SEMISOLID DRUG PRODUCTS-PERFORMANCE TESTS
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- SCOPE
- INTRODUCTION
- IVRT
- IVPT
- IN VITRO RELEASE TEST (IVRT)
- IN VITRO PERMEATION TEST (IVPT)
- IVRT Versus IVPT
- EQUIPMENT
- General Recommendations for Equipment Qualification and Requalification
- Vertical Diffusion Cell (VDC)
- General IVRT VDC Equipment Set Up
- Membrane mounting and dosing
- Dosing vdc with open donor compartments
- Dosing vdc model a diffusion cells
- Sampling vdc (general procedure for all models of vdc)
- Sampling by withdrawal and replacement
- Displacement sampling
- General IVPT VDC Equipment Set Up
- VDC Qualification
- VDC Preventative Maintenance Procedure and Requalification
- Immersion Cells
- General IVRT Immersion Cell Equipment Set Up
- Immersion Cell Qualification
- Flow-Through Diffusion Cell (FDC) for IVPT
- General IVPT FDC Equipment Set Up
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 SCOPE
This chapter provides general information about developing in vitro performance tests to evaluate drug release or skin permeation for topical and transdermal semisolid and liquid-based dosage forms, including but not limited to, creams, gels, ointments, pastes, suspensions, lotions, and foams. For information related to in vitro performance tests that evaluate drug release for transdermal delivery systems (TDS), refer to Drug Release 〈724〉.
In this chapter, the term “drug” is utilized to refer more generally to active ingredients, potentially including active ingredients that may not be considered drugs (e.g., sunscreen active ingredients).
For information related to product quality tests for topical and transdermal dosage forms, refer to Topical and Transdermal Drug Products-Product Quality Tests 〈3〉.
2 INTRODUCTION
This chapter provides general information for developing in vitro release test (IVRT) methods to assess the rate of drug release from topical and transdermal semisolid and liquid-based dosage forms, including but not limited to, creams, gels, ointments, pastes, suspensions, lotions, and foams applied on the skin and other mucosal membranes. Definitions and descriptions of these dosage forms can be found in Pharmaceutical Dosage Forms 〈1151〉. This chapter also provides general information for developing in vitro permeation tests (IVPT) to assess the rate and extent of drug permeation into and through the skin from semisolid and liquid-based drug products. The same equipment and similar methodological principles/procedures may be relevant for IVPT methods with other epithelial membranes (for information on appropriate mucosal membranes, see Biological Membrane below).
2.1 Drug Product Quality and Performance Tests
Drug product tests are divided into two categories: 1) those that assess general quality attributes i.e., product quality tests, and 2) those that assess product performance, e.g., using an IVRT/IVPT method. Product quality tests characterize the physicochemical and/or structural attributes (e.g., pH, particle size/morphology etc.) of the formulation. By contrast, product performance tests assess how a drug product functions under specified conditions, which may provide information relevant to its in vivo performance.
Product quality tests are generally useful to characterize and/or compare product quality attributes that can control the performance of the product, including, but not limited to, identity, strength, purity, uniformity, pH, particle size/morphology, apparent viscosity. Details about these product quality tests can be found in 〈3〉. The performance tests that are the focus of this general chapter are specifically useful to characterize and/or compare the linear (steady state) drug release rate (using an IVRT) or the dynamic rate and extent to which a drug permeates into and through the skin (using an IVPT) from semisolid and/or liquid-based dosage forms, under the conditions of the test.
Potential contexts for use of IVRT and IVPT methods are summarized below.
3 IVRT
- Characterizes the steady-state drug release rate for a product batch. When the performance (release rate) of a topical product has been characterized by a validated IVRT method using the same batch of product that supported a demonstration of the safety and/or efficacy of the product, that performance (release rate) may have the potential to serve as a basis of reference for the product in the future.
- Characterizes the influence of processes, formulations, and/or manufacturing differences on a drug product by comparing the steady-state drug release rate for the postchange (test) and prechange (reference) products, typically in the context of scale-up or post–approval changes for an approved drug product, which can support a demonstration of equivalence in certain situations.
- Characterizes the influence of processes, formulations, and/or manufacturing differences on a drug product by comparing the steady-state drug release rate for a prospective generic (test) product and an approved (reference standard) product, which can support a demonstration of bioequivalence in certain situations.
4 IVPT
- Characterizes the rate and extent to which a drug permeates into and through the skin for a product batch.
- Characterizes the influence of process, formulation, and/or manufacturing differences on a drug product by comparing the rate and extent to which a drug permeates into and through the skin. This may occur in the context of comparing prototype formulations of a topical product, or potentially in the context of comparing postchange (test) and prechange (reference) products.
- Characterizes the influence of process, formulation, and/or manufacturing differences on a drug product by comparing the rate and extent to which a drug permeates into and through the skin for a prospective generic (test) product and an approved (reference standard) product, which can support a demonstration of bioequivalence in certain situations.
(See the FDA Guidance for Industry, Nonsterile Semisolid Dosage Forms; Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Release Testing and In Vivo Bioequivalence Documentation; available at www.fda.gov/media/71141/download.)
5 IN VITRO RELEASE TEST (IVRT)
An IVRT is intended to characterize a steady-state drug release rate from a semisolid formulation under a specific set of conditions (i.e., a given set of method parameters). IVRT methods are typically precise and reproducible. However, if the parameters of the IVRT method change, then the release rate measured may also change. Notably, when the IVRT method is consistent, then changes in the release rate between a test and reference formulation are reflective of a difference in the physicochemical properties and/or the structural arrangement of matter in the formulations. Thus, IVRT methods can provide a sensitive and discriminating way to monitor for differences in the physicochemical or structural properties of a test versus the reference formulation.
The steady-state drug release kinetics are not representative of the finite dose (non-steady-state) kinetics of drug permeation through the skin. The drug release rate measured by an IVRT method should not be misconstrued to represent a “true” drug release rate for that formulation, or even a biorelevant release rate. For these reasons, IVRT methods are most appropriate to assess (compare) the "sameness" of formulations (e.g., before and after certain manufacturing or formulation changes).
If the drug release rate is found to be different between a test and reference formulation of comparable components and composition, it typically indicates that there is a difference in the physicochemical and/or structural attributes between the formulations. The IVRT methods discussed in this chapter are not expected to predict whether an observed difference in measured release rate may impact in vivo bioavailability, nor can they identify the exact nature of the difference. Instead, the utility of IVRT studies is that they can efficiently assess whether test and reference products perform in a manner that appears to be the same. When the drug release rates are equivalent for two formulations with similar components and compositions, it suggests that there may be a high degree of sameness in physicochemical and structural attributes between the formulations, thereby providing evidence that mitigates the risk of a potential difference in therapeutic performance. Consequently, IVRT studies may provide evidence to support a demonstration of equivalence between prechange and postchange batches or a demonstration of bioequivalence in certain situations.
5.1 Theory
After a short lag period, release of drug from the semisolid dosage form is kinetically described by diffusion of a chemical out of a semi-infinite medium into a sink, by the following equation:
m = √(2 × Q × Dm × Cₛ × t)
Where m is the amount of drug released, Q is total amount of the drug in solution and suspended in the matrix, D is the drug diffusion coefficient in the semisolid matrix, C is the drug solubility, and t is time.
A plot of m versus √t will be linear with a slope of:
m = √(2 × Q × Dm × Cₛ)
5.2 IVRT Method Development, Validation, and Transfer
IVRT method development should encompass key aspects that influence the IVRT results, including the IVRT receptor solution sample analysis method. IVRT method validation is outside of the scope of this chapter. An IVRT method transfer between laboratories should include a demonstration that the IVRT method and the associated IVRT receptor solution sample analysis method produces comparable, valid results when compared between the laboratory where the methods were validated and the laboratory to which the methods are transferred.
5.3 Synthetic Membrane
Filter membranes (e.g., 0.45-µm pore size) comprised of synthetic materials (e.g., mixed cellulose esters, nylon, polysulfone, polyethersulfone) are frequently suitable for IVRT methods as they are typically resistant to binding most drugs, and they permit drugs to diffuse through the membrane. Filter membranes comprised of a synthetic material, sometimes with different pore sizes, are commonly evaluated during IVRT method development. Other types of simple, monolayer synthetic membranes, like dialysis membranes, may also be suitable for an IVRT method. The use of biological membranes or synthetic membranes developed to emulate a biological membrane are not appropriate for an IVRT method because such membranes may inappropriately influence the apparent rate of drug release.
5.4 Diffusion Cells
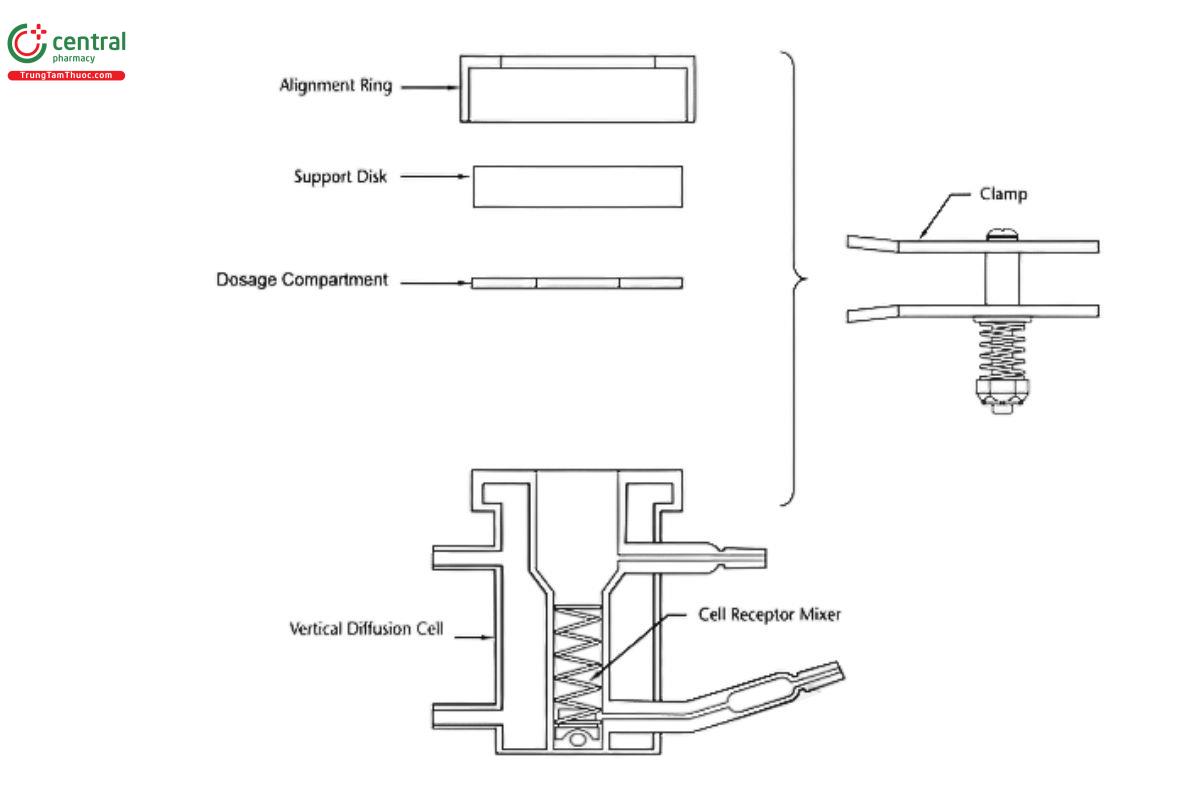
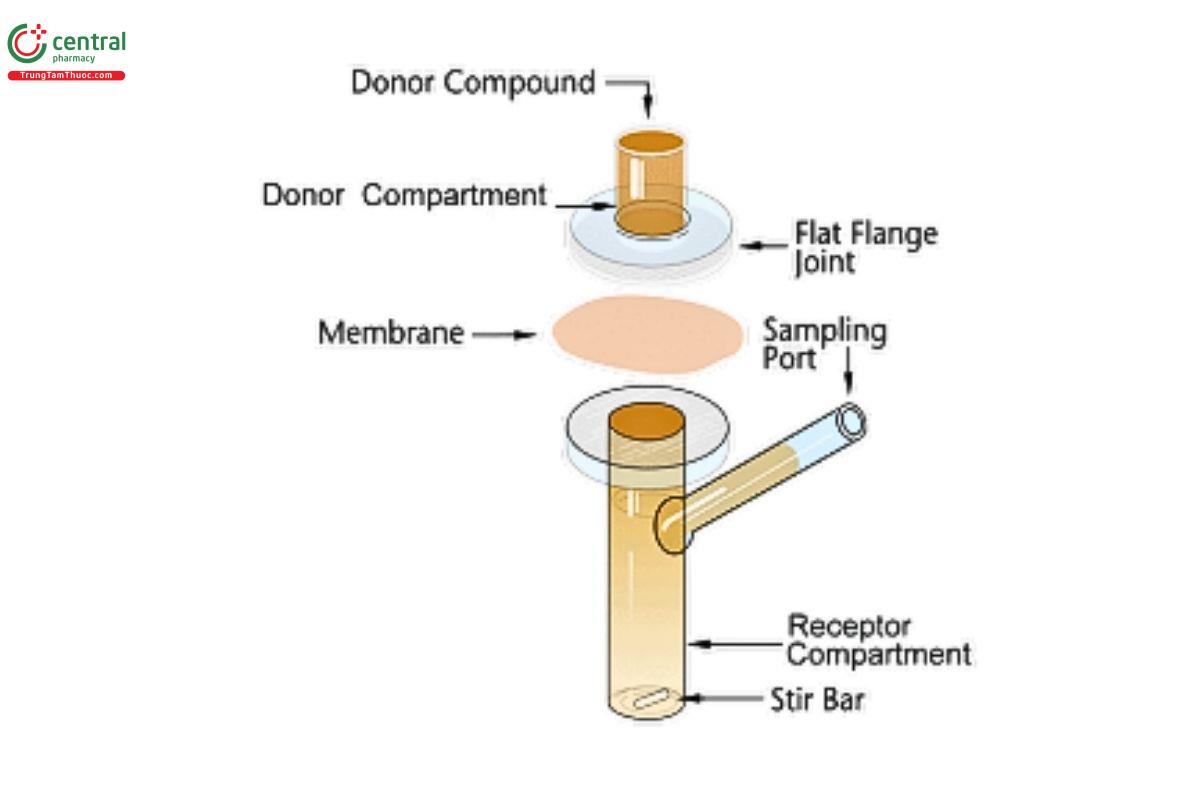
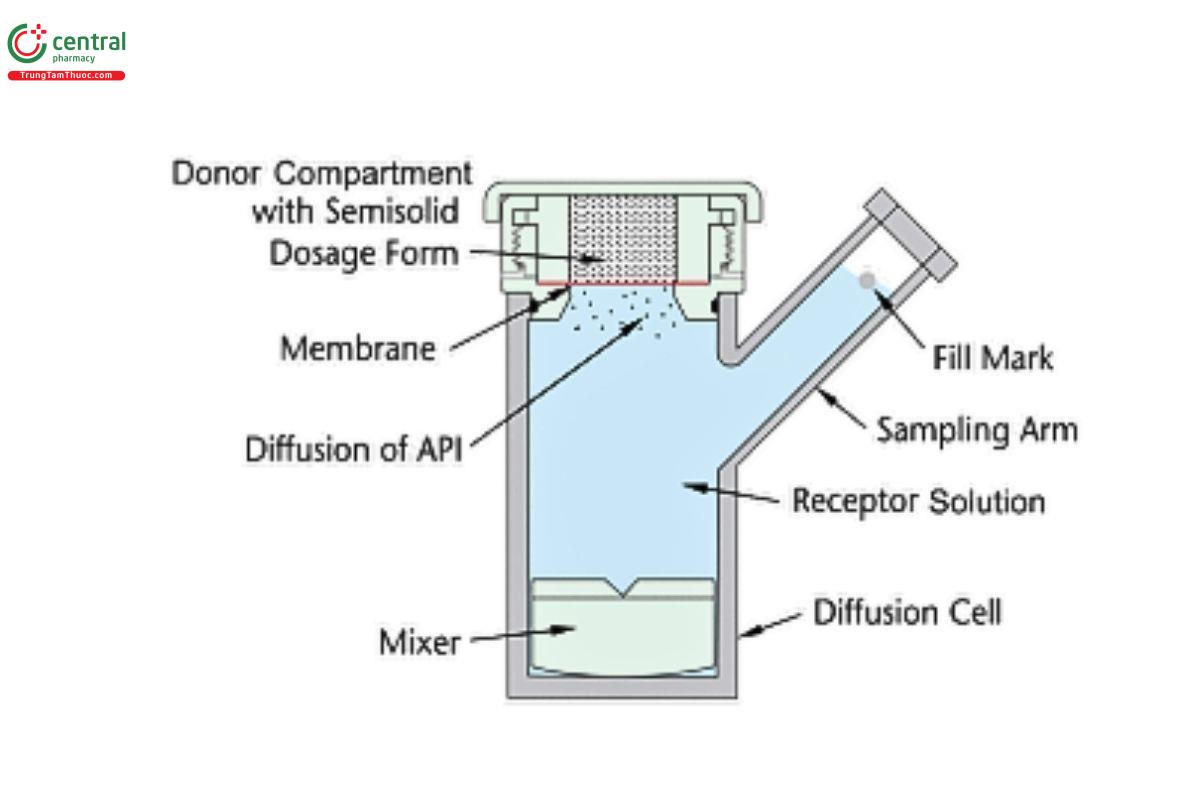
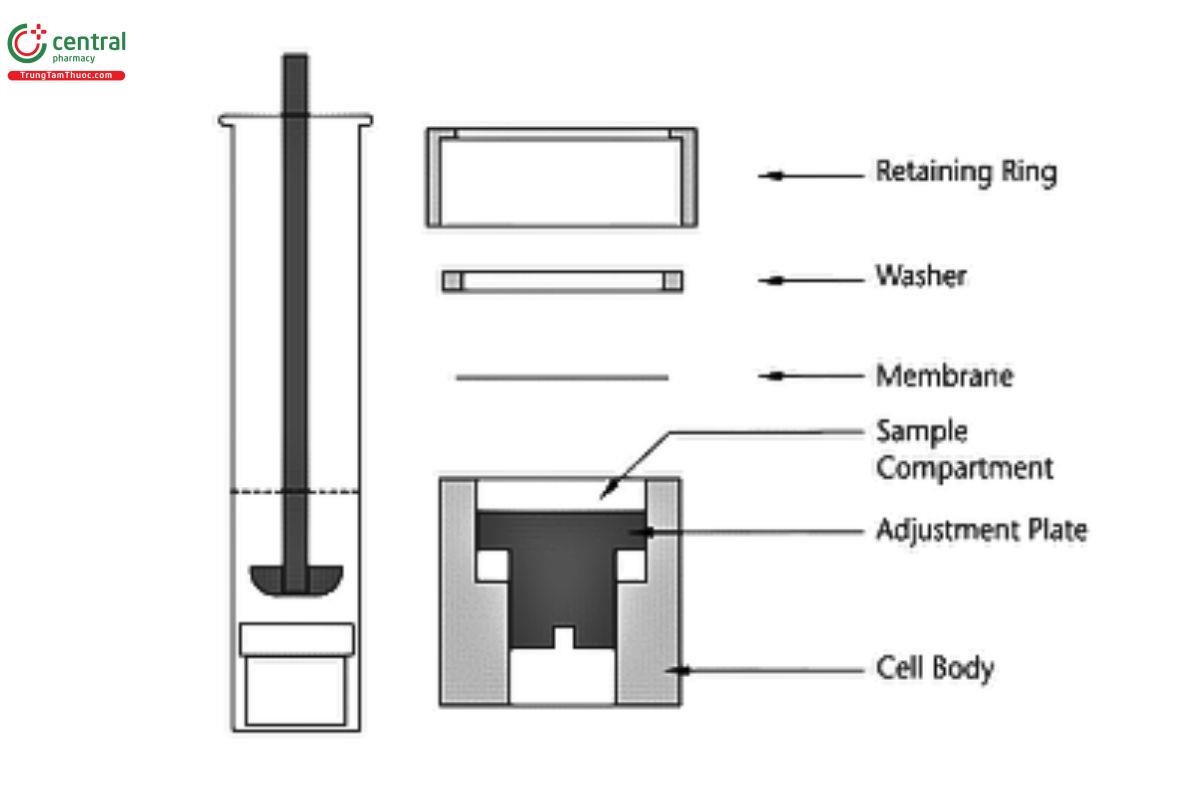
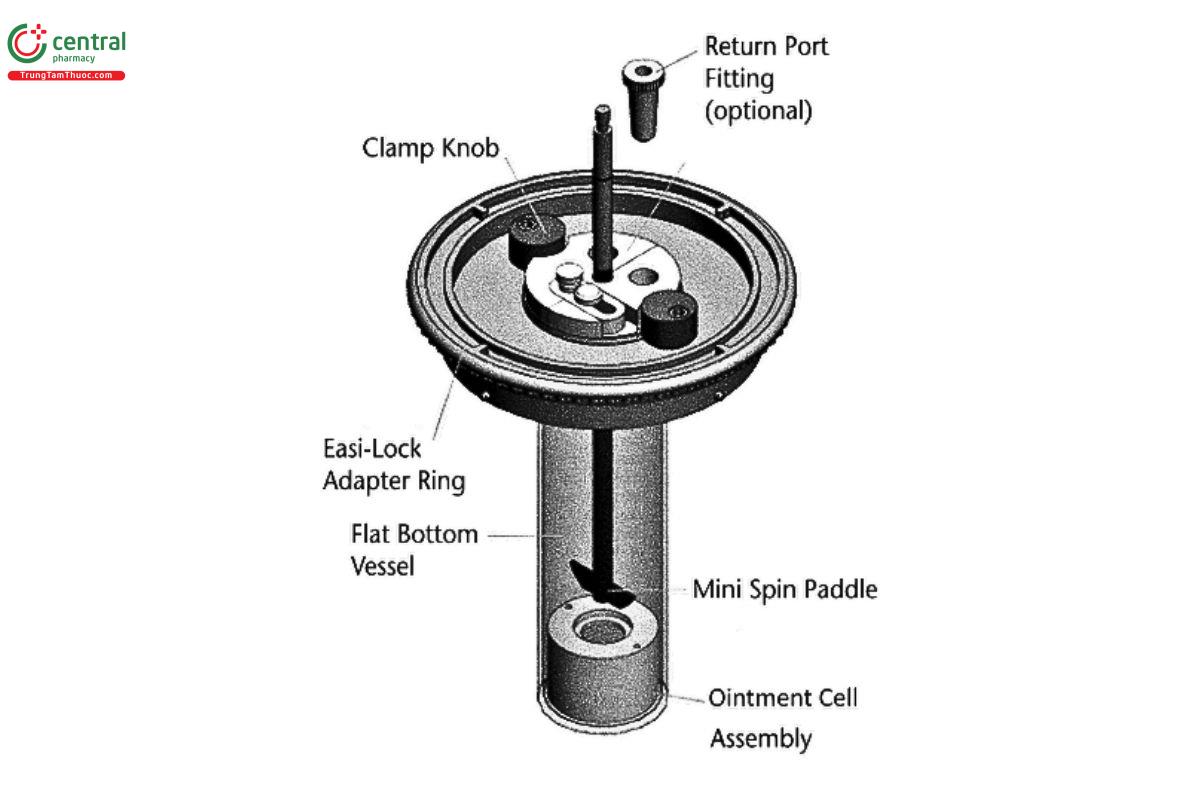
Suitable equipment for the IVRT method includes various models of vertical diffusion cell (VDC) and immersion cells. The most commonly used equipment for IVRT methods are VDC (see Figures 1–5), although immersion cells (Figure 6 and Figure 7) also have been used successfully. Equipment features (e.g., bubble-free cells or cells with a vent hole) other than the ones depicted below may be suitable once qualified. The total receptor compartment volume of VDC typically ranges from 5–15 mL, while the total vessel volume for the immersion cell typically varies between 50 and 200 mL; values outside of those typical ranges may be available depending on the manufacturer of the equipment. VDCs may offer advantages over immersion cells; for example, in the assessment of lower strength dosage forms and/or dosage forms with low amounts of drug release, VDCs provide less drug dilution in the receptor solution, minimizing potential analytical challenges regarding drug quantification. Also, many VDC models allow the user to adjust the amount of the dosage form utilized, which can allow users to increase the amount of the dosage form in the donor compartment, thereby reducing the percent dose depletion and sustaining the pseudo-in nite dose conditions that support steady-state drug release kinetics. See Equipment for further details on the equipment used in IVRT.
