Risks and Mitigation Strategies for the Storage and Transportation of Finished Drug Products
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
Proper storage and transportation of finished drug products are critical activities in an integrated supply chain. These finished drug products include but are not limited to temperature-sensitive small molecules, vaccines, biologics, biotechnological products, radiopharmaceuticals, and combination products. With the globalization of the pharmaceutical industry, various individuals and organizations from locations around the world can come into contact with the finished drug product. The storage and transportation processes for a drug product may involve complex movements with differences in documentation, handling requirements, and communication between the various entities throughout the supply chain.
Environmental controls play a key role in maintaining drug safety, quality, and efficacy. Temperature is one of the most important parameters to control. Drugs must be stored and transported according to predetermined conditions (e.g., temperature) as supported by stability data. Temperature excursions outside of their respective labeled storage conditions, for brief periods, may be acceptable provided that stability data and scientific/technical justification exist, demonstrating that product safety, quality, and efficacy is not affected. To maintain the original quality, every party involved in the storage and transportation of a finished product should have an in-depth understanding of the storage and transportation risks and have the appropriate mitigation strategies in place to control these risks. The intent of this chapter is to identify common risks in the storage and transportation of drug products and to recommend mitigation strategies. The chapter is not meant to prescribe specific approaches or discuss regulatory frameworks currently in place, but rather to focus on risks and mitigation strategies for quality processes to maintain product and supply chain integrity. The principles of this chapter can be used to facilitate the storage and transportation of drug products throughout a supply chain that is controlled, measured, and analyzed for continuous improvements while also maintaining the integrity of the drug product in its packaging during distribution.
2 SCOPE
This chapter applies to organizations and individuals involved in the storage and transportation of drug products, including but not limited to the following:
- Manufacturers of drug products, radiopharmaceuticals, biological products, and biotechnological products
- Repackaging operations in which the product may be owned by a company other than the primary manufacturer
- Healthcare providers and institutions such as hospitals; outpatient, ambulatory, and urgent care centers; home health providers; vaccine clinics; emergency departments; and medical, dental, and veterinary offices
- Pharmacies, including but not limited to retail, compounding (sterile and nonsterile), specialty, mail order, hospital, nursing home, and hospice
- Importers and exporters
- Wholesale distributors
- Third-party logistics providers, brokers, freight forwarders, consolidators, and other organizations involved in storage; road, rail, sea, and/or air transport services, or mail distributors that offer expedited or controlled-temperature shipping services
Manufacturers of active pharmaceutical ingredients, excipients, packaging materials, medical devices, and dietary supplements are not within the scope of this chapter. However, the concepts, risks, and mitigation strategies discussed in this chapter may be useful and can be applied in these cases, if desired.
3 RISK-BASED APPROACH TO THE STORAGE AND TRANSPORTATION OF FINISHED DRUG PRODUCTS
Figure 1 illustrates the risk-based approach of a quality management system (QMS). It represents how product knowledge and process knowledge facilitate the identification of risk. The gure also illustrates how mitigation strategies that are planned to reduce the identified risks, categorized in clusters, form the pillars of a QMS.
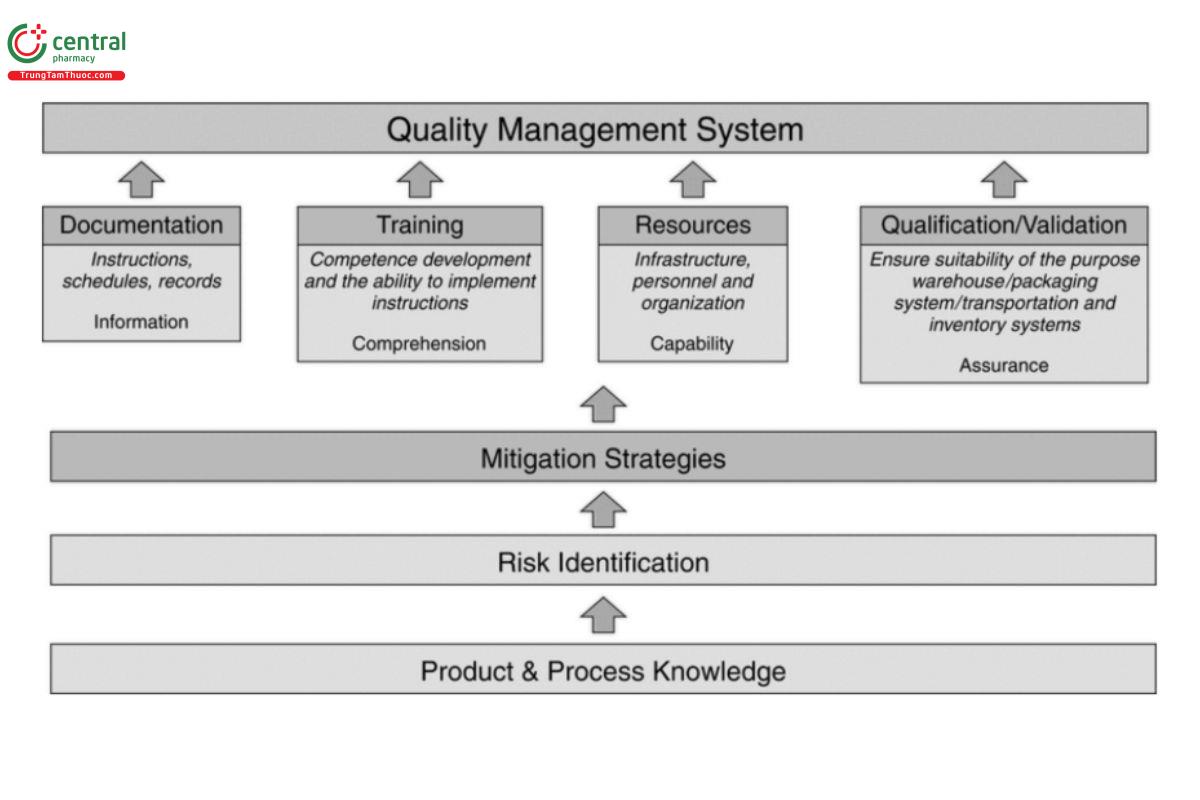
Product and process knowledge is the starting point in identifying risks related to the storage and transportation of drug products. Product knowledge includes but is not limited to the following: intended use; storage conditions; potential hazards to environment and personnel (e.g., hormones, cytotoxic drug products, and radiopharmaceuticals); and inherent vulnerability (e.g., high potential for abuse, high-value drugs, attractiveness of freight to criminals, counterfeiting, and diversion). Process knowledge includes but is not limited to the following: knowledge of supply chain partners; physical modes of transportation (air, sea, rail, road, or a combination of modes); transportation routes; and national and international regulation. Understanding these factors helps an organization identify their associated risks. Process mapping is a useful tool for organizations to gain further understanding of a particular process and/or operation (e.g., transport lane selection or loading/unloading patterns of warehouses and vehicles).
Risk identification is the systematic use of information to identify potential sources of harm (hazards). Information can include historical data, theoretical analysis, informed opinions, product and process knowledge, and the concerns of stakeholders. Risk identification addresses the question: “What might go wrong?” Mitigation strategies are part of the risk control process, specifically risk reduction. Risk reduction addresses the question: “What can be done to reduce or eliminate risks?” In this way, risk reduction can include actions taken to mitigate the severity or probability of harm. Processes that improve the detectability of hazards and quality risks can also be used as part of a risk control strategy.
Different stakeholders perceive risks differently; for example, they may assign different levels of risk based on their experience and knowledge or they may estimate the probability of risk to product differently. Regardless of where your organization fits into the supply chain, consideration of risks and mitigation actions taken should consider potential impact throughout the supply chain. The mitigation strategies can be divided into four categories that are fundamentals (or pillars) of a QMS (see Figure 1). These strategies, when implemented, give an organization the autonomy to plan, implement, measure, and improve their processes according to current regulations and associated risks. Generally, mitigation strategies fall within four categories related to: 1) documentation, i.e., providing instructions for a specific operation or process to standardize it and establish consistency; 2) training, i.e., ensuring competence; 3) resources, i.e., providing capability through infrastructure and human resources; and 4) qualification and validation, i.e., assurance that the resources and processes are reliable, reproducible, and robust.
Several informal and formal tools can be used to conduct risk assessments and control risk. Examples of tools used to perform risk identification include (but are not limited to): flow charts, process mapping, trends, historical data records (such as temperature records over a particular route), and observations. Other tools—such as failure mode effects analysis (FMEA), fault tree analysis (FTA), hazard analysis and critical control points (HACCP), hazard operability analysis (HAZOP), and preliminary hazard analysis (PHA)—can also be used for conducting risk management. [Note—See International Council for Harmonisation (ICH) in Additional Sources of Information].
Table 1 contains illustrative examples of the risks related to drug product storage and transportation, along with their mitigation strategies. The list presented below is not exhaustive and is meant to stimulate discussion and provide examples.
Table 1. Storage and Transportation Risks and Mitigation Strategies
Hazard | Effect | Mitigation Strategy | Mitigation Category |
General Risk | |||
Human error due to excessive duties or lack of training or competence | Mishandling along the supply chain, which can affect product quality and integrity and patient | Evaluate training effectiveness [are trainees competent in key aspects of the standard operating procedures (SOPs)?] | Training and Resources |
General Risk | |||
|
| Assign appropriate number of personnel to avoid excessive duties placed on one individual Assign employees to duties that they are qualified for based on education, experience, and competence |
|
Procurement and Sales | |||
Buy from or sell to unlicensed trading partners | Legal sanctions; patient safety | Supplier qualification Customer qualification Checks to ensure license is current and appropriate Quality agreements between supplier and trading partners | Documentation and Resources |
Receiving and Shipping | |||
Receive adulterated, falsified, or recalled product | Patient safety; introduction into legitimate supply chain of a product that is potentially substandard, illegal, or counterfeited | Quarantine Quality control test Packaging identification fingerprints Recall awareness Notify regulatory authorities or trading partners Qualification of supply chain partners and on going performance qualifications | Documentation and Training |
Receive product that was not ordered | Unmatched transaction (e.g., wrong paperwork or transaction data sent); introduction into legitimate supply chain of a product that is potentially substandard, illegal, or counterfeited | Adhere to receiving SOP | Documentation and Training |
Mix products with different status (rejected, recalled, or returned) | Patient safety; shipping or selling of inappropriate product | Product segregation, physical in the location and/or system Warehouse layout (logical ow and holding areas in order to avoid mix-ups) Adhere to receiving SOP | Storage, Documentation, and Resources |
General Risk | |||
Shipping and receiving delays due to inclement weather, natural disasters, trac disruption | Patient safety; arrival delays; temperature out of specification (temperature excursions, e.g., product freezes accidentally) | Reschedule the delivery Temporary parking (waiting for opportunity to unload) or off-loading to a temperature controlled facility or vehicle Recondition materials to ensure temperature maintenance during delay Rescue services Monitoring to demonstrate that the product integrity was not compromised; in cases when monitoring was not implemented due to qualifications and risk assessments, these qualification parameters delays might be out of scope due to the qualification parameters | Documentation, Training, and Resources |
Storage | |||
Improper entry into a materials management system: wrong batch number, wrong expiration date, wrong status (e.g., product was approved but should be quarantined), or wrong amount | Inaccurate stock; picking and/or shipping product that should have been quarantined but was marked approved | Adhere to stocking SOP Software validation | Documentation and Validation |
Product stored in wrong physical location | Patient safety because of picking error (software shows location, but staff can pick the wrong product if there is no check of the physical location) | Adhere to stocking SOP Automated checking system | Documentation, Training, and Validation |
Product exposed to temperature excursions | Refer to SOP showing list of products and their temperature specifications | Documentation | |
Legal sanctions for controlled substance; risk of diversion for controlled substance | Refer to SOP showing list of products and their license category (e.g., controlled, radiopharmaceutical) | Documentation | |
Environmental conditions out of specification | Affects product quality, product integrity, and patient safety (e.g., freezing of vaccine or biologic product); product loss causing financial loss | Warehouse, packaging, and transportation qualification (temperature mapping) Product storage identification
| Qualification and Validation, Training, and Documentation
|
Out-of-range cold or hot areas; product storage temperature excursion product loss; financial loss; patient product availability | Qualification (temperature mapping) Storage temperature monitoring program Homogenous airow Monitoring and alarms Adhere to excursion handling SOP | Documentation, Resources, and Qualification and Validation | |
Temperature monitoring device failure | Out-of-range cold or hot areas; product storage temperature excursion; product loss | Backup monitoring devices with independent power source Adhere to excursion handling SOP | Documentation and Resources |
Storage or temperature system failure due to: Loss of electrical power Failure of temperature control or air circulation systems Unusual weather event | Out-of-range cold, warm, or hot areas; product storage temperature excursion; product loss | Temperature and power alarms Backup power and coolant systems (redundant) and/or contingency storage Adhere to excursion handling SOP | Documentation and Resources |
Fear of reporting nonconformance and exception conditions | Affects product integrity and patient safety due to serious conditions not communicated | Independent quality reporting structure Education on product integrity and impact on patients and the supply chain | Training and Resources |
Picking | |||
Picking (human) error | Patient safety; wrong item shipped (product availability); product returns | Automated checking system or second person verification | Documentation, Training, and Qualification and Validation |
[Note—The effect of a hazard, if not mitigated, could impact product integrity and ultimately patient safety.]
Change to read:
4 RISK MITIGATION CATEGORIES AS QMS ELEMENTS
A QMS is necessary to implement, monitor, and maintain a robust storage and transportation process. Key elements of a QMS are management responsibility, documentation, training, resource management, complaint handling, deviation/excursion handling, returns and recalls management, qualification/validation, monitoring, audit, corrective action, preventive action, and continuous improvement. This chapter does not intend to provide a QMS framework; rather this chapter highlights how risk mitigation strategies are pillars of a QMS and how QMS elements ensure the quality and security of drug products throughout storage and transport.
The four categories of risk mitigation strategies are discussed in Table 2 , with a matrix that links the mitigation strategy to the role that an organization plays within the supply chain.
Table 2. Mitigation Strategies Used by Organizations within Supply Chain
Role of Organization within Supply Chain | |||||||||
Applicable Mitigation Strategies | Manufacturer | Wholesaler and Distributor | Pharmacy or Compounding Pharmacy | Hospital and Healthcare Provider | Broker | Logistics Service Provider (LSP) | |||
Documentation (Manuals, Procedures, Protocols, Records) | |||||||||
Quality manual | Yes | Yes | Yes | Yes | Yes | Yes | |||
Labeling | Yes | Yes | Yes | Yes | No | Yes | |||
Procurement | Yes | