SIGNIFICANT CHANGE FOR BULK PHARMACEUTICAL EXCIPIENTS
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 1. INTRODUCTION
1.1 Purpose
This informational general chapter is intended to establish a uniform approach to the evaluation of the significance of changes involving the manufacture and distribution of pharmaceutical excipients. The purpose of the evaluation is to consider the impact of the change on the excipient and to determine whether or not the excipient user and/or regulatory authority should be informed. It is recommended that users and excipient suppliers utilize this chapter as the basis for notification requirements in quality and/or supply agreements.
1.2 Scope
This chapter is applicable to all excipients used in the manufacture of pharmaceutical products. Although the principles of good manufacturing practices (GMP) as described in Good Manufacturing Practices for Bulk Pharmaceutical Excipients (1078) are the focus of this chapter, there are instances where the changes affect the principles and practices described in Good Distribution Practices for Bulk Pharmaceutical Excipients (1197).
This chapter is intended to guide the assessment of a change that affects the manufacture and/or supply of the excipient. All significant changes should be considered as requiring user notification. The level of change is determined by the type of change as well as the results of the evaluation. Conclusions regarding the level of change should be justified and documented.
The principles set forth in this chapter should be applied once the excipient manufacturer has determined that an excipient is intended for use as a component of a drug product. This chapter applies to excipients manufactured by either batch processing or continuous processing, and the use of the term “batch” or “lot” may refer to either type of processing.
1.3 Principles Adopted
This chapter is internationally applicable, reflecting the diverse nature of pharmaceutical excipients, which often have uses other than pharmaceutical applications. It provides minimum recommendations when considering the impact of a change on the excipient. This general chapter cannot specify all national legal requirements or cover in detail the particular characteristics of every excipient.
When considering how to use this chapter, each excipient manufacturer should consider how it may apply to their product’s manufacturing processes. The diversity of excipients means that some principles of the chapter may not be applicable to certain excipients products and processes. The terminology “should” and “it is recommended” does not mean “must” and common sense should be used in the application of this chapter. However, the excipient manufacturer has a responsibility to follow the principles and guidance provided herein and to inform (notify) the drug product manufacturer(s) of any significant change (as outlined in this chapter), enabling the appropriate evaluation of the potential impact on the end drug product(s) due to the change.
The underlying principle of this chapter is that all changes should be regarded as significant (Level 2) and thus notifiable unless otherwise scientifically justified and documented.
Level 2 changes are discussed further in 2.2 Change Risk Levels.
1.4 Layout
The first section of this chapter provides the background discussion necessary for evaluating a change and determining the need to inform the user and/or regulatory authorities. The second section defines the term “significant change” and is followed by guidance on determining the risk that a change will be significant. Notification processes to customers and possibly regulatory authorities follows, and the chapter concludes with a series of specific changes in which the classification possibilities are examined. The Glossary contains terms and definitions used in all sections of this chapter. Appendix 1 includes case studies to show how the significance of change can be determined. Appendix 2 provides decision trees useful in considering the potential impact of a change on excipient performance.
1.5 General Considerations
EXCIPIENT COMPOSITION
Excipients frequently function because of their heterogeneous composition. They may contain multiple components that are dependent on their source and manufacturing process and may be necessary for their established functions. Potential change in composition is an important consideration when assessing significance of change.
DIFFERENTIATION OF EXCIPIENT MANUFACTURE
Whereas drug substances are typically of high purity, well characterized, and used in a limited number of therapeutic applications, the pharmaceutical excipient is often a natural substance, mixture, or polymer whose chemical and physical properties are more difficult to quantify. Unlike drug substances, pharmaceutical excipients are often used with a broad range of active ingredients and in a variety of finished dosage forms. Evaluating the impact of a change in the manufacture of an excipient on its performance in the finished dosage form/medicinal product is complex. Compounding the complexity of such evaluations by the excipient manufacturer are the unlimited possibilities for excipient function relative to new combinations of excipients, drug substances, and/or processing conditions that users may choose to apply.
EXCIPIENT GMP
At some logical processing step, as determined by the excipient manufacturer, GMP as described in (1078) and the NSF/IPEC/ANSI 363: Good Manufacturing Practices (GMP) for Pharmaceutical Excipients (1) or in the EXCiPACT cGMP standard (2) should be applied and maintained. Judgment, based on risk assessment and a thorough knowledge of the process and intended application, is required to determine from which processing step GMPs should be applied. Generally, this chapter should be applied from the point at which GMPs are applied; nevertheless, it may be important to consider changes that occur prior to this point (e.g., in raw materials) and evaluate them for significance.
2 2. SIGNIFICANT CHANGE
2.1 Definition of Significant Change
Significant change is defined as any change that alters an excipient’s physical, chemical, or microbiological properties from the norm, and/or any change that has the potential to alter the excipient’s performance in the dosage form.
2.2 Change Risk Levels
In the evaluation of the impact of changes to the excipient, it is recognized that even with objective criteria some judgment may be necessary. To facilitate the decision as to the significance of a change and the potential impact on the pharmaceutical dosage form, the types of changes are classified using two levels (examples of two case studies can be found in Appendix 1). The impact of the change should be assessed against the guiding principles listed in 3.2 Guiding Principles for Evaluating Change Significance, which often reflect the potential impact of the change on the performance of the excipient. Evaluation according to the principles of this chapter, the types of changes, and, where appropriate, risk assessment principles will determine the classification. The risk assessment needs to take into consideration the complexity of the change and the ability to fully characterize the impact. The two levels of change are:
• Level 1: Not Significant
• Level 2: Significant
The notification of Level 1 changes is not mandatory, and it is up to the excipient supplier to determine whether to notify the user. All Level 2 changes require user notification and, where appropriate, regulatory authority notification (see 4. Notification Requirements).
Unless otherwise justified and documented, all changes should be regarded as Level 2.
