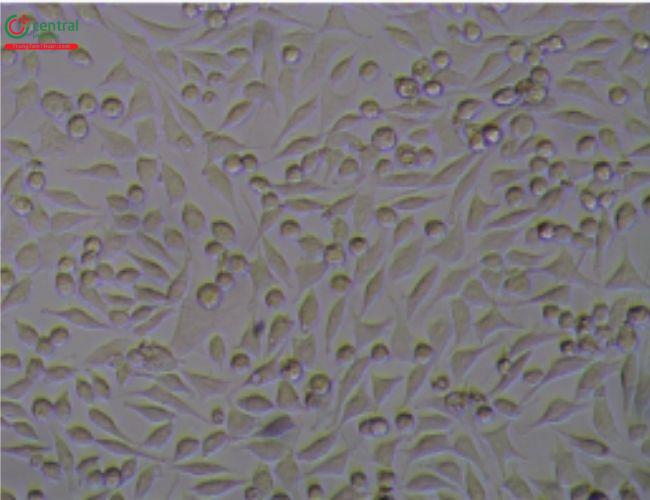
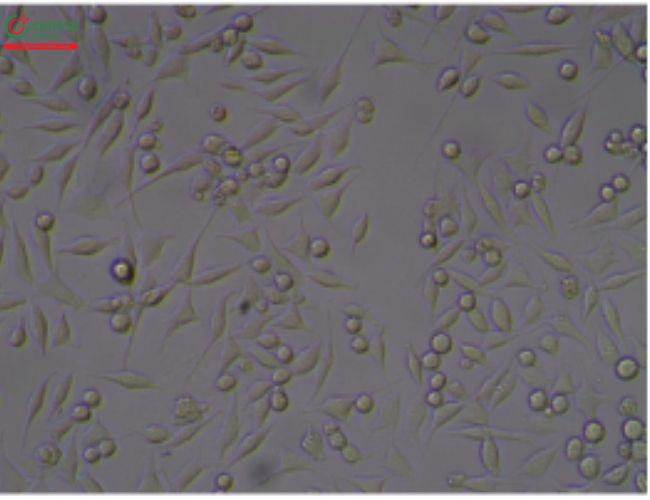
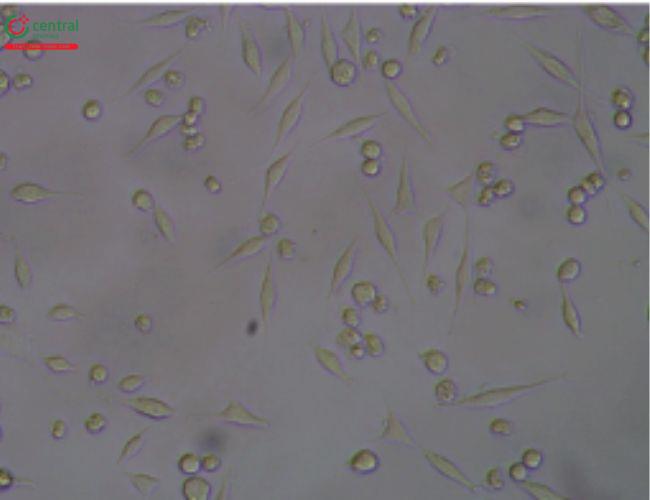
5.3 5.3 Illustrative Examples Cytotoxicity Reactive Grades
This test is described in 〈87〉 and allows for simultaneous extraction and testing of extracted chemicals from the specimen with a serum-supplemented medium. The biological reactivity (cellular degeneration and malformation) is described and rated on a scale of 0–4 (Table 4,Table 5).