5.5 Analytical Method
The analytical method should be precise, accurate, and specific for the drug in the receptor solution. The use of validated analytical methods with multipoint calibration curves is encouraged. The validation of an analytical method for IVRT sample analysis is done separately from the validation of an IVRT method, which follows specific procedures not described in this chapter.
5.6 Experimental Design
5.6.1 DOSE
When utilizing an appropriately selected receptor solution, membrane, equipment, dose, and sampling duration (as well as other potential method parameters) the release rate of the drug should be linear (when plotted as the amount of drug released versus the square root of time) and reproducible. The amount of drug product in the donor compartment should ensure that dose depletion does not occur to an extent that alters the linear (steady state) release kinetics during the duration of the test (i.e., a pseudo-infinite dose amount should be utilized). Excessive dose depletion is characterized by a nonlinear region in the plotted cumulative amount of drug released curve (when plotted as the amount of drug released versus the square root of time).
The dose application can be done by different methods, such as, but not limited to, dispensing directly on the membrane from a tube, transferring and spreading on the membrane with a spatula, or transferring and dispensing with a positive-displacement pipette or a disposable harvester (which minimizes shear stress during dosing). The shear stress imposed upon the dosage form during the dispensing (e.g., from a product tube) and dosing (e.g., using a positive displacement pipette) should be considered, and the dosing method should preferably minimize and routinely control the shear stress in a consistent manner for all diffusion cells. It is also important that a consistent amount of the dosage form is applied uniformly upon the membrane, without air pockets, making a compact cylinder of the formulation. The diffusional surface area of the product on the membrane needs to be consistent and well controlled as it is a factor in the calculation of the release rate. The donor compartment should be occluded to avoid evaporation of volatile components (including water) from the dosage form that is in contact with the membrane, because that evaporative metamorphosis may lead to changes in the composition of the drug product and may alter its linear (steady state) drug release kinetics.
5.7 Receptor solution
The selection of the receptor solution is guided by the solubility of the drug substance(s) in potential solvents/solvent combinations. Most commonly, combinations of aqueous and alcoholic/organic solvents are used, and the solubility of the drug in the receptor solution should exceed the highest sample concentration in the IVRT study, ideally by an order of magnitude, but demonstrably sufficient to facilitate a linear (steady state) release rate for the duration of the study (even when evaluating a relatively higher release rate of a formulation that is 150% of the nominal reference strength).
The receptor solution should be adequately mixed, typically by stirring at a constant and well-controlled speed throughout the experiment. The stirring speed or the flow rate should also be part of system qualification and is further described under Equipment.
Another factor to consider is the stability of the drug in the receptor solution at the temperature of the test during the IVRT experiment.
5.8 System equilibration
The system should be appropriately equilibrated under the predefined experimental conditions prior to the beginning of the IVRT run. No air bubbles should be present in the receptor solution (under the membrane) immediately prior to, or during the experiment; if appropriate, the receptor solution can be degassed. The system temperature (including the membrane surface temperature and the circulating water bath temperature, if applicable) should be stable and well controlled. IVRT membranes can be presoaked with receptor solution prior to assembling in the diffusion cell or equilibrated with the receptor solution in situ within the diffusion cell prior to dosing (e.g., for 30 min at a membrane temperature of 32°) depending upon the method.
5.9 Experiment length and sampling
The test duration should be suitable to characterize the linear (steady state) drug release kinetics of the product formulation. This is typically demonstrated based upon a sustained steady-state release between 4–6 h. The data obtained during the first hour may not represent the steady-state release kinetics, and for many IVRT methods steady-state release kinetics are calculated based upon timepoints from 1 h onward (e.g., at 1, 2, 3, 4, and 5 h, or at 2, 3, 4, 5, and 6 h). At least 5 sampling time points should be planned to obtain a well-characterized release rate. Sampling durations of at least 4 h are suitable to assess steady-state release rates, whereas shortened (e.g., 2 h) sampling durations may not be representative of the steady-state release kinetics. The specific sampling times may be varied depending on the formulation (e.g., every 30 min, or every hour); however, caution should be taken regarding the precise timing of each sample collection to ensure that sample collections happen within the predefined tolerance range. Samples should be withdrawn within a tolerance of ±15 min or ±2% of the nominal time, selecting the tolerance that results in the narrowest time interval.
Sampling is dependent upon the equipment used and should follow instructions provided by the manufacturer, when appropriate. The receptor solution used to replace the volume removed during sampling may be pre-warmed to the temperature of the receptor solution in the receptor compartment, and it is important to ensure that air bubbles are not present beneath the membrane after sampling and refilling the receptor solution at each time point.
5.10 Number of replicates
The test is often conducted with a group of 6 or 12 cells per test run. The results from 6 diffusion cells dosed with a specific product are routinely sufficient to characterize the performance (release rate) of that product using that specific IVRT method. In instances when two products are compared, the results from 6 cells of a test product are often compared with the results from 6 cells of a reference product during the first stage of data analysis. See Data Reporting for more details on the use of additional replicates in instances when a second stage of testing is performed in a comparative IVRT study.
5.11 Membrane temperature
The test is conducted at 32 ± 1° for products applied to the skin (measured at the surface of the membrane, when possible, or inferred based upon the temperature of the receptor solution for immersion cells or VDC Model A). The test is conducted at 37 ± 1° for products intended for internal application (e.g., rectal and vaginal products).
During the entire test, the nominal system conditions, like the temperature of the receptor solution, should be maintained so that the temperature at the membrane remains within specified parameters (e.g., 32 ± 1°) for the duration of the IVRT. The temperature control qualification is described under Equipment.
5.12 Data reporting
For each cell, the amount of drug released (typically in µg/cm²) at each sampling time (t₁, t₂, etc.) is determined, and the cumulative amount released is plotted versus √t. The slope of the resulting line is a measure of the rate of drug release.
For each cell, the individual amount of drug released is plotted versus the square root of time. The slope of the resulting line is the rate of drug release.
IVRT is a useful tool during semisolid product development to assess whether there may be differences in the arrangement of matter within the semisolid product matrix of compositionally identical (or similar) test and reference products, which may alter their rates of drug release, and that may have the potential to alter product performance in clinical use. When test and reference products exhibit equivalent drug release rates based upon a validated IVRT method, it can mitigate the risk of potential differences in product performance. Therefore, evidence from an IVRT can support a demonstration of bioequivalence, along with other evidence that collectively mitigates the risk of a difference in product performance between a reference product and a prospective generic (test) product.
IVRT is commonly used to assess the sameness of a drug product after postapproval changes. Because common testing artifacts, such as air bubbles and membrane defects, yield measurements that are not normally distributed, a nonparametric statistical technique is used to evaluate the test results. The Mann-Whitney U test is used to calculate the 90% confidence interval for the ratio of the slopes between the test and the reference batches. This is illustrated by the following example in which the initial drug product batch is referred to as the reference batch (R) and the changed or subsequent batch is referred to as the test batch (T). The individual amounts of drug released from R are plotted versus the square root of time, and the resulting slopes are determined. Those are the reference slopes. The process is repeated for the test batch (T).
The T/R slope ratios are calculated for each test-to-reference combination of all pairs of T/R slopes. This procedure is facilitated with a table where the values for the slopes for T are listed down the left side of the table and the slopes for R are listed across the top of the table. The T/R slope ratios are then determined. See Table 1.
Table 1. Comparison of Test Slope (TS) and Reference Slope (RS) Ratios
| RS1 | RS2 | RS3 | RS4 | RS5 | RS6 | |
| TS1 | TS1/RS1 | TS1/RS2 | TS1/RS3 | TS1/RS4 | TS1/RS5 | TS1/RS6 |
| TS2 | TS2/RS1 | TS2/RS2 | TS2/RS3 | TS2/RS4 | TS2/RS5 | TS2/RS6 |
| TS3 | TS3/RS1 | TS3/RS2 | TS3/RS3 | TS3/RS4 | TS3/RS5 | TS3/RS6 |
| TS4 | TS4/RS1 | TS4/RS2 | TS4/RS3 | TS4/RS4 | TS4/RS5 | TS4/RS6 |
| TS5 | TS5/RS1 | TS5/RS2 | TS5/RS3 | TS5/RS4 | TS5/RS5 | TS5/RS6 |
| TS6 | TS6/RS1 | TS6/RS2 | TS6/RS3 | TS6/RS4 | TS6/RS5 | TS6/RS6 |
After the T/R ratios have been calculated, they are ordered from the lowest to the highest. The 8th and 29th T/R ratios are identified and converted to percent (multiplied by 100). These values represent the 90% confidence interval for the ratio of test to reference release rates. To pass first stage testing, those ratios must be within the range of 75%–133.33%.
If the results do not meet this criterion, 4 additional tests (2 reference and 2 test) of 6 cells each should be performed, resulting in 12 additional slope determinations for each product tested. The T/R slope ratios for all 18 slopes for each product tested are determined. All 324 individual T/R slope ratios are ordered from the lowest to the highest. To pass this second stage testing, the 110th and 215th slope ratios, representing the 90% confidence interval, must be within the range of 75%–133.33%.
6 IN VITRO PERMEATION TEST (IVPT)
The in vitro permeation test (IVPT) is intended to characterize the rate and extent to which a drug applied on the surface of a biological membrane permeates into and through it, using method parameters aimed to simulate in vivo conditions. IVPT methods can utilize a variety of biological membranes, including excised human skin, which can exhibit natural variations in permeability that are reflective of the variability observed in vivo. This variability can be substantial; it is not uncommon to observe a 10-fold difference in skin permeability for a given compound (in the same formulation) between individuals in the population or between different anatomical regions on the same individual. Experimental variability may also be a result of the physicochemical properties of a molecule. For all products or treatments compared in an experiment, the replicate skin sections used should be sourced from the same donor (or the same set of donors), the same anatomical site (e.g., abdomen, back, etc.), the same source (e.g., elective surgery or cadavers) and manner of preparation (e.g., dermatoming, freezing, etc.) to minimize variability.
An IVPT study can be sensitive and discriminating to differences in the rate and extent to which compounds applied on the skin from different formulations become available in and through it. Notably, unlike an IVRT study, differences in permeation that are observed in an IVPT study comparing a test versus reference product, if any, may correlate with and/or be predictive of differences in bioavailability in vivo. The IVPT is routinely carried out to guide semi-solid topical formulation development.