Guidance on specific changes is given in 5. Specific Changes, which includes examples that reinforce the position that certain changes are always notifiable (i.e., Level 2).
3 3. DETERMINATION OF SIGNIFICANCE/RISK ASSESSMENT
3.1 General
It is recommended that the evaluation of changes and the processes for the determination of significance are integrated into the documented procedure for change management. If the level of change is not specifically defined in 5. Specific Changes, further assessment is needed utilizing the risk assessment principles described in this section of the document.
All changes should be assumed to be Level 2 unless otherwise scientifically justified and documented.
3.2 Guiding Principles for Evaluating Change Significance
The following principles should be considered in determining the significance of the change with or without the use of a formalized risk assessment approach:
1. Complexity of the change(s) (including possible cumulative effects)
2. Level of understanding of historical norms
3. The ability to fully characterize the impact of the change on the:
• Excipient properties (i.e., chemical, physical, microbiological, composition profile, etc.)
• Intended excipient performance
• Equivalency of the composition profile comparing pre-change and post-change batches
○ No new component is present at or above 0.10%; neither has a component that was previously present at this level disappeared (3). Minor components, including residual solvents and elemental impurities, remain within historical norms for the batches produced before the change.
4. The ability to assess the change in trial batches and/or model products
5. Manufacturer’s understanding of the users’ application(s) and use(s) of the product or critical material attributes
6. The content and requirements of any quality or technical agreements that are in place
7. In the case of raw material changes—the level of knowledge, understanding, credibility, and reliability of the raw material manufacturer and the relationships that exist within the raw material supply chain
8. The content of regulatory documents used in the applications under the excipient manufacturer [drug master file (DMF), Certificate of Suitability to the monographs of the European Pharmacopoeia (CEP)] or submitted to customers for their own regulatory applications (technical dossier)
3.3 Change Management Documentation
The change management documentation should describe the nature of the change, the reason it may be significant, the testing to be performed to evaluate the change, the criteria for determining the significance, and the final decision on the level of the change.
The associated risk assessment and decisions made should be documented.
Evidence may be obtained after implementation or testing that requires the original decision on the level of change to be re-evaluated. Under such circumstances, the reasons for the re-evaluation and the decision based on the re-evaluation should be scientifically justified and fully documented.
3.4 Justification for Level 1 Change
Level 1 changes that are specifically given in this section or in the decision tree do not need further justification. However, a Level 1 change that is determined through risk assessment should be justified and documented. It is recommended that the justification includes a detailed rationale explaining the conclusion that the change does not pose a significant risk to the medicinal drug product quality.
3.5 Testing
The results from the testing of an appropriately determined number of pre- and post-change batches of excipient, or results from predefined operational time periods, should be compared for evaluation of the change prior to final implementation.
Where the manufacture of a predetermined number of multiple post-change batches for evaluation is not practical, concurrent evaluation of batches produced after the change has been implemented should be compared to historical data from a predetermined and sufficient number of batches manufactured before the change.
One method to compare the new data with historical data is the two one-sided test (TOST) for demonstrating equivalence (see Analytical Data—Interpretation and Treatment (1010)). As a further check on consistency, the new batch property specifications can be plotted on standard statistical quality control (SQC) charts, along with the batch results from the selected pre-change batches or operational time periods and in-process testing and controls. The choice of statistical test, if used, should be justified and documented.
Samples for comparison purposes must be suitable to evaluate the impact of the change. Consideration should be given to the stability of the samples since the batch was produced. The comparison should encompass, where appropriate, chemical and physical properties, microbiological properties, composition profile, stability, and performance. Sample types could include retention samples or other samples that have been stored under appropriate conditions.
Chemical and physical properties lend themselves to quantitative measurement. Often these properties are part of the specification for the excipient. As such, for those properties potentially affected, there should be a large body of historical data for comparison with the corresponding data for the excipient made after the change. However, there may be additional properties that should be assessed on the basis of the type of change being made.
Where appropriate, the process validation should be updated to reflect the changed process.
4 4. NOTIFICATION REQUIREMENTS
Level 2 changes always require user notification. The user should be given as much advance notice of impending changes as is reasonably possible. The timing of the notification will rely on the specifics of the particular change being made. The notification should include the date of implementation and the urgency of the change.
The user may require time to complete the evaluation of the impact of the change on drug products. During this period the user may request inventory of the excipient produced before the change was made. Where possible, the manufacturer should plan for the change with this eventuality in mind and collaborate with the user to develop an appropriate implementation plan.
It is recommended that a summary of the changes and any supporting data and information be provided to the user to aid in the evaluation. As further applicable data become available (e.g., stability studies), they should be communicated.
On occasion, there may be a need for emergency changes. In such cases, it should be understood that notice periods may be very short and supporting data and information, as detailed above, may not be available at the time of initial notification.
If a regulatory filing exists, such as an excipient DMF or CEP, the authorities may require notification of significant changes involving the manufacture of excipients. Holders of United States DMFs should consult the FDA Guideline for Drug Master Files (4) for more details.
5 5. SPECIFIC CHANGES
The types of change described in this section should drive decisions on the significance and determination as to whether a change is Level 1 or Level 2. The following information should be considered when assessing the types of change. If a decision cannot be made by using the guidance in this section, then the risk assessment approach in 3. Determination of Significance/Risk Assessment should be used to make a decision.