Table 4. Reactivity Grades for Direct Contact Test
| Grade | Reactivity | Conditions of All Cultures |
|---|---|---|
| 0 | None | No detectable zone around or under specimen
|
| 1 | Slight | Some malformed or degenerated cells under specimen
|
| 2 | Mild | Zone limited to area under specimen
|
| 3 | Moderate | Zone size up to 1.0 cm
|
| 4 | Severe | Zone extends further than 1.0 cm beyond specimen
|
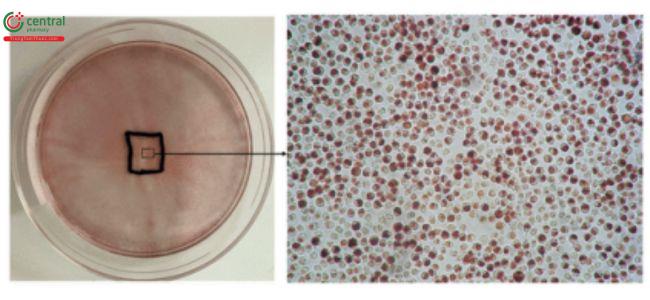
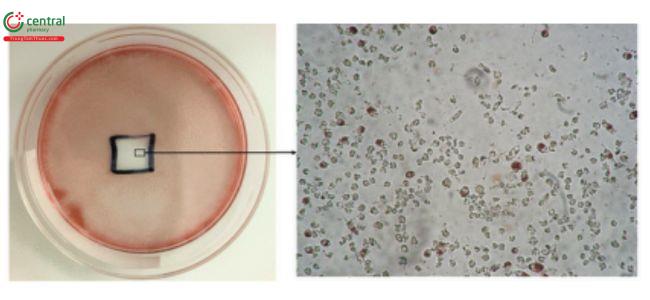
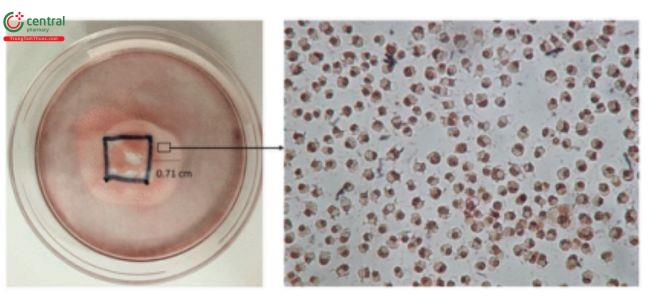
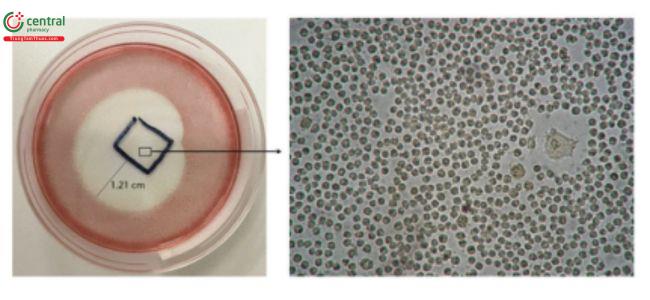
Table 5. Reactivity Grades for Elution Test
| Grade | Reactivity | Conditions of All Cultures |
|---|---|---|
| 0 | None | Discrete intracytoplasmic granules; no cell lysis, no reduction of growth
|
| 1 | Slight | NMT 20% of the cells are round, loosely attached, and without intracytoplasmic granules or show changes in morphology; occasional lysed cells are present; only slight growth inhibition observable
|
| 2 | Mild | NMT 50% of the cells are round, devoid of intracytoplasmic granules; no extensive cell lysis; no more than 50% growth inhibition observable
|
| 3 | Moderate | NMT 70% of the cell layers contain rounded cells or are lysed; cell layers are not completely destroyed, but more than 50% growth inhibition observable
|
| 4 | Severe | Nearly complete or complete destruction of cell layers
|
5.4 5.4 Factors to Consider When Investigating a Biological Reactivity Test Failure
Investigation of a biological reactivity test failure may consider several factors before implicating physicochemical attributes of the component or material tested. These may include the test article, prepared sample, testing and laboratory equipment, the laboratory environment in which testing is conducted, the laboratory personnel conducting the testing, and proper and consistent performance of the actual testing. Figure 4 is an Ishikawa diagram illustrating some key factors potentially in fluencing the performance and outcome of biological reactivity testing.