6.1 IVPT Method Development and Validation
IVPT method development should encompass key aspects that influence the IVPT results (some of these aspects are discussed below), including the bioanalytical methodology used to quantify the amount of the drug in the IVPT study samples. The scope of the IVPT method development is dependent upon the purpose of the study. For example, a bioequivalence study is likely to require a larger number of skin donors in order to adequately power a statistical analysis, compared to a study intended to qualitatively evaluate different formulation prototypes. Further, whereas IVPT studies designed to support a demonstration of bioequivalence generally rely upon assessments of the rate and extent to which drugs permeate the skin based upon receptor solution results, the selection of new chemical entities or formulation prototypes may also employ assessments of tissue distributions. A comprehensive discussion on IVPT method validation is outside of the scope of this chapter.
6.2 Biological Membrane
Dermatomed human skin (split-thickness, typically around 250–500 µm), sourced from cadavers or elective surgery (typically abdominoplasties or breast reductions), is often the most appropriate membrane of choice due to its intrinsic relevance to a human drug product development, as well its ease of handling during experimental procedures. The thickness of the dermatomed skin should be confirmed prior to assembling the membrane in the diffusion cell, as significant variability in thickness may increase the variability of IVPT results. Epidermal preparations (sheets of epidermis that have been separated from the dermis at the dermal-epidermal junction) require technical experience to process, have less structural integrity compared to dermatomed skin, and may tear or lose barrier integrity more easily. However, epidermal preparations are often more consistent in thickness than dermatomed skin, and the magnitude of permeation for some compounds may be greater through epidermal preparations than through dermatomed skin. Therefore, while dermatomed skin is a good choice to evaluate in most cases, in some situations (e.g., when the drug of interest is not feasible to quantify permeating through dermatomed skin) epidermal preparations may be useful to evaluate during IVPT method development.
Dermatomed porcine skin has a relatively similar morphology to human skin and can be used as a secondary option (as a surrogate for human skin) for research and development studies, unless regulatory guidance recommendations specify otherwise. The use of rodent skin (as a surrogate for human skin) is discouraged due to significant dissimilarity to human skin in terms of morphology and stratum corneum structure and thickness, which can make the results difficult to interpret in terms of their relevance to human skin permeation. The use of cultured skin constructs (reconstructed human epidermis) is not ideal as a surrogate for natural excised human skin because the permeability properties of such tissues is currently not representative of excised human skin. The use of any synthetic membrane (including the types developed to emulate a biological membrane) is not appropriate for an IVPT method; the results of tests with any synthetic membrane may not reflect the rate and extent to which a drug permeates into and through skin and may be misleading.
The use of human or porcine full-thickness (non-dermatomed) skin can pose experimental challenges and is generally not recommended. Other membranes such as rectal, vaginal, or corneal epithelial tissue may also be employed, depending on the intended route of administration for the drug product under evaluation. In such circumstances, several parameters of the test method may differ from those used for an IVPT with skin due to specific considerations for modeling the relevant in vivo conditions. For example, the appropriate temperature at which to maintain the membrane may be different.
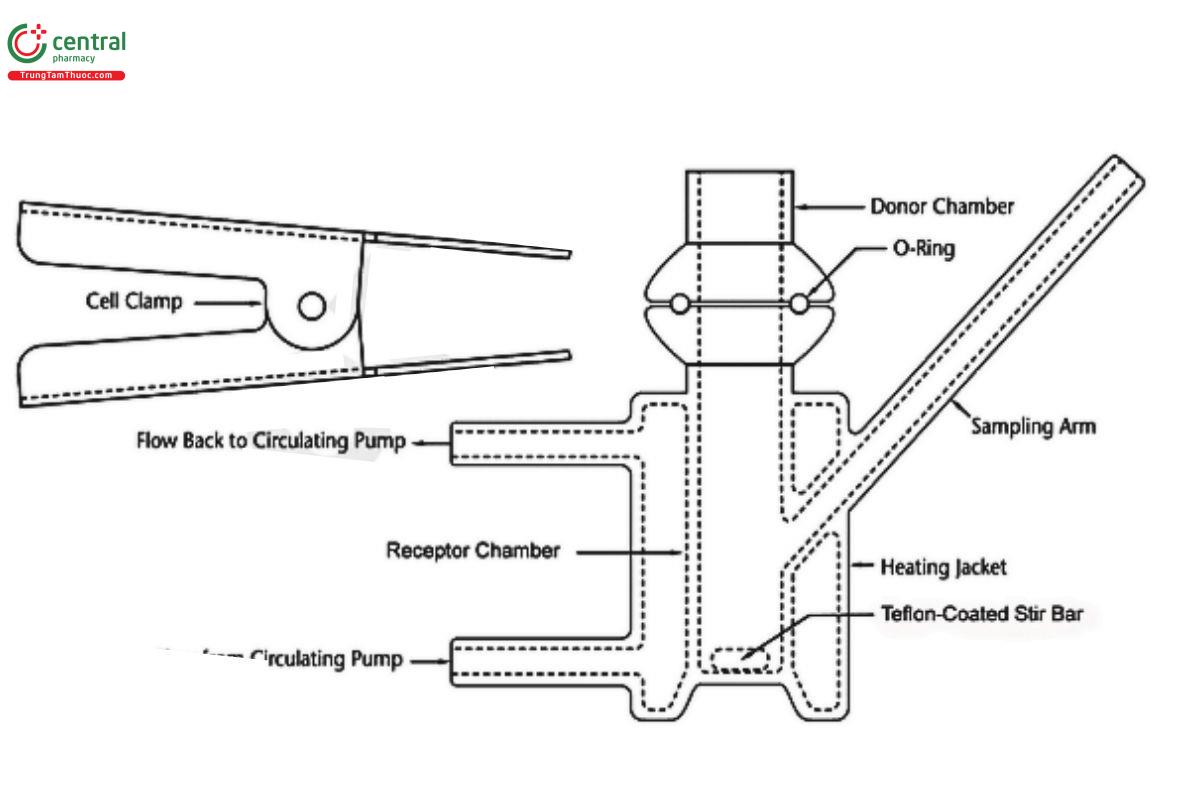
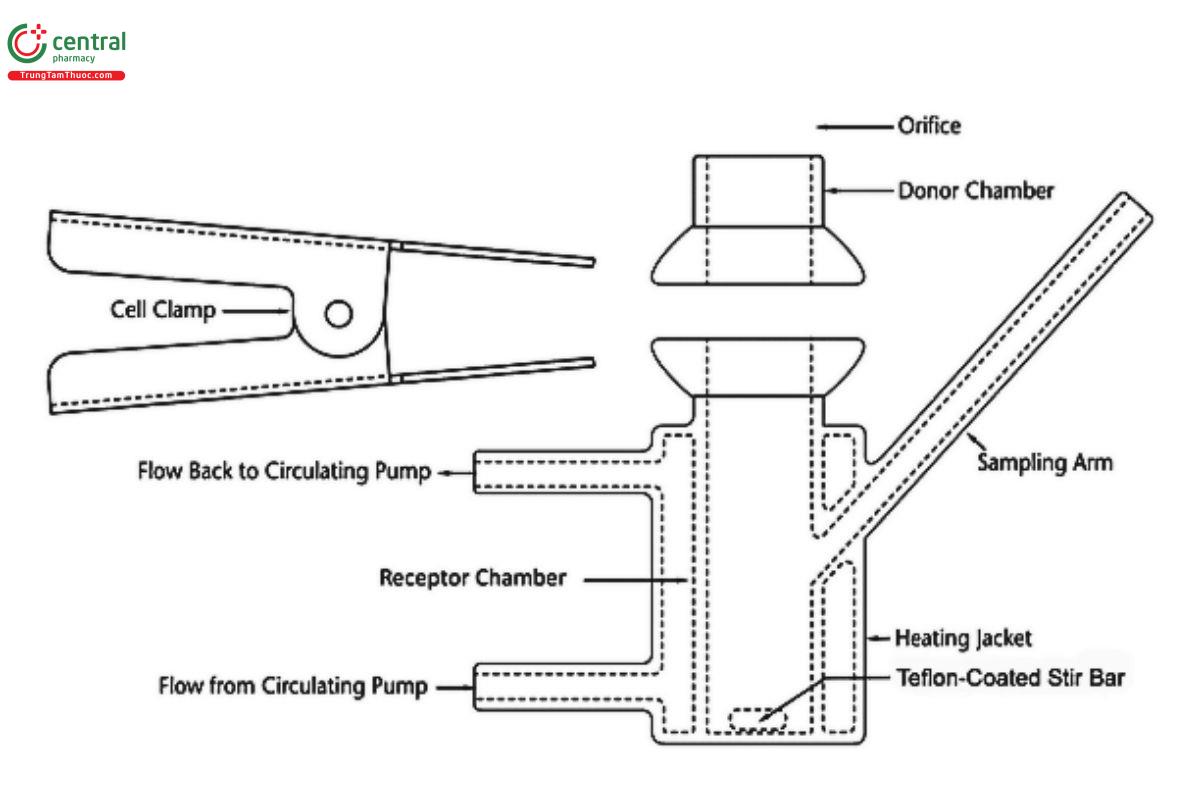
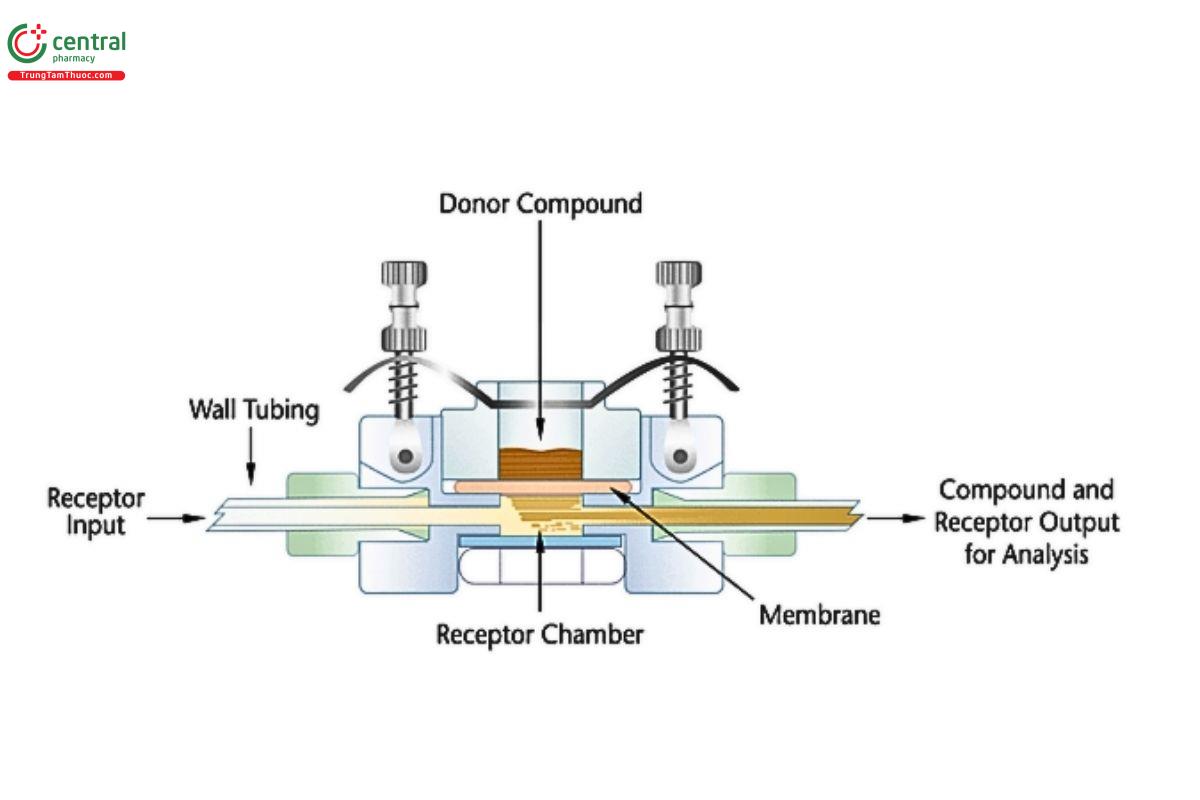
6.3 Diffusion Cells
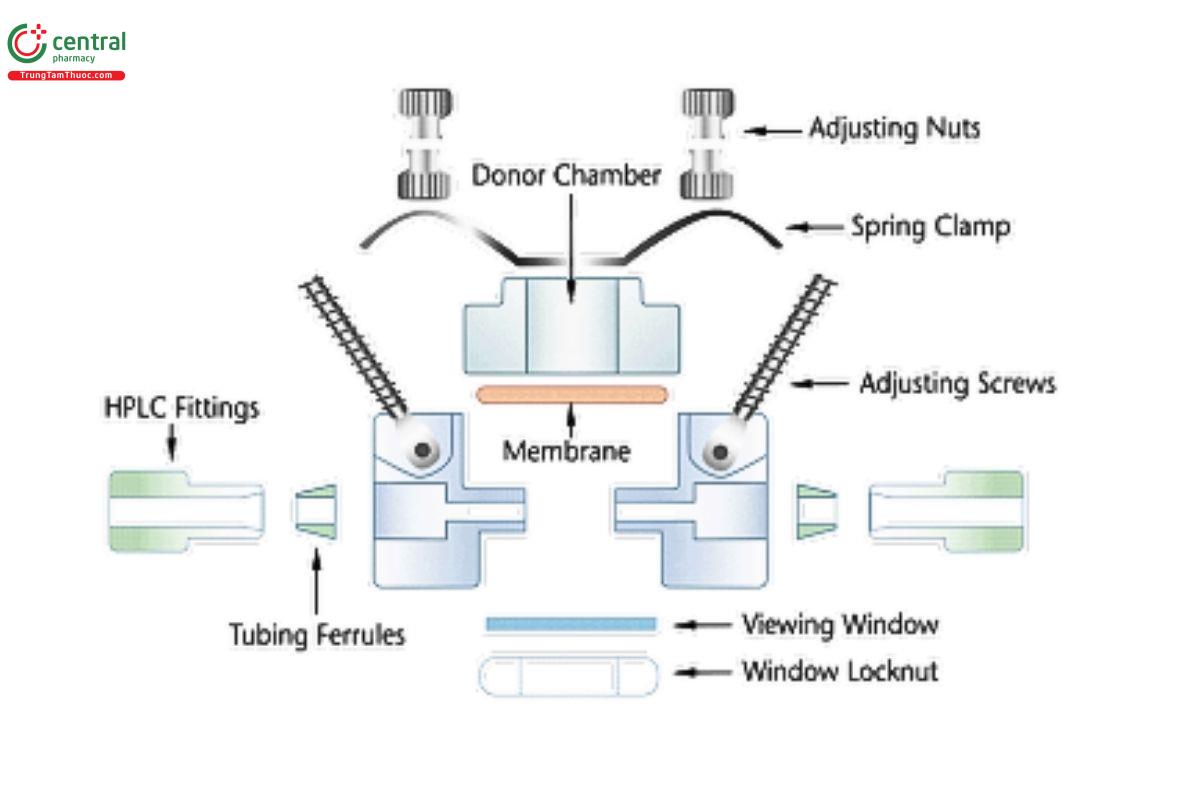
Two types of diffusion cells are commonly used for IVPT studies: the vertical diffusion cell (VDC) (see Figures 1–5) and the flow-through diffusion cell (FDC) (see Figures 8–10). Equipment features (e.g., bubble-free cells or cells with a vent hole) other than the ones depicted below may be suitable once qualified. VDCs are well characterized and can be utilized for both, IVRT and IVPT studies. FDCs are used exclusively for IVPT studies (not IVRT studies) and have the advantage that the sampling is more easily automated, which can make it easier to acquire samples at times when investigators are typically not in the laboratory (e.g., the middle of the night). VDCs and FDCs can both produce excellent results when the IVPT method is appropriately developed, and each type of cell has its advantages.
6.4 Bioanalytical Method
The choice of analytical method should consider the physicochemical properties of the analyte(s) present in the formulation and the actual formulation composition; these factors combined will directly affect the ability of a drug to cross the stratum corneum, therefore impacting drug concentrations in each skin layer and in the receptor solution. Liquid chromatography (LC) coupled with a mass spectrometer (MS) or a tandem mass spectrometer (MS/MS) detector provides excellent sensitivity and selectivity, albeit at the expense of higher complexity in terms of method development/validation, troubleshooting, and higher maintenance costs. LC coupled with an ultraviolet (UV) detector, diode array detector (DAD), or fluorescence detector (FLD) offers lesser technical complexity but is generally less sensitive and can pose selectivity challenges for analytes present in biological samples.
The use of validated bioanalytical methods with multipoint calibration curves is encouraged. The validation of an analytical method for IVPT sample analysis is done separately from the validation of an IVPT method, each of which follow specific procedures not described in this chapter.
6.5 Experimental Design
6.5.1 Dose
The thermodynamic activity of the drug(s) present in a formulation will affect the rate and extent of skin permeation. It is prudent to understand the drug(s) solubility in the solvents/excipients of interest, as well as in the formulation prototypes under evaluation.
The finite dose typically ranges from 2–15 mg of formulation per square centimeter of dosing area, with a dose of 5–10 mg/cm² often used as a starting dose for method development. Ensuring consistency in the dispensing of the formulation (e.g., from the product tube) and uniform dose administration (spreading/coverage) on the skin section placed in the diffusion cell can help minimize experimental variability. The use of positive-displacement pipettes is recommended to ensure dosing precision. For semisolid dosage forms such as ointments, creams, and certain gels that do not flow like a solution, a glass rod or the bottom of a glass vial can be used to evenly spread the formulation within the dosing area.