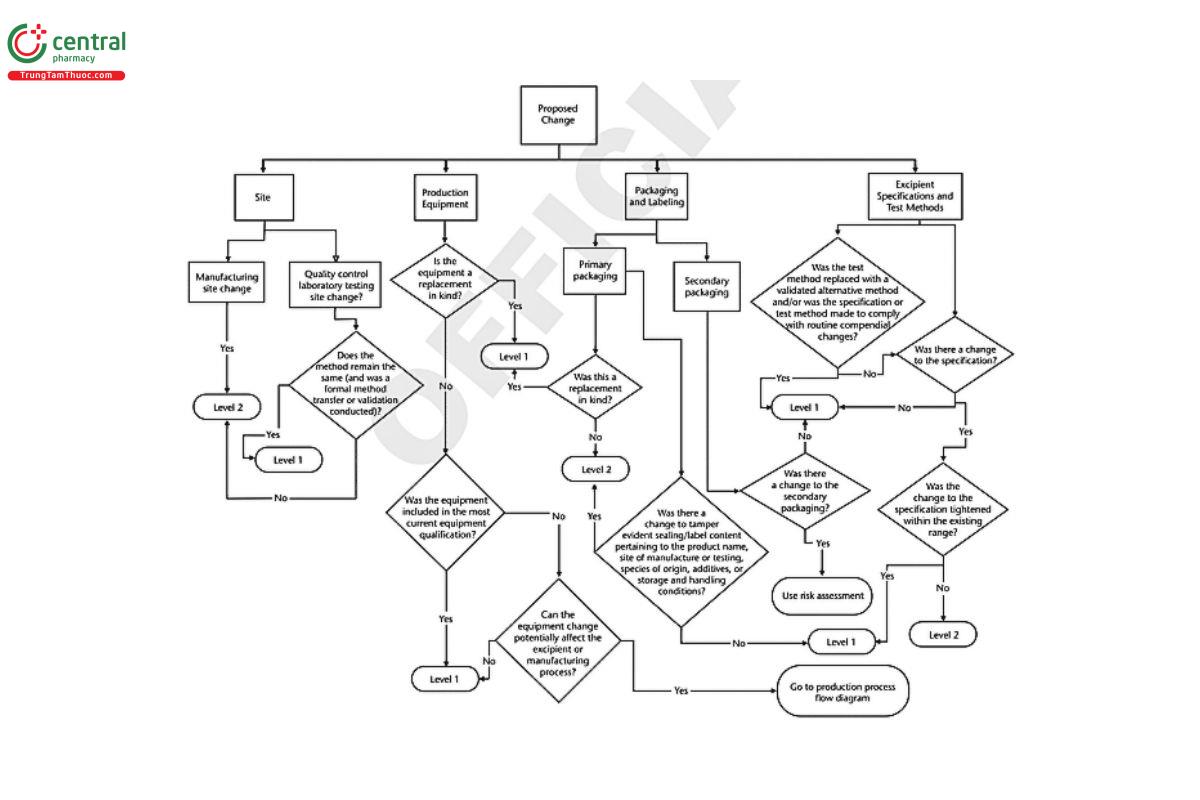
5.1 Changes to the Site, Infrastructure Used to Manufacture, and Distribution of the Excipient
SITE CHANGE
A change in manufacturing site could be for either the production or packaging of the excipient. A change in the manufacturing site is a Level 2 change.
If the change involves the site of the quality control laboratory, then the impact hinges on the test method. If the method remains the same, the change is Level 1 provided a formal method transfer or validation is conducted. If the new laboratory uses a different analytical technique, then the change is Level 2.
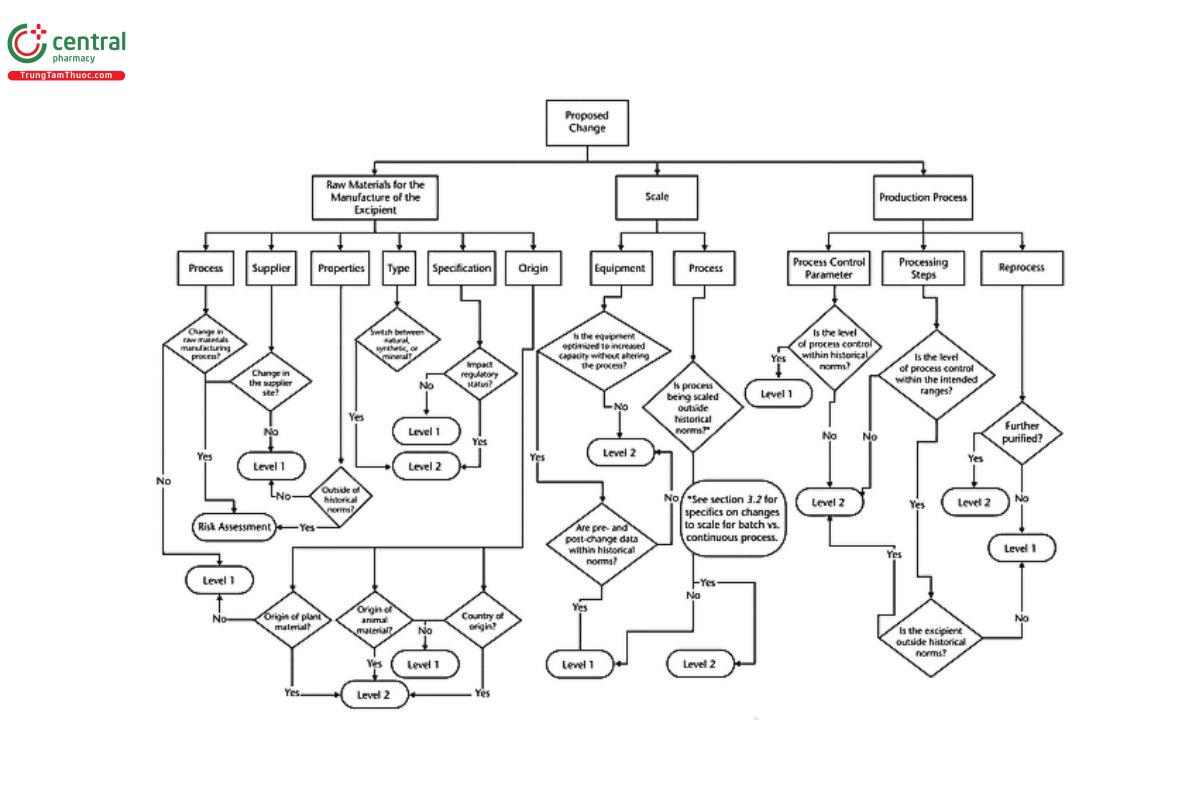
SCALE
Manufacturers may change the scale of their production. If the process is being scaled outside the historical norms, and the excipient as evaluated is also outside the historical norms, the change is significant and thus Level 2. If the existing equipment is optimized to increase capacity without altering the process, often found in continuous processing, then the change is Level 1 provided that a comparison of pre- and post-change data is within historical norms. However, careful consideration should be given to changes that can clearly impact the properties and/or functionality of the excipient.