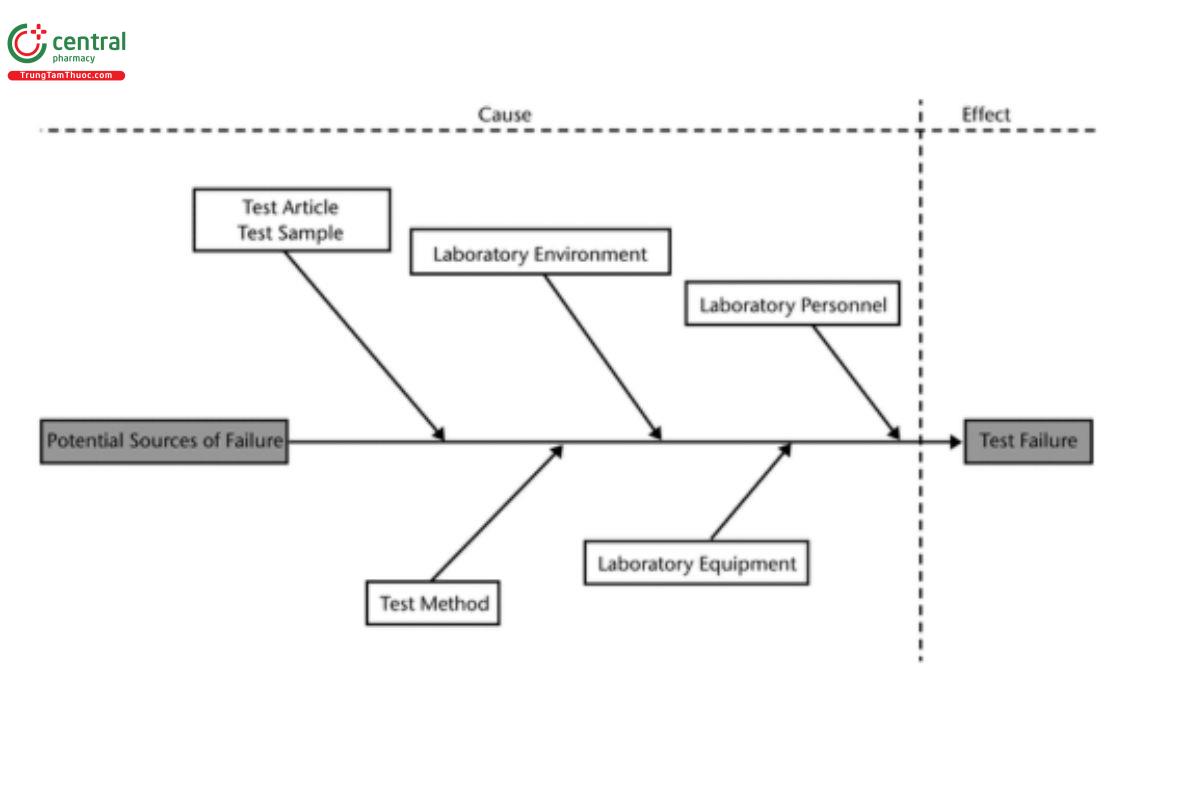
These factors include:
- Test Article: The integrity and condition of the test article being evaluated is critical to a reliable test outcome. The identity of the test article should be confirmed, and the shipping or transfer conditions should not in any way contribute to degradation or decomposition of the test article. There should be a comprehensive understanding of the composition, handling, and any processing steps (e.g., molding, cleaning, sterilization) that may have been applied. If possible, a comprehensive description of any processing or treatment applied to components during pre- and post-manufacturing should be determined, including any processing or treatment during or following the final assembly of the system.