6.5.2 Receptor solution
The receptor solution should ensure sufficient solubility of the drug(s) of interest throughout the length of the study, thus not limiting the rate of diffusion/partitioning of the drug(s) from the skin to the receptor solution. The solubility of the drug in the receptor solution should exceed the highest sample concentration in the IVPT study, ideally by an order of magnitude, if possible. The inclusion of 0.1% (w/v) polyoxyethylene (20) oleyl ether is ideal to enhance the solubility of physiological buffer based (aqueous) receptor solutions for hydrophobic drugs. If additional solubility is needed, small increases in the concentration of polyoxyethylene (20) oleyl ether [e.g., from 0.1% (w/v) to 0.2% (w/v)] is typically adequate for most hydrophobic drugs. Higher concentrations of polyoxyethylene (20) oleyl ether may be evaluated, as needed, but should not exceed 6% because higher concentrations may alter the skin barrier. Other solubility modifiers may also be effective but may not be suitable for studies assessed by a regulatory agency unless the impact of that solubility modifier on the permeability of the skin barrier has been adequately characterized. Other strategies to improve the solubility of the drug in the receptor solution that may have the potential to alter the permeability of the skin (e.g., inclusion of organic solvents and alcohols in the receptor solution) are not prudent and may invalidate the IVPT method. Additionally, labile compounds may require modifications in the receptor solution, including the addition of antioxidants and/or chelating agents (e.g., ethylene diamine tetra acetic acid; EDTA) or the use of acidified water (e.g., pH 4.0) instead of phosphate-buffered saline (PBS). The addition of antibacterial/antimycotic agents [e.g., 0.1% (w/v) sodium azide; 0.01% (w/v) Gentamicin sulfate] in the receptor solution is also important to prevent microorganism growth and membrane tissue degradation.
6.5.3 System equilibration
The system should be appropriately equilibrated under the predefined experimental conditions prior to the beginning of the IVPT run. No air bubbles should be present in the receptor solution (under the membrane) immediately prior and during the experiment; if appropriate, the receptor solution can be degassed. The system temperature (including the membrane surface temperature and the circulating water bath temperature, if applicable) should be stable and well controlled. Frozen biological membranes should be thawed at ambient temperature prior to assembling on the diffusion cells. Once mounted, the skin sections should be allowed to equilibrate to a physiologically relevant state of hydration and temperature (e.g., for 30 min at a membrane surface temperature of 32°).
Experiment length and sampling
The test duration should be sufficient to characterize a suitable permeation profile. For IVPT studies that are intended to support a demonstration of bioequivalence, the selected sampling schedule and study duration should be sufficient to characterize the cutaneous pharmacokinetics of the drug, which ideally includes a sufficiently complete flux profile to identify the maximum (peak) flux and a decline in the flux thereafter across multiple subsequent time points. IVPT studies typically range from 12–72 h in length; IVPT bioequivalence studies may require longer periods of time to observe a sufficient decline in the skin flux to adequately characterize the cutaneous pharmacokinetics.
The IVPT method development should support the selection of an appropriate sampling schedule, intended to provide suitable resolution for the flux profile. A study with fewer than 8 nonzero sampling time points may not provide adequate characterization of the flux profile.
6.6 Number of donors and replicates
Depending on the stage of method development, the number of donors may vary. A study with fewer than 4 donors and 4 replicates per donor per treatment may be difficult to interpret. IVPT bioequivalence studies may require a larger number of donors and replicates to adequately power a statistical analysis. The inclusion of a nondosed control (no formulation) is recommended and may help ensure the skin source(s) and receptor solution are absent of contaminants that may influence the results.
6.7 Membrane temperature
The surface of each skin section should be kept constant at 32 ± 1° throughout the length of the study. Temperature fluctuations of the skin surface will affect drug diffusion and may increase experimental variability. The use of a thermoregulated diffusion cell is encouraged to control the temperature of the membrane surface.
6.8 Membrane integrity
The skin barrier integrity in each diffusion cell should be confirmed prior to dosing by using techniques such as transepidermal water loss (TEWL), electrical impedance/conductance or tritiated water permeation; only skin sections with acceptable barrier integrity results should be dosed. TEWL is often a preferred method because, unlike the other methods, it measures the flux of water through the skin barrier while it is dry and in contact with the air, as it is in the normal in vivo state, and TEWL is relatively rapid and convenient.
6.9 Drug tissue distribution and mass balance
Most IVPT experiments report skin flux and the total cumulative amount of drug that has permeated into the receptor solution (or as a percentage of drug from the total applied dose). Depending on the purpose of the study, an evaluation of drug tissue distribution (amounts in the stratum corneum, viable epidermis, and dermis) and total mass balance may provide additional insights.
Full mass balance requires more intensive bioanalytical method development to ensure that the drug(s) of interest are efficiently extracted from each sample matrix type, such as cotton swabs used to remove the excess of formulation present on the skin surface at the end of the experiment, tape strips used to remove stratum corneum, and homogenized viable epidermis and dermis.
Epidermis and dermis heat splitting can be accomplished by placing the skin sections (harvested at the end of the IVPT study) on a sheet of aluminum foil and incubating it (dry heat) at 60° for 2–3 min. After this step, the epidermis can be separated (by scraping) from the dermis by using forceps. Because the drug levels in the epidermis are generally higher than the dermis, the splitting should be done carefully to avoid cross contamination, and clean forceps should be used in between samples.
6.10 Data reporting
Depending on the purpose of the study (bioequivalence, formulation development, prototype screening, etc.), data may be reported and assessed using different parameters (endpoints). For example: a bioequivalence study will focus on the cutaneous pharmacokinetic endpoints of maximum flux (Jmax), and total cumulative amount (AMT) of drug permeated. Studies conducted in early stages of drug development, such as those intended to select a new chemical entity (NCE), or to rank different formulation prototypes, may report tissue (epidermis and dermis) concentrations in addition to the parameters (endpoints) above, and may include full mass balance data.