A change in batch size for a continuous process does not necessarily mean a change in scale. If the same process and equipment train is used and there is no change in the process and control parameters (i.e., simply a longer time of running to define a batch), then the change is Level 1. If the process equipment, process parameters, or control parameters are changed, then the change is Level 2.
PRODUCTION EQUIPMENT
The evaluation of equipment change is predicated on whether the new equipment is equivalent to the equipment it replaces. Generally, equipment that is a replacement in kind is a Level 1 change. If the new equipment is not a replacement in kind but was included in the most current equipment qualification, then the change is still Level 1. If the new equipment is not a replacement in kind and was not included in the most current equipment qualification, then the change is Level 2. If an equipment change could potentially affect the excipient or manufacturing process, it should be evaluated using the risk assessment approach in 3. Determination of Significance/Risk Assessment.
PRODUCTION PROCESS
A change in production process involves changes to the synthetic route or target levels for parameters such as temperature, pressure, flow rate, the processing aids to be used, the sequence of operating steps, and the operation to be performed. Each type of process change is further described in Appendix 2.
If there is a change in a process parameter within the intended range, such as operating at a new target within that range, then the change is Level 1.
If a process parameter or processing step is outside the intended range (i.e., validated range and/or design space) and the excipient as evaluated is also outside the historical norms, then the change is Level 2.
If a change in the production process is made that increases the level of process control within historical norms, then the change is Level 1.
Introduction of new products into production equipment that was, until that point, dedicated to one excipient is a Level 2 change.
PACKAGING, LABELING, AND DOCUMENTATION
Any change in the primary or barrier packaging that is a replacement in kind is Level 1. Replacement in kind applies to packaging constructed of the same materials and sealed in the similar manner and liners made of the same materials such that the protection provided to the excipient by the packaging system (container/closure system) is the same as before the change. Any change that is not a replacement in kind is Level 2.
Any change to seals that are intended to be tamper evident is Level 2.
Any change to labeling or documentation pertaining to the company name, product name, batch/lot numbering scheme, site of manufacture or testing, species origin, additives, or storage and handling conditions is a Level 2 change.
Changes in secondary packaging—packaging materials that do not have direct contact with the excipient and are not considered barrier packaging components—should be assessed using the risk assessment principles outlined in 3. Determination of Significance/Risk Assessment to determine the appropriate level of change.
EXCIPIENT SPECIFICATIONS AND TEST METHODS
Changes to excipient specifications are Level 2 unless the specification is tightened within the existing range.
Any change to an excipient specification or test method made to comply with routine compendial changes is Level 1.
Replacement of an excipient test method with an equivalent validated alternative method is a Level 1 change.
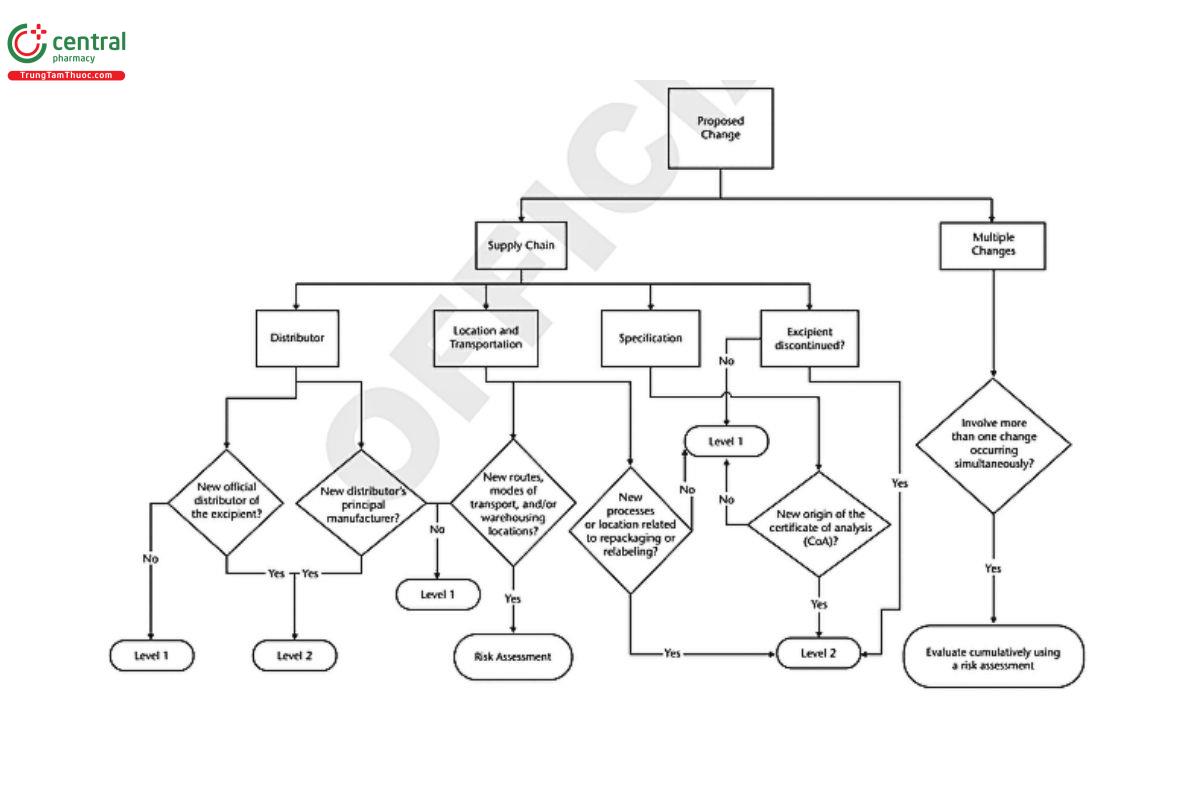
SUPPLY CHAIN
Changes by the excipient manufacturer of their official distributor(s) are Level 2.
