Tamsulosin Hydrochloride
If you find any inaccurate information, please let us know by providing your feedback here

Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
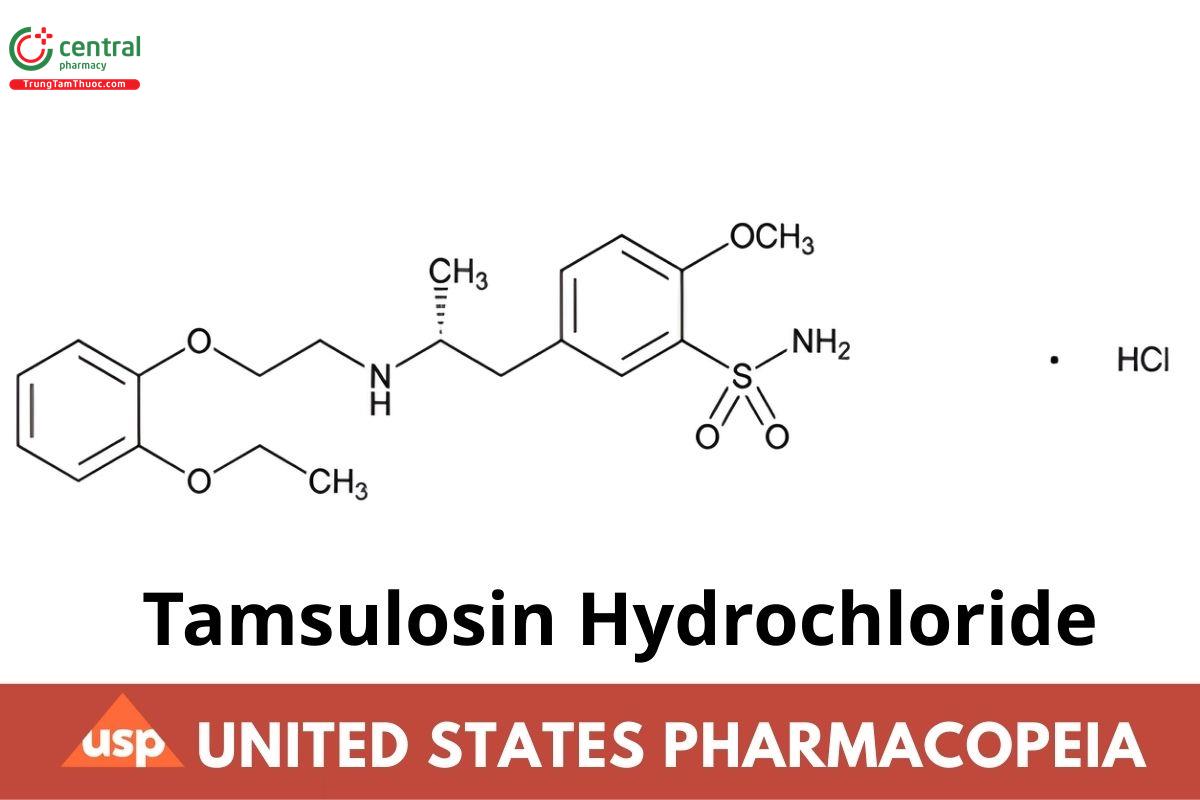
C20H28N2O5S · HCl 444.97
Benzenesulfonamide, 5-[2-[[2-(2-ethoxyphenoxy)ethyl] amino]propyl]-2-methoxy-, monohydrochloride, (R)-;
(−)-(R)-5-[2-[[2-(o-Ethoxyphenoxy)ethyl]amino]propyl]-2-methoxybenzenesulfonamide monohydrochloride CAS RN®: 106463-17-6; UNII: 11SV1951MR.
1 DEFINITION
Tamsulosin Hydrochloride contains NLT 98.0% and NMT 102.0% of tamsulosin hydrochloride (C20H28N2O5S · HCl), calculated on the dried basis.