The flux (rate of drug permeation) should be calculated for each time point in units of mass/area/time (e.g., ng/cm²/h) on the y-axis versus time in the x-axis. The extent of drug permeation should also be reported in units of mass/area (e.g., ng/cm²) in the y-axis versus time in the x-axis—the slope of this curve can help determine the overall drug flux through the skin at different points in time. Drug levels in the viable epidermis and dermis can be reported as absolute mass amount (e.g., nanograms or micrograms) or concentration (e.g., nanograms or micrograms of drug per milligram of tissue [ng/mg or µg/mg]).
Full mass balance results are generally reported as percentage of drug(s) in each compartment (e.g., stratum corneum, viable epidermis, dermis and receptor solution) relative to the total amount applied at the beginning of the experiment.
7 IVRT Versus IVPT
The selection between IVRT or IVPT should be based on the intended objective(s) of the formulation assessment, as described in Table 2. Intrinsic experimental differences between the two methodologies are further described in Table 3.
Table 2. Guide for Selecting an Appropriate Test Method (IVRT or IVPT) Depending Upon the Goal
| Evaluation/Goal | Test |
| Assessment of in vitro/in vivo correlation | IVPT |
| Assessment of the ability of a drug in a formulation to cross the stratum corneum and of the distribution of the drug in the epidermis, dermis, and/or receptor solution | IVPT |
| Selection of a new chemical entity (NCE) for further development as a semisolid formulation | IVPT |
| Comparison of the permeation kinetics of different topical semisolid formulations (different composition of different dosage forms, such as solutions, gels, creams, ointments, etc.) | IVPT |
| Comparison of the rate and extent to which a topically administered drug permeates through the skin and becomes available at or near a site of action from a reference and a test product formulation | IVPT |
| Evaluation of the effect of inactive ingredients on the rate of drug release from the formulation matrix | IVRT |
| Assessment of sameness in the formulation matrix between formulations with the same amount of an active ingredient and the same or similar composition of inactive ingredients | IVRT |
| Comparison of the effect of critical manufacturing/process steps on the microstructure of semisolid formulation | IVRT |
| Compare batch-to-batch variability in drug release rate at the time of manufacture, and during stability | IVRT |
Table 3. Key Differences Between IVRT and IVPT Methodologies
| Parameter | IVRT | IVPT |
| Membrane | Synthetic (e.g., mixed cellulose esters, nylon, polysulfone, polyethersulfone) | Biological (e.g., ex vivo human epithelial tissue such as skin, rectal or vaginal tissue, corneal tissue, nails, other mucous membranes) |
| Receptor solution | Aqueous and aqueous–organic combination, may include high percentages (≥30%) of alcohols and/or organic solvents (e.g., Ethanol, isopropyl alcohol, acetonitrile) | Aqueous, typically phosphate buffered saline; low levels (typically ≤5%) of additives may be added to enhance solubility of hydrophobic compounds (see the IVPT Receptor Solution section) |
| Sampling | Aliquot sampling of a portion of the receptor solution at designated sample time points | Removal of the entire volume of the receptor solution, or sampling of relatively large volume aliquots of the receptor solution for VDCs; FDCs offer a different sampling methodology due to the continuous flow of receptor solution into the collection vials |
| Apparatus | VDC or immersion cell | VDC or FDC |
| Dose | Pseudo-infinite, occluded | Finite (typically ≤15 mg/cm²), unoccluded |
| Experimental duration | Typically ≤6 h | Typically ≥24 h |
| Receptor solution drug levels | Microgram to milligram range | Picogram to nanogram range |
| Analytical technique | HPLC/UPLC with detection by UV, DAD, or FLD | HPLC/UPLC with MS or MS/MS detection |
| Key data obtained | Release rate (slope) | Flux profile including peak flux (Jmax) and cumulative amount (AMT) permeated; drug distribution in the different layers of the skin may also be assessed in certain study designs |
8 EQUIPMENT
8.1 General Recommendations for Equipment Qualification and Requalification
The VDC, immersion cells, and FDC should match the general descriptions provided below and may have design variations of the types shown among specific examples illustrated in Figures 1–5 for VDCs, Figures 6 and 7 for immersion cells, or Figures 8–10 for FDCs. The VDC, immersion cell, and FDC components should be manufactured with inert materials that do not adsorb, absorb, bind, or react with the analyte. The diffusion cell and its components should not alter the amount of diffusing drug that is measured, either by adsorbing, absorbing, binding, or reacting with the drug, or by releasing drug that was adsorbed, absorbed, bound, or reacted with in a previous experiment.
The operating principles and specific test procedures differ among the various equipment; relevant procedures from the manufacturer may be used for installation, operational, and performance qualifications, if available.
However, regardless of what qualification information is provided by a diffusion cell manufacturer, the laboratory performing the test should perform an initial qualification of each diffusion cell. Qualified diffusion cells can be used in numerous experiments, and do not need to be qualified again (i.e., requalified) each time. The condition of the orifices and diffusion cells should be ascertained prior to each test. Certain diffusion cell components such as O-rings or tubing may require re-evaluation and occasional replacement. Automated systems are too varied to be covered in any detail within this chapter; however, they should be requalified routinely (e.g., every 6–12 months) based on the manufacturers' recommendations.
The initial qualification of each diffusion cell should, at minimum, include:
- Measurements of the diffusional area of the orifices of the donor and receptor compartments between which the membrane is mounted
- Empirically measured volume of the receptor solution compartment/vessel for each VDC or immersion cell, or the empirically measured outflow tube length for each FDC
- Stability of the temperature measured at the membrane surface (e.g., 32 ± 1°) across a relevant duration (e.g., 6 h for IVRT or 48 h for IVPT)
- Rate of stirring or agitation for VDCs or immersion cells, or the flow rate for FDCs, as applicable
In addition to the quantitative assessments described above, qualitative assessments should verify that diffusion cells do not leak, and that the membrane can be mounted securely in the diffusion cell so that there is no bulk flow of a dosage form from the donor compartment to the receptor compartment that circumvents the membrane. Several other considerations relevant to a well-controlled test may be included in the qualification.
1. Qualification of Orifice Diameter: The orifice diameter for the donor and receptor compartments can be determined using vernier calipers. The orifice should be inspected for any chips or other damage which may alter the surface area.
2. Qualification of Volume of Receptor Solution Compartment/Vessel: In the case of a VDC, the volume of the receptor solution should be empirically determined for each cell, individually, and recorded in a manner that can be uniquely associated with each diffusion cell thereafter, for use in calculations. The volume should be filled in a consistent manner for all diffusion cells (some cells may have a fill line marked on the cell which can be utilized) and the procedure used should consider whether the displacement by a stir bar (or other mechanism) may impact the volume measured such that the measured volume would not be relevant to that diffusion cell if a different size stir bar was used during the test. The nominal volume of the diffusion cells provided by the manufacturer should be used only as a guide.
For immersion cells, the volume of receptor solution to be filled in the dissolution vessel should be accurately measured with the appropriate volumetric glassware and transferred to the dissolution vessel.
3. Qualification of Temperature Control: The temperature of each cell should be equilibrated to provide the target temperature at the membrane, and the initial qualification of the equipment should verify that the temperature can be maintained for the duration of the test when using the relevant equipment and method parameters. Depending on the VDC or FDC equipment design, a measurement of the membrane temperature can often be made conveniently using an infrared thermometer, or using a thermocouple mounted with the membrane; for immersion cells, the temperature at the membrane may be assumed to be the same as the bulk volume of the receptor solution in the dissolution vessel, once equilibrated and stabilized. The temperature of each cell should typically remain within ±1° of the target temperature (typically 32° or 37°) during the test.
4. Qualification of Stirring/Agitation/Flow Rate: The stirring rate should be verified with a suitable device, such as a photo tachometer. For VDC, stirring rates in the range of 400–600 rpm are common. The stirring rate measured is typically applicable to the specific location where the stirring impeller is (e.g., a specific position in a rack that holds multiple VDC, or a specific stir plate). Stirring rate can be measured using a surrogate stir bar if the stir bar used in the diffusion cell is too small to obtain an accurate measurement. The paddle stirring rate for immersion cells of 50 or 100 rpm is common and should be verified and maintained during the test. Stirring rates are relevant for VDC, but not necessarily for FDC or immersion cells. In case of FDC, the flow rate of the receptor solution should be set at a specific value and maintained throughout the test. The flow rate (typically expressed in units of milliliters per minute) can be determined by setting the desired flow rate and collecting a sample into a tared vial for a specific duration. A suitable flow rate allows for a sample of sufficient size to ensure an accurate weighing and should not be substantially impacted by normal variability that may arise due to a drop that fails to fall from the tip of the dispensing tube. It is essential that the lengths of all the dispensing tubes are exactly the same, because differences in dispensing tube lengths can alter the void volume and lag time for samples to be dispensed into collection tubes.
8.2 Vertical Diffusion Cell (VDC)
A VDC consists of two compartments (a donor compartment and a receptor compartment with a sampling arm) separated by a membrane and held together by a clamp, screw top, or other means (see Figures 1–5). The VDC body (i.e., donor and receptor compartment with associated cell structures like the sampling arm and water jacket, if present) is usually made from borosilicate glass, although different materials may be used to manufacture the body and other parts of the VDC assembly. Other diffusion cells that are similar in design principles and features to those depicted in Figures 1–3 can generally be used.
The diameters of the orifices of the donor compartment and receptor compartment, which influence the diffusional area for the test, should be sized within ±5% of the specified diameter. The diameter of the donor and receptor compartment orifices may vary for different diffusion cell models. The receptor compartment orifice should be the same size as the donor compartment orifice. The design of the VDC should facilitate proper alignment of the orifices in the donor and receptor compartments. The membrane should be held in a horizontal plane between the donor and receptor compartments. Also, the membrane should be mounted so that it is flat, with no folds or wrinkles. The receptor compartment in each unit of a given model of VDC should be manufactured consistently, with a uniform height and geometry. All the cells (units) of the same VDC model should have the same nominal volume, and the actual volume should be measured and recorded for each individual cell. Care should be taken to minimize the intercell volume variability (e.g., not more than ±5%).
When conducting IVRT or IVPT studies, the VDC units are typically positioned in a stirrer rack (not depicted) that holds multiple VDC units (e.g., in sets of 6 or 7) in the correct orientation, providing magnetic stirring at a calibrated rate.
A magnetic nonstick (e.g., Teflon-coated), inert stir bar or similar stirring/agitation device in the receptor compartment is used as the internal stirring mechanism. The stirring bar or device should provide enough circulation to ensure contents of the receptor compartment are uniform. Samples of the receptor solution are removed at intervals throughout the test, and regardless of whether the entire receptor compartment volume or only an aliquot is sampled, an equivalent volume of stock receptor solution is replaced at each sampling event. The aliquots should be removed from the well-mixed center of the receptor compartment, unless sufficient proof exists that the entire receptor solution volume, including the portion in the sampling arm, are homogeneous and at the same concentration. For most VDC models, it is ideal for the volume of receptor solution in the sampling arm to be sampled and dispensed (repeatedly, a few times) into the well-mixed center of the receptor compartment, to ensure that the entire volume of the receptor compartment is homogeneous.
8.3 General IVRT VDC Equipment Set Up
Observational checks should be performed prior to testing. The donor and receptor compartment orifices should be evaluated for any chips or other damage which may affect the orifice area. Chipped or damaged cells should not be used.