[Note—For distributors, any change to their supply source is Level 2. Changes due to excipient discontinuation are Level 2.]
Changes to distribution and warehousing locations should be evaluated using risk assessment principles (see 3. Determination of Significance/Risk Assessment).
Changes to processes or locations related to repackaging or relabeling are Level 2.
For more information on good distribution practices, please refer to (1197).
5.2 Determination of Impact of Changes on Excipient Quality and Performance
INTRODUCTION
It is important to give careful consideration to any processing changes after the excipient has been synthesized or isolated but prior to packaging. However, it must be recognized that a change made earlier in the process can result in a change in the excipient performance and it is recommended that such changes also be considered.
When determining impact of changes, as a minimum, the criteria below should be evaluated. For those identified as applicable, all conclusions or decisions should be scientifically justified and documented.
The following represents the minimum criteria that should be used for evaluating the impact of change:
1. Change in the physical properties of the excipient
2. Change in the chemical properties of the excipient
3. Change in the microbiological properties of the excipient
4. Change in the composition profile of the excipient
5. Change in the origin, type, or site of any raw materials
6. Change in the distribution of the excipient
7. Change in the origin and/or type of packaging and/or labeling
8. Change in excipient stability
9. Change in the regulatory status of the excipient
10. Change in the compliance with compendia or other regulation
11. Potential to change the intended performance of the excipient based on the excipient manufacturer’s understanding
Additional guidance on these points is provided in the following sections. Changes to any of these attributes may impact the excipient quality and/or performance in the dosage form, and it is important to identify objective criteria for evaluation. An assessment of the impact of such changes provides the excipient manufacturer with the rationale for determining the significance of the change to the user of the excipient and the justification for notifying the user and/or the regulatory authorities.
PHYSICAL PROPERTIES
Evaluation of the physical properties of an excipient should include, at a minimum, all applicable specifications and other relevant parameters that define the physical properties of the excipient. Physical properties should be considered based upon the physical form of the excipient. The number of batches chosen for evaluation should be justified. A comparison of these test results for the excipient pre- and post-change should be carried out to determine if there has been a change from historical norms as well as to assess the likely impact of such change(s). For example, the following properties should be considered if relevant:
• Bulk density (loose and tapped)
• Surface area
• Particle shape and structure
• Particle size distribution
• Color
• pH
• Viscosity
• Molecular weight distribution
CHEMICAL PROPERTIES
Evaluation of the chemical properties of an excipient should include, at a minimum, all applicable specifications and other relevant parameters that define the chemical attributes of the excipient. The number of batches chosen for evaluation should be justified. A comparison of these test results for the excipient pre- and post-change should be carried out to determine if there has been a change from historical norms and to assess the likely impact of such change(s).
MICROBIOLOGICAL PROPERTIES
Change in processing steps, raw materials, water, or equipment, can impact control of microorganisms in the excipient. Therefore, the effect of the change on the microbiological properties should be evaluated, particularly for excipients susceptible to microbial growth.
When the risk-based evaluation determines that testing is required, a comparison of the microbiological properties pre- and post-change should be carried out to determine if there has been a change from historical norms and to assess the likely impact of such change(s). The number of batches chosen for evaluation should be justified.
POTENTIAL IMPACT ON THE INTENDED PERFORMANCE OF THE EXCIPIENT BASED ON THE EXCIPIENT MANUFACTURER'S UNDERSTANDING
Although performance/functionality is often defined by the previous parameters (physical, chemical, and compositional properties), objective criteria for evaluating other potential changes to excipient performance or functionality are desirable. However, the nature of this type of study can vary broadly based upon the excipient, its application in the dosage form, and the capabilities of the excipient manufacturer. It must also be recognized that the excipient manufacturer may not always be aware of all applications of the excipient. Therefore, this chapter cannot provide objective criteria for such studies but stresses the importance of such consideration by the excipient manufacturer. If there is a potential that the performance or functionality of the excipient may be impacted by the change, users should be notified. Material samples should be provided if requested so the user can determine the impact of the change on their finished pharmaceutical product(s). Excipient Performance (1059) can provide guidance in this area.
COMPOSITION PROFILE
Objective criteria are also necessary when considering the impact on the composition profile for an excipient as a result of changes (3). The composition profile may include, if relevant:
• Identified organic components
• Unidentified organic components
• Residual solvents
• Identified inorganic components
• Water content
The feasibility of developing a composition profile will vary with the nature and origin of the excipient. It is important to note that the presence of impurities and concomitant components in some excipients is extremely difficult to quantify. Thus, an excipient manufacturer may not have developed a complete quantitative composition profile. In such cases, it is important for the excipient manufacturer to document efforts to identify and quantify the concomitant components that may be present so as to justify the limited results and to justify other means by which changes may be evaluated.
A comparison of the composition profile of the excipient pre- and post-change should be carried out to determine if there has been a change from historical norms. The number of batches chosen for evaluation should be justified.
Changes in the levels of residual solvents should be considered when determining the significance of a change. See Residual Solvents (467) for details.
Changes in the levels of elemental impurities should be considered when determining the significance of a change. Water content can have an impact on excipient performance in the preparation of the pharmaceutical dosage form and the performance of the dosage form in vivo. Therefore, a change in the water content beyond the range typical for excipient production, even if within the compendial or specification limit, can impact the stability and performance of the drug product and/or end use of the excipient.