- Test Sample: There should be sufficient knowledge and understanding of the sample to ensure compatibility with the test conditions (e.g., temperature, time, extraction solvent, exposure). Sample preparation, handling, and storage (e.g., duration, temperature, relative humidity, light exposure), degradation, and possible contamination of the prepared sample should be considered such that the integrity of the sample being tested is not compromised.
- Test Method: Performance of the testing method by trained laboratory personnel per instructions specified in a testing protocol is critical to successful generation of a set of reliable results. Important considerations are the inclusion of controls along with prepared samples as appropriate to ensure that a specific test or analysis is performing reliably and the assurance that test materials (reagents, media, cells, etc.) have been appropriately stored and maintained and not utilized past expiry, contaminated, or degraded in any way.
- Laboratory Environment: The environmental conditions associated with the testing laboratory should be appropriate for the testing conducted and limit potential for contamination or degradation of the material or component being tested. The conditions should be consistent with temperature, humidity, and lighting requirements for a particular test method or technique.
- Laboratory Equipment: Any laboratory equipment involved with biological reactivity testing and chemical analysis should be appropriately maintained, calibrated, and demonstrated to be in acceptable operating condition. This not only includes sophisticated analytical instrumentation but also equipment involved with sample preparation and storage (e.g., pipettes, incubators, pH meters).
- Laboratory Personnel: The personnel performing any laboratory testing should demonstrate sufficient knowledge, training, and technical competency to ensure consistent and reliable generation and reporting of test results to support scientifically sound conclusions and decisions. Not to be overlooked is the capacity and work flow management appropriate to enable trained laboratorypersonnel to perform reliable testing with minimal opportunity for error.
5.5 5.5 Risk-Based Approach to Investigating a Failed Biological Reactivity Test
Application of a risk-based approach to investigating failure of in vitro and/or in vivo biological reactivity testing can assist with flunderstanding the nature, cause, and potential impact of the test failure.
The initial consideration for investigating a biological reactivity test failure is ensuring that there is full understanding of the advantages and limitations of the specific biological reactivity test(s) in scope, clear interpretation of the type of failure, and understanding of the potential impact of the failure. Depending on the specific in vitro or in vivo test performed, the failure may indicate potential for a tested component or material to elicit a biological effect such as:
- Cytotoxicity
- Irritation
- Acute toxicity
- Genotoxicity
The impact of the failure may include patient safety, product quality, and relevant considerations for use of a unique component or material with a drug product.
Determining the root cause(s) for a specific biological reactivity test failure may be facilitated with an investigation involving the assessment of several characteristics of a tested component or material including the composition of the material, preparation or processing applied to the component or material prior to biological reactivity testing, and the identification and assessment of extractable chemical substances. It is important to understand the biological reactivity test as well as the chemical assessment extraction and analysis conditions with the goal of establishing a correlation between the detected chemical substances and the biological reactivity test results.
This may assist in providing a scientifically sound rationale for the observed test failure, including assessment of toxicological significance of detected chemical substances. Emphasis should be placed here on the highly complex nature of biological systems, which implies that sound and thoughtful scientific judgment be applied when attempting to correlate either a sole chemical entity or multiple chemical substances with a biological reactivity test result. It is important to bear this in mind because it may not be possible to ascertain with certainty whether the observed biological reactivity test result is due to chemical substances acting upon a system independently, collectively (additively), or cooperatively.
Investigating a biological reactivity test failure includes understanding any controls that are applied, are modifiable, and are potentially contributing to biocompatibility of a component or material. These may include the material formulation, additives and processing aids, component processing conditions (e.g., molding, cleaning, sterilization), and the drug product storage and use conditions. This consideration is important when developing and applying potential corrective action and understanding factors that may require modification in mitigating the biological reactivity test failure. Some examples include replacement or modification of a component or material, reduction or elimination of specific chemical substances used to process or impart desired properties to a material or component, and modification of molding, processing, cleaning, or sterilization conditions.