2 IDENTIFICATION
Change to read:
A. Spectroscopic Identification Tests 〈197〉, Infrared Spectroscopy: 197A or 197K. (CN 1-May-2020)
B. Identification Tests—General 〈191〉, Chloride
Sample solution: 7.5 mg/mL in water, using heat to dissolve the sample. In an ice bath, cool 5 mL of the solution. Add 3 mL of diluted nitric acid, and shake until mixed thoroughly. Allow to stand for 30 min at room temperature, and fIlter.
C. The retention time of the major peak of the Sample solution corresponds to that of the Standard solution, as obtained in the Assay.
3 ASSAY
3.1 Procedure
Buffer: Transfer 0.1 g of octanesulfonic acid sodium salt and 1.0 mL of phosphoric acid into 1 L of water, and mix.
Mobile phase: Acetonitrile and Buffer (20:80)
Diluent: Acetonitrile and water (20:80)
Standard solution: 0.5 mg/mL of USP Tamsulosin Hydrochloride RS in Diluent
Sample solution: 0.5 mg/mL of Tamsulosin Hydrochloride in Diluent
3.2 Chromatographic system
(See Chromatography 〈621〉, System Suitability.)
Mode: LC
Detector: UV 225 nm
Column: 4.6-mm × 15-cm; 3.5-μm packing L1
Column temperature: 35°
Flow rate: 1.5 mL/min
Injection volume: 10 μL
3.3 System suitability
Sample: Standard solution
3.4 Suitability requirements
Relative standard deviation: NMT 0.85% for six replicate injections
3.5 Analysis
Samples: Standard solution and Sample solution
Calculate the percentage of tamsulosin hydrochloride (C20H28N2O5S · HCl) in the portion of Tamsulosin Hydrochloride taken:
Result = (rU/rS) × (CS /CU) × 100
rU = peak response of tamsulosin from the Sample solution
rS = peak response of tamsulosin from the Standard solution
CS = concentration of USP Tamsulosin Hydrochloride RS in the Standard solution (mg/mL)
CU = concentration of Tamsulosin Hydrochloride in the Sample solution (mg/mL)
Acceptance criteria: 98.0%–102.0% on the dried basis
4 IMPURITIES
Residue on Ignition 〈281〉: NMT 0.1%
Organic Impurities, Procedure 1: Use for impurities eluting before tamsulosin
Buffer: Dissolve 8.7 mL of perchloric acid (70%) and 3.0 g of sodium hydroxide in 1900 mL of water. Adjust with 1 N sodium hydroxide to a pH of 2.0, and add sufficient water to make 2000 mL.
Mobile phase: Acetonitrile and Buffer (3:7)
System suitability solution: 25 μg/mL of Tamsulosin Hydrochloride and 50 μg/mL of Propylparaben in Mobile phase
Sample solution: 5.0 mg/mL of Tamsulosin Hydrochloride in Mobile phase
Standard solution: 10 μg/mL of Tamsulosin Hydrochloride from the Sample solution in Mobile phase
Chromatographic system
(See Chromatography 〈621〉, System Suitability.)
Mode: LC
Detector: UV 225 nm
Column: 4.6-mm × 15-cm; 5-μm packing L1
Column temperature: 40°
Flow rate: 1.3 mL/min
Injection volume: 10 μL
[Note—Record the chromatogram for NLT 1.5 times the retention time of tamsulosin.]
System suitability
Samples: System suitability solution and Standard solution
Suitability requirements
Resolution: NLT 12 between tamsulosin and propylparaben, System suitability solution. [Note—The elution order is tamsulosin followed by propylparaben.]
Relative standard deviation: NMT 4% for six replicate injections, Standard solution
Analysis
Samples: Sample solution and Standard solution
Calculate the percentage of any individual impurity eluting before the tamsulosin peak in the portion of Tamsulosin Hydrochloride taken:
Result = (rU/rS) × (CS /CU ) × 100
rU = peak response of each impurity eluting before tamsulosin from the Sample solution
rS = peak response of tamsulosin from the Standard solution
CS = concentration of the Standard solution (mg/mL)
CU = concentration of the Sample solution (mg/mL)
Acceptance criteria: The reporting level for impurities is 0.05%.