CHANGE IN THE ORIGIN, TYPE, OR SITE OF RAW MATERIALS
It is recommended that excipient manufacturers and their raw material suppliers agree to a change notification process wherein they are notified of significant changes to the raw materials. Changes in the properties of the raw materials outside of historical norms should be evaluated using risk assessment principles (see 3. Determination of Significance/Risk Assessment).
Changes to the specifications of the raw materials that may impact the regulatory status of the excipient are Level 2.
Changes in the type of raw material (natural, synthetic, or mineral) are Level 2.
Changes to the origin of plant materials (e.g., corn vs. potato) are Level 2.
Changes to the origin of animal materials (e.g., bovine vs. porcine) are Level 2.
Changes in the country of origin of raw materials, which may impact regulatory status of the excipient, are Level 2.
Changes in the manufacturing process of the raw material should be evaluated using risk assessment principles (see 3. Determination of Significance/Risk Assessment).
Changes to the supplier or site of the raw material manufacturer should be evaluated using risk assessment principles (see 3. Determination of Significance/Risk Assessment).
The origin of the raw material includes the country of origin, geological origin, and species (animal or plant) origin. The type of raw material includes whether the material is natural or synthetic, the physical form and/or preliminary extraction of the raw material, and/or processing prior to delivery to the excipient manufacturing site. The site of the raw materials includes the actual manufacturing site or distribution points.
Changes in the species of origin, from animal to animal or from plant to animal, may cause a change in the viral and microbiological safety profile of the excipient.
A change in the country of origin of a raw material can impact the status of the excipient as it relates to the potential presence of bovine spongiform encephalopathy (BSE) or transmissible spongiform encephalopathies (TSE) material or genetically modified organisms (GMO). The country of origin of animal-derived raw material, or components used in the manufacture of the raw material, can result in noncompliance with relevant TSE regulations (5–7). These aspects may have impact on the regulatory status (as discussed further in this section and in 4. Notification Requirements).
Switching raw material from animal-derived to plant-derived, or from one plant species to another, raises the potential for the presence of plant-based allergenic material in the excipient. Changes to plant-derived raw materials can also affect the GMO status of the excipient.
A change in the geological origin of mineral-based excipients can alter the composition of the excipient. Geological formations containing the same mineral can still differ in their chemical composition (particularly relating to minor concomitant components), crystalline structure, density, inorganic components, etc. A change in geological origin of a raw material can impact the excipient chemical or physical properties, the composition profile, or excipient performance/functionality.
Changes to or additions of a further site of manufacture, even from the same supplier, can result in changes to the raw material that can impact the properties of the excipient. The equipment and processes between sites may differ. Changes in the distribution points of the raw material supply chain may impact its quality.
CHANGE IN THE DISTRIBUTION OF THE EXCIPIENT
Assurance of the quality (purity, integrity, safety) of the excipient may be impacted by how the excipient is transported from the manufacturer to the end user, considering the key distribution points within the supply chain. Each partner in the supply chain has the potential to affect the quality of the excipient. For example, storage and transportation conditions may affect excipient quality, excipient stability, or the potential to become contaminated. Therefore, changes in the distribution or supply chain can be important. It is not anticipated that all changes in carriers must be a notifiable change; however, the excipient manufacturer must evaluate any known carrier changes to be assured that there will be no changes in storage and/or transportation conditions.
CHANGE IN THE ORIGIN OR TYPE OF PACKAGING OR LABELING
A change in the primary or barrier packaging components can involve the manufacturer, country of origin, or materials of construction. The evaluation of the primary packaging should include the impact on the composition profile, excipient stability (see Impact on Excipient Stability), and interactions between the excipient and the packaging (leachables/extractables). The evaluation of barrier packaging, if separate from primary packaging, should include, as a minimum, the impact on excipient stability.
A change in the labeling may impact information that the user needs to properly identify or use the excipient. In some cases minor labeling changes that involve simple things such as graphic design may not be significant. However, if information on the label changes from what was previously provided, this must be carefully assessed to determine the level of notification necessary.
Such changes may necessitate notifying the regulatory authority, if the excipient company has filed information with regulators that would require notification (such as a DMF or CEP).
IMPACT ON EXCIPIENT STABILITY
An assessment should be made for the potential of the change to impact the stability of the excipient. Where this potential is identified, stability studies should be initiated as part of the evaluation of change. If the risk assessment shows that stability implications are predictable, stability studies may be done concurrently with notification and implementation of the change (8).
CHANGE IN THE REGULATORY STATUS OF THE EXCIPIENT
Changes can occur in regulations, guidelines, and directives that may affect the regulatory status of the excipient. An evaluation of the change(s) should be carried out for the potential of the change to impact registration dossiers, such as drug master files, certificates of suitability, drug import/export licenses, and manufacturing authorization registrations (as applicable).
CHANGE IN COMPLIANCE WITH A COMPENDIUM OR OTHER REGULATION
When changes to compendial monographs or regulations occur, evaluation should be carried out to confirm continued compliance with these requirements.
Removal of an existing compendial claim is a significant change; however, the expansion of claims to include compliance with additional regulatory requirements is not necessarily a significant change.
5.3 Multiple Changes
Multiple changes involving more than one type of change, as discussed here, may occur simultaneously. Where Level 2 changes have been identified, user notification should proceed without delay. The other changes should be evaluated cumulatively using risk assessment principles (3. Determination of Significance/Risk Assessment) to determine the appropriate level for the totality of changes.
5.4 Discontinuation of an Excipient
If an excipient is to be discontinued, this will have a significant impact on the user since the user will have to qualify an alternative excipient and/or supplier. Depending on the particular use of the excipient, the new excipient source may well have a significant impact on performance and will require careful evaluation by the excipient user.