Regardless of actions taken to correct a biological reactivity test failure, circumstances may exist under which it is not possible to obtain a successful result for one or more tests employed. In this event, it is important to consider all factors potentially impacting patient safety, including the dose amount, route of exposure, frequency and duration of administration, and application of appropriately relevant thresholds.
Ultimately, a scientifically sound assessment of risk/bene fit impact to patient safety and drug product quality should be applied in the interpretation and investigation of a biological reactivity test failure and in any decision to employ appropriate countermeasures and corrective action.
5.6 5.6 Role of Chemical Assessment in Assessing Biological Reactivity
Assessment of biocompatibility or biological reactivity may be both complemented and supplemented by chemical characterization and CSA of extractables from the packaging system and also can play a complementary role in investigating and providing a rationale for a failed biological reactivity test.
A CSA can play a complementary role in biocompatibility evaluation, in that it may:
- Provide an understanding of packaging components and their materials of construction as well as the chemical entities that can potentially leach from them
- Play an important role in supporting the elimination of unnecessary in vivo testing
- Inform additional appropriate biocompatibility end point evaluation
- Assist with biocompatibility test failure analysis by potentially providing a clear and scientifically sound rationale for such failures
The following points should be considered when correlating the chemical assessment with the biological reactivity test results (see 〈1663〉):
- Tests should be performed on finished components including any treatment and processing (e.g., molding conditions, cleaning, surface treatment, coating, sterilization) that would be representative of the final component as intended for use in a packaging system.
- Biological reactivity testing solvents and media are not necessarily compatible with appropriate analytical techniques to permit comprehensive chemical assessment.
- Analytically expedient solvents are necessary to permit detection and assessment of diverse classes and structures of chemical substances. Ideally, sample preparation and solvent selection can be chosen to represent as closely as possible, or more conservatively, extraction characteristics of solvents used in biological reactivity testing (particularly for failure mode analysis).
- Surrogate extraction solvents for correlating chemical assessment with biological reactivity testing may be selected for correlating biological reactivity tests with chemical assessment - see ISO 10993-18 (10).
- Furthermore, there may exist unique instances under which other extraction conditions (e.g., differing surface area or volume or alternate solvents) may be considered to achieve appropriate analytical sensitivity and facilitate a meaningful correlation; these should be justified with a scientifically sound rationale on a case-by-case basis.
6 6. OVERALL BIOCOMPATIBILITY EVALUATION
Biocompatibility is established based on evaluation of the potential to elicit an irritating, sensitizing, systemically toxic, and/or mutagenic effect for a given intended use. Leachables from a material of construction/component or packaging system can affect the safety, efficacy, quality, and/or stability of the drug product. Contact between a packaging system or its components and the human tissue may produce adverse reactions. Therefore, acceptability of chemical exposure risk through leaching is established based on qualitative evaluation against USP–NF compendial standards, and, if indicated, chemical safety assessment results.
The biocompatibility evaluation should be performed by a multidisciplinary team of appropriately qualified experts capable of interpreting the relevant material of construction and component or packaging system requirements. Evaluation takes into consideration:
- The intended use
- The patient population
- The duration of use
- The review of all data such as:
- Biological reactivity testing
- Chemical characterization with associated chemical safety assessment
- Physicochemical testing
- Literature and previous clinical experience
When legacy testing results are considered, the relevance of the methods to current practice should also be evaluated.
The biocompatibility evaluation should consider both the chemical exposure risk and the biological reactivity of the material of construction and component or packaging system. If a specific exposure risk is identified from the chemical safety assessment or failure occurs during biological reactivity testing, the anticipated impact should be assessed and continued use of the sample in a packaging material justified.
Points to consider in the assessment are as follows:
- Replacement or modification of raw material and component
- Evaluation of chemical characterization results for correlation with failed biological reactivity result
- Consideration of additional in vitro and/or in vivo test(s) to mitigate risk
Collectively, this information and additional mitigation steps are used in the biocompatibility evaluation process to develop a weight-of-evidence assessment for the drug product.