Any individual impurity: NMT 0.10%. [Note—If present, the des-ethoxy and methoxy impurities eluting at the relative retention time of about 0.8 are not separated by this method, and should be integrated together to determine conformance. (Des-ethoxy impurity is 2-methoxy-5- [(2R)-2-[(2-phenoxyethyl)amino]propyl]benzenesulfonamide, and methoxy impurity is 2-methoxy-5-[(2R)-2-[[2-(2- methoxyphenoxy)ethyl]amino]propyl]benzenesulfonamide.) NMT 0.15% of the sum of des-ethoxy and methoxy impurities is found.]
Organic Impurities, Procedure 2: Use for impurities eluting after tamsulosin
Buffer, Sample solution, and Standard solution: Prepare as directed in Procedure 1.
[Note—Use the Mobile phase in Procedure 1 to prepare the Sample solution and Standard solution.]
Mobile phase: Acetonitrile and Buffer (1:1)
Chromatographic system
(See Chromatography 〈621〉, System Suitability.)
Mode: LC
Detector: UV 225 nm
Column: 4.6-mm × 15-cm; 5-μm packing L1
Column temperature: 40°
Flow rate: 1.0 mL/min
Injection volume: 10 μL
[Note—Record the chromatogram for NLT 5 times the retention time of tamsulosin.]
System suitability
Sample: Standard solution
Suitability requirements
Resolution: Use a column that meets the resolution requirements in Procedure 1.
Relative standard deviation: NMT 4% for six replicate injections
Analysis
Samples: Sample solution and Standard solution
Calculate the percentage of any individual impurity eluting after the tamsulosin peak in the portion of Tamsulosin Hydrochloride taken:
Result = (rU/rS) × (CS /CU) × 100
rU = peak response of each impurity eluting after tamsulosin from the Sample solution
rS = peak response of tamsulosin from the Standard solution
CS = concentration of the Standard solution (mg/mL)
CU = concentration of the Sample solution (mg/mL)
Acceptance criteria: The reporting level for impurities is 0.05%.
Any individual impurity: NMT 0.10%
Total impurities: NMT 0.2%, including all impurities in Procedure 1 and Procedure 2
Enantiomeric Purity
Mobile phase: Hexane, dehydrated alcohol, methanol, and diethylamine (650:200:150:1)
System suitability solution: 40 μg/mL of USP Racemic Tamsulosin Hydrochloride RS in methanol
Sample solution: 2.0 mg/mL of Tamsulosin Hydrochloride in methanol
Standard solution: 2 μg/mL of Tamsulosin Hydrochloride from the Sample solution in methanol
Chromatographic system
(See Chromatography 〈621〉, System Suitability.)
Mode: LC
Detector: UV 225 nm
Column: 4.6-mm × 25-cm; packing L51
Column temperature: 40°
Flow rate: 0.5 mL/min
Injection volume: 10 μL
System suitability
Sample: System suitability solution
[Note—The relative retention times for the S-enantiomer and tamsulosin are 0.8 and 1.0, respectively.]
Suitability requirements
Resolution: NLT 2 between the S-enantiomer and tamsulosin
Analysis
Samples: Sample solution and Standard solution
Calculate the percentage of the S-enantiomer in the portion of Tamsulosin Hydrochloride taken:
Result = (rU/rS) × (CS /CU) × 100
rU = peak response of the S-enantiomer from the Sample solution
rS = peak response of tamsulosin from the Standard solution
CS = concentration of the Standard solution (mg/mL)
CU = concentration of the Sample solution (mg/mL)
Acceptance criteria: NMT 0.3% of the S-enantiomer
5 SPECIFIC TESTS
Loss on Drying 〈731〉
Analysis: Dry at 105° for 2 h.
Acceptance criteria: NMT 0.5%
6 ADDITIONAL REQUIREMENTS
Packaging and Storage: Preserve in tight containers, and store at controlled room temperature.
USP Reference Standards 〈11〉
USP Tamsulosin Hydrochloride RS
USP Racemic Tamsulosin Hydrochloride RS