GLOSSARY
Bovine Spongiform Encephalopathy (BSE): A slowly progressive, degenerative, fatal disease affecting the central nervous system of adult cattle. The exact cause of BSE is not known but it is generally accepted by the scientific community that the likely cause is an infectious form of a type of protein, called a prion, that is normally found in animals. In cattle with BSE, these abnormal prions initially occur in the small intestines and tonsils. They are found in central nervous tissues, such as those of the brain and spinal cord, and other tissues of animals experiencing later stages of the disease. A disease similar to BSE, called Creutzfeldt-Jakob Disease (CJD), is found in people. A variant form of CJD (vCJD) is believed to be caused by eating contaminated beef products from BSE-affected cattle.
Bulk Pharmaceutical Excipient (BPE): See Excipient.
Chemical Property: A quality parameter that is measured by chemical or physicochemical test methods.
Certificate of Suitability to the monographs of the European Pharmacopoeia (CEP): Certificate granted by the European Directorate for the Quality of Medicines (EDQM) to manufacturers of active ingredients or excipients confirming that the applicable European Pharmacopoeia (Ph. Eur.) monographs and general chapters are adequate to control the chemical purity of the material. A CEP also can be granted to confirm that the material conforms to the Ph. Eur. general chapter 5.2.8 “Minimising the risk of transmitting animal spongiform encephalopathy agents via medicinal products” even if the material itself does not have a Ph. Eur. monograph.
Concomitant Component: A minor component of an excipient that accompanies the nominal component, which is identified either in the title or definition of a monograph. Concomitant components are characteristic of many excipients and are not considered to be impurities if there is no negative impact on drug products. Some, but not all, concomitant components are defined or specified in excipient monographs. Added substances are not considered concomitant components. (Any component that can be considered a toxic impurity because of significant undesirable biological effects is not considered to be a concomitant component.)
Continuous Process: A process that continually produces material from a continuing supply of raw material.
Decision Tree: A visual presentation of the sequence of events that can occur, including decision points.
Drug Master File (DMF): Detailed information concerning a specific facility, process, or product submitted to a drug regulatory authority (such as the US Food and Drug Administration, Health Canada, and Japan's Pharmaceuticals and Medical Devices Agency) and intended for incorporation by reference into a new drug application, supplemental new drug application, abbreviated new drug application, investigational new drug application, or biological license application.
Drug Substance: Any substance or mixture of substances intended for use in the manufacture of a drug product that, when used in the production of a drug, becomes an active ingredient of the drug product. Such substances are intended to furnish pharmacological activity or other direct effect in the diagnosis, cure, mitigation, treatment, or prevention of disease or to affect the structure or any function of the body of humans or animals.
Excipient: Substances other than the drug substances that have been appropriately evaluated for safety and are intentionally included in a drug delivery system.
Foreign Substance: A component present in the bulk pharmaceutical excipient but NOT introduced into the excipient as a consequence of its synthesis or purification and not necessary to achieve the required functionality (formerly referred to as contaminant).
Functionality: A desirable property of an excipient that aids and/or improves the manufacture, quality, or performance of the drug product.
Genetically Modified Organism (GMO): An organism, with the exception of human beings, in which the genetic material has been altered in a way that does not occur naturally by mating and/or natural recombination.
Impurity: Any substance that detracts from the quality of the excipient (i.e., that is not the substance appearing in the official name, or concomitant component, or an added substance as defined above).
Packaging: The container and its components that hold the excipient for transport to the customer.
Physical Property: A quality parameter that can be measured solely by physical means.
Process: The combination of operating steps including synthesis, isolation, purification, packaging, etc., that produces the finished excipient.
Process Parameter: A measurable operating condition.
Process Validation: A documented program that provides a high degree of assurance that a specific process will consistently produce a result that will meet predetermined acceptance criteria.
Raw Material: A general term used to denote starting materials, reagents, and solvents intended for use in the production of intermediates or excipients. Raw material and starting material are not equivalent; starting material has a different meaning in a regulatory context.
Replacement In Kind: Manufacturing equipment that uses the same operating principles and is of similar construction; packaging components that are made with the same materials of construction and sealed in a similar manner.
Residual Solvent: Organic chemical solvents that are used or produced in the manufacture of active substances or excipients or in the preparation of medicinal products.
Significant Change: Any change that alters an excipient’s physical, chemical, or microbiological properties from the norm, and/or any change that has the potential to alter the excipient’s performance in the dosage form.
Site: A defined location of the equipment where the excipient is manufactured. It may be within a larger facility. A change in site may be to a different part of the existing facility, but in a different operational area, or to a remote facility including that of a contract manufacturer.
Solvent: An inorganic or organic liquid used as a vehicle for the preparation of solutions or suspensions in the manufacture of an excipient.
Specification: A list of tests, references to analytical procedures, and appropriate acceptance criteria, which are pre-established numerical limits, ranges, or other criteria for the tests described, that the material is required to meet.
Starting Material: A raw material or intermediate defined at the starting point for excipient GMPs and used in the production of an excipient that is incorporated as a significant structural fragment or that is purified to meet the quality requirement of an excipient.
Statistical Quality Control (SQC): The plotting of sequential test results to show their variation relative to the specification range and their normal variation.
Transmissible Spongiform Encephalopathy (TSEs): TSEs are rare forms of progressive neurodegenerative disorders that affect both humans and animals and are caused by similar uncharacterized agents that generally produce spongiform changes in the brain. Specific examples of TSEs include: scrapie, which affects sheep and goats; BSE, which affects cattle; transmissible mink encephalopathy; feline spongiform encephalopathy; chronic wasting disease (CWD) which affects mule deer, white-tailed deer, black-tailed deer, and elk; and Creutzfeldt-Jakob disease, variant Creutzfeldt-Jakob disease (vCJD), Gerstmann-Sträussler syndrome, fatal familial insomnia, and kuru, which affect humans.
6 APPENDICES
Appendix 1: Case Studies
CASE STUDY EXAMPLES
The two examples below describe changes that are indeterminate and require risk assessment of the significance of change.