7 GLOSSARY
Chemical safety assessment (CSA):
A toxicological assessment to evaluate risks from exposure to a substance during product use. The assessment includes a comprehensive review of available literature, QSAR analysis, regulatory limits, and application to the product-specific conditions of use.
Endpoint:
Event or outcome that can be measured objectively.
In silico:
A computer-assisted analysis or simulation performed to obtain a toxicological endpoint.
Pharmaceutical grade polymeric packaging material: Polymeric materials of construction and polymeric components for pharmaceutical packaging/delivery systems and for packaging of combination products that are in accordance with 3.1 Pharmaceutical Grade Polymeric Packaging Materials.
QSAR:
Structure–activity relationship that describes the quantitative relationship between the chemical or its structure and its biological activity.
Sample:
A representative portion of the test article selected and prepared for a specific test or set of tests. Any criteria applicable to the selection, handling, storage, preparation, and disposition of a sample should be described and observed (e.g., in a test method or protocol).
SAR:
Structure–activity relationship that describes the qualitative relationship between the chemical or its structure and its biological activity.
Test article:
Material of construction, component, group of components, or packaging system on which a test is performed.
Three Rs:
A set of principles to guide the ethical evaluation of animal use: refine animal procedures to reduce pain and distress; reduce number of animals used to obtain a scientifically valid result; replace animal use, where possible, with non-animal alternative systems (e.g., computer programs). The 3 Rs have been modernized from those originally proposed by Russell and Burch (7).
8 APPENDIX
Chemical Safety Assessment Case Studies
Case Study A: Biological reactivity testing employed to mitigate risk for a potential safety risk during clinical development
Product X is a dry powder inhaler used to treat chronic obstructive pulmonary disease. The dry powder is packaged in a multilaminate blister that is inserted into an inhalation delivery device. Product X is administered once daily and may be used over the lifetime of a patient.
Extraction studies on the blister (assumed commercial packaging system) were performed in parallel to clinical development of the product. Only one chemical was identified that posed a potential safety concern.
On the basis of extraction studies, maleic anhydride was calculated to be present in Product X at a worst-case calculated dose of 16 μg/day. In silico analysis determined that maleic anhydride had the potential to be a respiratory irritant and/or sensitizer. A literature search confirmed that anhydrides can be mild-to-severe irritants and/or sensitizers, and given that the drug containing this potential leachable was indicated to treat bronchospasm, the toxicological profile of the extractable was deemed concerning.
Leachable studies had not yet been started and Product X was in use in ongoing clinical trials. To determine if Product X in the current blister posed any safety issue, an in vivo 〈88〉 study was conducted on the oldest available Product X samples. Product X complied with〈88〉 and clinical trials continued. However, as a result of the chemical safety assessment, the project team determined that an alternative multilaminate blister was required for the commercial product to eliminate any potential irritation and/or sensitization risk.
Case Study B: Drug product risk analysis without biological reactivity testing
Product Y is a monoclonal antibody used to treat breast cancer and is administered intravenously with a week separating each dosing cycle. Leachable studies were conducted, and several leachables were identified from the packaging system, comprising of a 30-mL borosilicate glass vial with an elastomeric stopper. All elastomeric components used in the packaging system were demonstrated by their supplier to be 〈87〉 compliant.
Based upon the analytical results of the leachables study and a maximum daily dose of 560 mg/day for Product Y, total daily intake (TDI) values were calculated for a number of leachables, and those that surpassed the Product Quality Research Institute (PQRI)-derived qualification threshold of 5 μg/day (12) were submitted for chemical safety assessment. Each leachable was evaluated based on published and database sources, with emphasis on information via the intravenous route. Where workplace exposure limits have been recommended or established by scientific or regulatory bodies, the resulting human daily dose levels (mg/kg) were calculated from an atmospheric concentration reported in the literature in parts per million (ppm) or mg/L (13).