8.3.1 Membrane mounting and dosing
Dosing procedures may be dictated by the requirements of the test method and/or the design of the diffusion cell. In general, two approaches are used: 1) the membrane is mounted and equilibrated in the diffusion cell prior to dosing the cell, or 2) the membrane is mounted and dosed during the assembly of the diffusion cell (e.g., VDC Model A in Figure 1), before the membrane comes into contact with the receptor solution. The dosing of the membrane prior to mounting on the diffusion cell in VDC Model A is acceptable because VDC Model A incorporates unique design elements to control the consistency of the dose and its alignment with the diffusion cell components. For all other VDC models, the membrane should be mounted and equilibrated in the diffusion cell prior to dosing the cell.
8.3.2 Dosing vdc with open donor compartments
It is conventional to install the membrane and then fill the receptor compartment with receptor solution. Any bubbles present underneath the membrane should be removed by tipping the cell and allowing the bubble to rise through the arm.
The system should be equilibrated, and the temperature should be measured at the membrane. The membrane should be within ±1° of the target temperature (typically 32° or 37°) prior to applying the dosage form. The stirrers should be turned on during the equilibration period.
For specific dosing techniques to apply the product to the membrane, refer to In Vitro Release Test (IVRT), Experimental Design, Dose.
8.3.3 Dosing vdc model a diffusion cells
Model A VDC are intended to have the membrane prepared and the dosing performed as part of the assembly of the cell top, which is separate from the cell bottom of the Model A VDC. In this model, the unique design and operation of the Model A VDC, as defined by the manufacturer, should be taken into consideration along with requirements of the test method. This method should not be used with conventional VDC designs like VDC models B and C.
During the preparation and dosing of the membrane in the Model A VDC cell top assembly, the membrane should be placed below the Model A VDC donor compartment’s dosage wafer on a flat and firm surface. The dosage form is then applied to the membrane within the dosage wafer. The Model A VDC donor compartment (dosage wafer) is filled to capacity; a small spatula can be used to dispense and spread the dosage form evenly within the dosage wafer and to remove any excess formulation.
The receptor compartment of the Model A VDC should be filled with receptor solution that is equilibrated to the target temperature (typically 32° or 37°) during the preparation and dosing of the membrane in the cell top assembly. The stirrers can be run during this period to speed up temperature stabilization. The receptor solution temperature should be measured and ensured to be within ±1° of the target temperature prior to mounting the cell top assembly. The stirrers can be run to help speed up temperature stabilization.
To start the test, the donor compartment cell top assembly is mounted on the cell bottom assembly of the Model A VDC so that the membrane is facing the receptor compartment, and the membrane separates the dosage form from the receptor solution. After assembly of the Model A cell top and bottom, the fully assembled cell should be checked to ensure that no bubbles are present underneath the membrane, and that the membrane has not slipped out of position whereby the dosage form might flow directly into the receptor compartment, circumventing the membrane.
8.3.4 Sampling vdc (general procedure for all models of vdc)
VDC are typically sampled one of two ways for IVRT, either withdrawal and replacement, or displacement. Cells which use withdrawal and replacement will typically only have one arm leading to the receptor compartment. Cells which are sampled by displacement will have two arms leading to the receptor compartment. For cells sampled by different methods it is recommended to refer to the manufacturer’s documentation.
8.3.5 Sampling by withdrawal and replacement
The receptor solution volume in the arm of many VDC may not mix adequately with the volume of receptor solution in the stirred receptor compartment. To ensure that the entire volume of the receptor compartment is homogeneous, it is ideal for the volume of receptor solution in the sampling arm to be sampled (drawn out) from the arm and re-dispensed into the well-mixed center of the receptor compartment multiple times prior to collecting a sample of the receptor solution for analysis. A precise volume of the sample should be withdrawn from the well-mixed center of the receptor compartment, typically using a precise syringe or pipette. Typical sample amounts are 200 to 500 µL. The same amount of receptor solution is then replaced through the same arm. Care should be taken to ensure that a bubble is not introduced during the sampling withdrawal and replacement process. If a bubble is present, it should be removed by tipping the cell and guiding the bubble up the arm. Depending upon the test method, the stirring may continue throughout the sampling procedure, or prior to the sample time point, the stirring may be stopped and then resumed shortly after the volume of receptor solution is replaced.
8.3.6 Displacement sampling
Cells with two sampling arms are often sampled by displacement. Prior to the sample time point, all stirring is stopped. The replacement receptor solution is slowly added to the receptor compartment to prevent mixing. The receptor solution displaces the sample out the other sampling arm for collection. Once the sample is collected the stirring is resumed. Typically, a rinse volume is required in the process for displacement sampling to ensure that the current sample does not contain remnants of a previous sample. This is especially important for automated systems.
8.4 General IVPT VDC Equipment Set Up
Observational checks should be performed prior to testing. The donor and receptor compartment orifices should be evaluated for any chips or other damage which may affect the orifice area. Chipped or damaged cells should not be used.
Receptor solution can either be preheated or allowed to equilibrate to temperature in the receptor compartments of the diffusion cells.
Skin should be cut to the appropriate size for the diffusion cell (larger than the diffusional area of the orifice) to ensure that the orifice is completely covered.
Ideally, the skin should be gently stretched to ensure that it is flat (with no folds or wrinkles) when mounted upon the diffusion cell with the stratum corneum of the skin facing toward the air. An inert support membrane (i.e., a synthetic membrane validated for use with an IVRT method for the same drug product) may be used beneath the skin, as appropriate, as long as it does not impede the diffusion of the drug from the skin to the receptor solution. The donor compartment should then be installed on top of the skin ensuring that the skin is mounted securely in the diffusion cell. The receptor compartment may be filled with receptor solution either before or after mounting of the skin. Any bubbles underneath the skin should be removed by tipping the cell and guiding the bubble up the sampling arm. The thermoregulatory mechanism of the VDC system should then be utilized to equilibrate the skin to the target temperature.
Once the hydration and temperature of the skin has equilibrated (i.e., after 30 min) the skin surface temperature should be measured. The skin surface temperature should be within ±1° of the target temperature (32°) prior to applying the dosage form. The temperature of the receptor solution, or the circulating water bath, or the set point of a dry heat system, or any other component of the system should not be assumed to be the same as the skin surface temperature. Instead, the skin surface temperature should be directly measured (with an infrared thermometer or a thermocouple), and the thermoregulatory mechanism of the VDC system should be adjusted as needed to produce the target temperature of 32° at the skin surface. The stirrers can be run during the duration of the equilibration to speed up temperature stabilization.
For specific dosing techniques to apply the product to the membrane, refer to In Vitro Permeation Test, Experimental Design, Dose.
One approach to sampling involves draining the receptor compartment of all the receptor solution and refilling it completely with fresh receptor solution. Removal of the entire receptor solution volume and full volume replacement of the receptor solution at each time point may provide optimal solubility sink conditions. Although the entire receptor solution volume is sampled, an aliquot is typically used for analysis.
Another approach to sampling involves the removal of only a partial amount (i.e., an aliquot) of the entire receptor solution volume, leaving behind a partial volume of the receptor solution at each sampling. This approach can allow the concentration of a compound to increase above a quantification limit during time periods of low flux. However, the sampling of relatively small aliquots of the receptor solution for an IVPT study may introduce anomalous measurements of apparently negative flux in certain regions of the IVPT study and produce flux profiles that are difficult to interpret. Therefore, when sampling partial volumes, it is prudent to maximize the volume of the sample aliquot, in order to minimize the potential for apparent negative flux results.
8.5 VDC Qualification
VDC systems should generally match the description of the equipment described in this chapter. Other designs can be used with sufficient scientific justification so long as they consist of a donor compartment separated from a receptor compartment by a membrane.
VDCs should be individually qualified and uniquely identifiable either by a manufacturer issued serial number or other means. The orifice area for the donor and receptor compartments can be determined using vernier calipers. Receptor compartment volume should be determined with all components (e.g., stir bars) in place. The receptor compartment volume should be determined (gravimetrically or volumetrically) to a precision of 0.01 mL. The nominal receptor compartment volume (e.g., 7 mL) should never be used in place of an accurately determined VDC volume (e.g., 6.89 mL). Using the nominal volume fails to account for variations of the receptor compartment volume due to manufacturing which could affect the accuracy and precision of test results.
8.6 VDC Preventative Maintenance Procedure and Requalification
All VDC systems should be properly maintained and requalified on a regular basis (as described previously). The relevant VDC systems (heating/thermoregulation, water bath and circulation tubes/manifolds, magnetic impeller/stirring, mechanical sampling, etc.) and other components of the VDC system should be kept clean and their operation should be evaluated to ensure that each diffusion cell can maintain the skin surface temperature within the target range (32 ± 1°), maintain the stirring speed within the specified range, and support the performance of the test within the specifications of the test method.
Any stirring device that is not stirring properly with the receptor solution present should be repaired or replaced. Since direct measurement of the stirring device used for testing is not always feasible, the stirring system (e.g., the rotating magnetic impeller) can be evaluated with a surrogate. For example, if stirring within a VDC is done with a 5 mm stir bar, the VDC can be temporarily removed, and a 25 mm stir bar can be placed in the cell block which can be measured to determine the stirring speed.
Sampling devices, such as pipettes, syringes, or automated systems should be evaluated to ensure that they are withdrawing and replacing sample volumes of the receptor solution with sufficient accuracy and precision.
Requalification of automated systems should be performed (e.g., every 6–12 months) based on considerations including the extent of equipment usage, the risk tolerance of the laboratory performing the test, and manufacturer’s recommendations. Requalification should also be performed if the system is relocated or undergoes a major repair. In situations where a new VDC unit is incorporated into a previously qualified VDC system (with a set of specific VDC units), the elements of VDC qualification relevant to the new VDC unit should be performed, and the elements of VDC qualification relevant to the (previously qualified) VDC system should be requalified, as relevant (e.g., this may include a requalification of the skin surface temperature control for the new/replacement VDC introduced into the previously qualified VDC system).
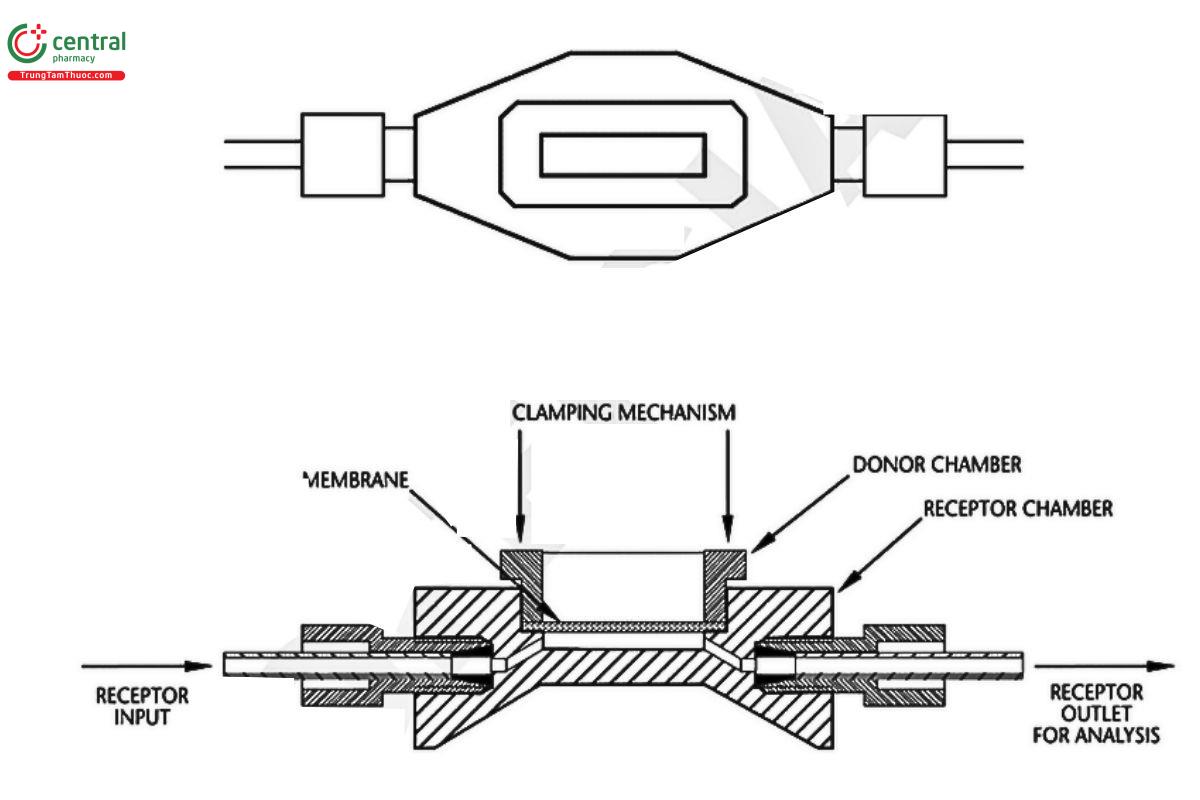
8.7 Immersion Cells
For immersion cells, USP Apparatus 2 should be qualified according to the procedure described in Dissolution 〈711〉.
The immersion cell system consists of the following components:
- immersion cell which is an inert housing that holds a membrane and donor compartment.
- a smaller version of USP Apparatus 2 (see 〈711〉) with vessel volumes that vary from 50–200 mL; however, 150- or 200-mL vessels are typically used.
An example immersion cell configuration can be seen in Figure 6 and Figure 7.
The components of the immersion cell are listed in Figure 6. Formulation is placed in the sample compartment and sealed to prevent leaks by the membrane, washer, and retaining ring. The diameters of the orifices of the donor compartment, which define the dosage delivery surface area for the test, should be sized within ±5% of the specified diameter. The membrane should be held horizontal and faced up toward the paddle. Also, the membrane should be mounted so that it is flat, not wrinkled from overtightening the retaining ring. Donor compartments range from holding 300 mg to 4 g of the formulation; some models are adjustable.
The immersion cell can be used with a smaller version of USP Apparatus 2 (see 〈711〉) with vessel volumes that vary from 50–200 mL; however, the 150- or 200-mL vessels are more commonly used. A flat-bottom variation of the 150- or 200-mL vessel should be used to avoid the issue of dead space under the cell and standardize the orientation of the immersion cell in the vessel. Flat bottom vessels with a small peak at the base, to keep the immersion cell centered, may also be used.
Use of the 150- or 200-mL vessel with USP Apparatus 2 requires the following modifications:
- holders for the small-volume vessels (e.g., 200 mL vessels instead of standard 900-mL vessels)
- adapter plate to position the small-volume vessel in the center of the spindle
- smaller size shaft/paddle [2/3 cm (1/4 in)] to fit in the small-volume vessels
8.8 General IVRT Immersion Cell Equipment Set Up
Immersion cell sample compartment volume should be set to a constant volume (compartment volume can be adjusted using an adjustment tool). Alignment of the retaining ring over the sample compartment is ensured using an alignment tool for the specific surface area of the immersion cell.
Observational checks should be performed prior to testing. The donor and receptor compartment orifices should be evaluated for any damage which may affect the orifice area. Damaged cells should not be used.
Before loading the cells and placing the receptor solution in the vessel, set the paddle height, which is 10 ± 2 mm above the surface of the membrane. All other operational parameters, such as level, vibration, wobble, etc., should be set at the same conditions defined for USP Apparatus 2. The small-volume condition is qualified by first using the standard Apparatus 2 setup and Performance Verification Test, Apparatus 1 and 2 (see 〈711〉).
Fill the reservoir dosage area with the sample under test. Ensure that the reservoir is filled to the top to minimize the possibility of air bubble formation between the surface of the sample and the membrane. A uniform surface can be obtained with the aid of a spatula. The typical quantity of sample is between 300 mg and 4 g, depending on the type of immersion cell used. Using forceps or tweezers, remove the membrane from the soaking receptor solution and place it over the top of the sample compartment by rolling it onto the surface to avoid entrapment of air. Ensure that the membrane is free of wrinkles. Assemble the immersion cell components as specified by the device manufacturer. Carefully place the completed assembly into the bottom of the dissolution vessel with the membrane facing up. The appropriate preheated receptor solution may be preloaded in the vessel or can be added after immersion of the immersion cell to start the test.
At the end of the test period, dismantle the cell and examine the contents for anything unusual that could explain any anomalous data (e.g., leaks, bubbles, etc.).
8.9 Immersion Cell Qualification
Immersion cells should match the description of one of the instruments listed in this chapter. Other designs can be used with sufficient scientific justification so long as they consist of a donor compartment separated from a receptor compartment by a membrane.
At a minimum, the orifice area and donor compartments volume must be certified. Additional dimensions should also be certified when possible. Due to the uniformity in manufacturing immersion cells, individual identification of the cells and components is recommended, but not required.
Orifice area for the donor and receptor compartments can be determined using vernier calipers. Volume of the donor compartment can be calculated using the donor compartment diameter and height.
A known volume of receptor solution should be transferred to the vessel when testing by suitably accurate method or device.
Immersion cells should only be used on suitably qualified dissolution apparatus.
Requalification should be performed every 6–12 months, based on equipment usage, risk tolerance, and manufacturers recommendation. Requalification should also be performed if the system is relocated or undergoes a major repair.
8.10 Flow-Through Diffusion Cell (FDC) for IVPT
The components of the FDC are displayed in Figures 8–10. The skin is placed above the receptor compartment and kept in place by the donor compartment and a clamping mechanism. The skin should be held in a horizontal plane between the donor and receptor compartments. The donor and receptor compartments should be made out of an inert material. The surface area of the orifices of the donor compartment, which define the dosage delivery surface area for the test, should be sized within ±5% of the nominal value.
The FDC is connected to a pump which provides a constant or pulsation flow of receptor solution that passes through the receptor compartment, into a sample line then collected by a sample vial. Typically, the receptor compartment within the cell holds relatively small volumes (≤0.5 mL) as sink conditions are controlled, in part, by the flow rate of the pump. Flow rates in the range of 2–200 µL/min are common, however some methods may have flow rates in the thousands-of-microliters-per-minute range (i.e., in the milliliter-per-minute range), and the appropriate flow rate for each IVPT method may need to be optimized during method development depending upon the solubility of the drug in the receptor solution, the permeation rates during relevant sampling durations, and the limit of quantification of the receptor sample solution analysis method. The cells are typically placed on the arm of a fraction collector which will align the sample lines with vials or test tubes for each sample point. The FDC is heated to allow the membrane to be maintained at appropriate temperature.
9 General IVPT FDC Equipment Set Up
Observational checks should be performed prior to testing. The donor and receptor compartment orifices should be evaluated for any damage which may affect the orifice area. Damaged cells should not be used.
Ideally, the skin should be gently stretched to ensure that it is flat (with no folds or wrinkles) when mounted upon the diffusion cell with the stratum corneum facing the air. An inert support membrane (e.g., a synthetic membrane validated for use with an IVRT method for the same drug product) may be used beneath the skin, as appropriate, as long as it does not impede the diffusion of the drug from the skin to the receptor solution. The donor compartment should then be installed on top of the skin ensuring that the skin is mounted securely in the diffusion cell. The receptor compartment may be filled with receptor solution either before or after mounting of the skin. Any bubbles underneath the skin should be removed by tipping the cell and/or facilitating the removal of the bubble via the outflowing receptor solution. The thermoregulatory mechanism of the FDC system should then be utilized to equilibrate the skin to the target skin surface temperature of 32°.
The FDC is connected to a pump and inflow as well as outflow tubing. When all the FDC for testing are connected to pumps, air bubbles may need to be purged from the receptor compartments. A typical method is the cells are tipped upward, with the outflow sample line raised, while the pump is run at a high speed which facilitates the removal of the air bubble from under the skin via the outflowing receptor solution. The pump can be stopped after all air has been purged from the receptor compartment. The cell should be lowered to the testing position and allowed to equilibrate.
Once the hydration and temperature of the skin has equilibrated (e.g., after 30 min) the skin surface temperature should be measured. The skin surface temperature should be within ±1° of the target temperature (32°) prior to applying the dosage form. The temperature of the receptor solution, or the circulating water bath, or the set point of a dry heat system, or any other component of the system should not be assumed to be the same as the skin surface temperature. Instead, the skin surface temperature should be directly measured (with an infrared thermometer or a thermocouple), and the thermoregulatory mechanism of the FDC system should be adjusted as needed to produce the target temperature of 32° at the skin surface.
Collection vials or test tubes should be weighed when empty to determine the exact volume of sample present after collection has completed.
For specific dosing techniques to apply the product to the skin, refer to the In Vitro Permeation Test, Experimental Design, Dose.
Set the pump to the flow rate to be used for testing and align the outflow sample tubes (which should all be the same length) with the first set of vials or test tubes.
After samples have been collected, the vials or test tubes should be weighed to determine the exact volume of receptor solution collected. This volume should be used when determining the total amount of drug present in each sample.
9.1 FDC Qualification
FDC systems should generally match the description of the equipment described in this chapter. Other designs can be used with sufficient scientific justification so long as they consist of a donor compartment separated from a receptor compartment by the skin.
FDCs should be individually qualified and uniquely identifiable either by a manufacturer issued serial number or other means. The orifice area for the donor and receptor compartments can be determined using vernier calipers. It may be appropriate to qualify the lengths of (inert) tubing for each of the flow-through diffusion cells, and their associated dead volumes, to accurately calculate the lag time before a sample elutes through the tubing and is collected.
The flow rate of the pump should be qualified for the rate used during testing. For flow rates in the microliters-per-minute range, it is recommended to extend the sampling time to reduce the effects of droplets adhering to the sample line. If an automated system is used it should be ensured that the sample lines are properly aligned over the vials.
Skin surface temperature should be measured using an IR thermometer. The temperature of the heating block must be set higher than the desired membrane temperature (e.g., 33° for a 32° skin surface temperature).
9.2 FDC Preventative Maintenance Procedure and Requalification
All FDC systems should be properly maintained and requalified on a regular basis (as described previously). The relevant FDC systems (heating and thermoregulation, pumps, tubes, mechanical sampling systems, etc.) and other components of the FDC system should be kept clean and their operation should be evaluated to ensure that each diffusion cell can maintain the skin surface temperature within the target range of 32 ± 1°, maintain the flow rate within the specified range, and support the performance of the test within the specifications of the test method.
The pump should be evaluated to ensure it is working properly. If a peristaltic pump is used, the tubing should be unclamped once the test has completed to prevent unnecessary wear on the tubing, which may cause variation in the flow rate. Peristaltic pump tubing should be replaced at regular intervals to ensure the flow rate remains consistent across all the cells.
For automated sampling systems, sample tube alignment should also be checked to ensure that all samples are entirely collected into the vials or test tubes, without leakage, failure to capture samples, or sample loss for other reasons.
Requalification of automated systems should be performed (e.g., every 6–12 months) based on considerations including the extent of equipment usage, the risk tolerance of the laboratory performing the test, and manufacturer’s recommendations. Requalification should also be performed if the system is relocated or undergoes a major repair. In situations where a new FDC unit is incorporated into a previously qualified FDC system (with a set of specific FDC units), the elements of FDC qualification relevant to the new FDC unit should be performed, and the elements of FDC qualification relevant to the (previously qualified) FDC system should be requalified, as relevant (e.g., this may include a requalification of the skin surface temperature control for the new/replacement FDC introduced into the previously qualified FDC system).