EXAMPLE 1
The excipient is a polymer. The processing involves taking the raw materials and polymerizing them, then applying a finishing process leading to the final product. The conditions for the polymerization are to be changed. No other aspects of processing are to be changed.
The processing changes lead to an improved control of the polymerization process. However, the product still meets the existing compendial requirements, and none of the measured parameters are out of trend as a result of the change. Schematically the process is as follows:
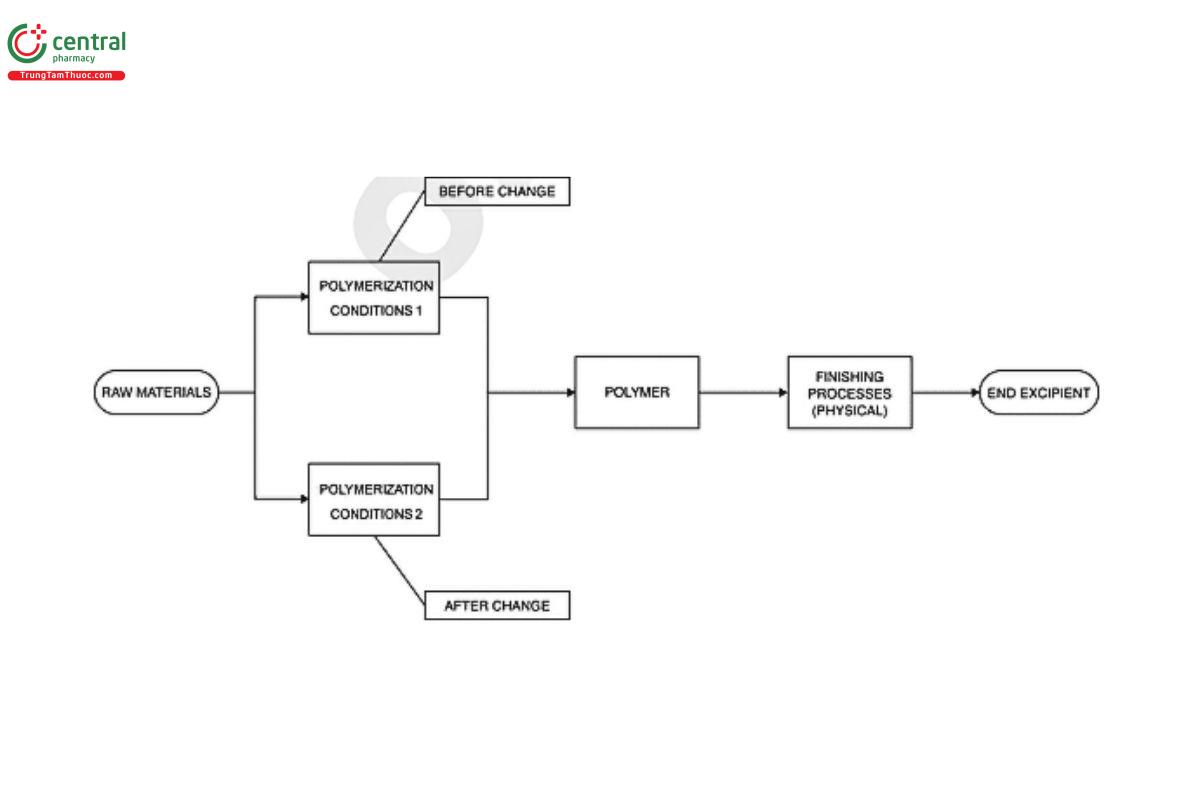
Applying the guidance in this general chapter, this is not clearly a Level 2 change to warrant user notification, neither is it a case where the change is automatically a Level 1 change.
Key to the assessment of this change is to determine if there are any other changes to the characteristics of the product resulting from the change. Changes in the method of polymerization can lead to different molecular weight distributions (which may not be a compendial test) and differences in the composition profile of the excipient. Therefore, these aspects of the excipient need to be assessed against the historic norms for the original process before a decision can be made. The difference in composition is especially relevant because minor
components and even impurities resulting from the original process, which were contributing to the performance and functionality of the excipient, have been removed from the excipient after the change.
If there is no evidence to indicate that the excipient has changed in any specification parameter or within the other assessments that the manufacturer can define, then the change is classified as Level 1. The manufacturer will document this rationale in the management of change system.
If no justification can be determined for designating this a Level 1 change, then this is a Level 2 change requiring user notification.
EXAMPLE 2
An excipient that is a proprietary blend of ingredients is prepared by a continuous manufacturing process involving a high temperature step. The proposed change is to increase the flow rates within the originally defined equipment capability (i.e., within the overall process design space) although these flow rates are outside the current operating ranges. No other aspects of processing are to be changed—only this step is being altered—and all raw materials and final processing steps are the same.
The increased flow rate is desired for economic reasons. The product made after the implementation of this change still meets the existing selling specification. Minor degradation of one of the components is technically unavoidable in the manufacturing process at the temperatures required for processing. As a result of the increased flow rates, the residence time at high temperature is reduced and the levels of degradants, though within historical ranges, are consistently toward the low end of that range.
Applying the guidance of this chapter, this is not automatically a Level 2 change to warrant user notification nor is it a case where the change is automatically a Level 1 change. Given the circumstances above, a reasonable justification for this being a Level 1 change can be made.
However, the manufacturer has information that the degradants may have an impact on some user applications. Although the degradants remain within typical historical ranges, there is reason to believe they will now trend lower within the permitted ranges due to the changed process. With this additional information, the change becomes Level 2 and requires user notification.
Appendix 2: Decision Tree
A decision tree has been developed to graphically aid and clarify the change levels in this chapter. The decision tree begins with the proposed change and guides the manufacturer to an indication of the likelihood the change will impact the excipient user. The decision tree classifies the types of change that occur in excipient manufacture as involving the site of manufacture, processing steps, packaging, or testing and quality control.
For convenience, the decision tree has been split into three parts (see Figure 1, Figure 2, and Figure 3).