Subsequently, the atmospheric concentration was converted into a dose, considering exposure time, minute volume, and body weight based upon data reported by Connelly et al (14). The total daily intake calculations applicable for some common leachables and the minute volume and standard body weight are listed in Table A.1 and Table A.2.
Table A.1 Total Daily Intake Calculations for Common Leachables
| Analyte | CAS # | Maximum TDI (µg) |
|---|---|---|
| Acetone | 67-64-1 | 7.7 |
| Hexane | 110-54-3 | 8.4 |
| Isopropanol | 67-63-0 | 31.7 |
| Butylated hydroxytoluene | 128-37-0 | 20.1 |
| Ethanol | 64-17-5 | 287.8 |
Table A.2 Human Daily Dose Level Based on Atmospheric Concentration
| Species | Minute Volume (L/min) | Standard Body Weight (kg) |
|---|---|---|
| Human | 20 | 50 |
The following dose calculation was utilized to calculate an mg/kg dose of a given chemical based upon the data in Table A.1 and Table A.2.
Concentration (mg/L) × Exposure Time (min) × Minute Volume (L/min) × Body Weight (kg)
As a result of this analysis, the presence of leachables in Product Y intravenous formulation at the μg/day levels, summarized in Table A.1, posed negligible risk to humans. No biological reactivity testing beyond the already performed 〈87〉 was indicated in the overall risk analysis of Product Y.
9 REFERENCES
1. Use of International Standard ISO 10993-1, "Biological Evaluation of Medical Devices - Part 1: Evaluation and testing within a risk management process". Guidance for Industry and Food and Drug Administration Staff. September 2020.
2. Guidance for industry. Container closure systems for packaging human drugs and biologics. Rockville, MD: US Food and Drug Administration; May 1999. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/container-closure-systems-packaging-human-drugs-and-biologics.
3. International Conference on Harmonisation. ICH M7(R1). Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. 2018. https://www.ich.org/page/multidisciplinary-guidelines.
4. Stults CLM, Wolff R, and Ball DJ. The chemistry and toxicology partnership: extractables and leachables information sharing among the chemists and toxicologists. In: Ball DJ, Norwood DL, Stults LM, Nagao LM, eds. Leachables and Extractables Handbook: Safety Evaluation, Qualification, and Best Practices Applied to Inhalation Drug Products. Hoboken, NJ: Wiley & Sons; 2012:93–116.
5. International Conference on Harmonisation. ICH Q9: Quality risk management. 2005. https://www.ich.org/page/quality-guidelines.
6.Registration, Evaluation, Authorization and Restriction of Chemicals (REACH) Regulation EC 1907/2006.
7. Russell, WMS and Burch, RL, (1959). The Principles of Humane Experimental Technique, Methuen, London. ISBN 090076778.
8. Organization for Economic Cooperation and Development (OECD) Test No. 439. In Vitro Skin Irritation: Reconstructed Human Epidermis Test Method. June 17, 2021.
9. International Organization for Standardization. ISO 10993-23:2021. Biological evaluation of medical devices. Part 23 Tests for Irritation.
10. International Organization for Standardization. ISO 10993-18:2020. Chemical characterization of medical device materials within a risk management process.
11. International Conference on Harmonisation. ICH Q10: Pharmaceutical Quality System. 2008. https://www.ich.org/page/quality-guidelines.
12. Ball D, et al. Development of safety qualification thresholds and their use in orally inhaled and nasal drug product evaluation. ToxicolSci. 2007;97(2):226–236.
13. Derelanko MJ. Inhalation Toxicology. In: Toxicologist’s Pocket Handbook 3rd Edition. New York, NY: CRC Press; 2017.
14. Connelly JC, Hasegawa R, McArdle JV, and Tucker ML. ICH Guideline—Residual solvents. Pharmeuropa. 1997;9(1):S1–S68.
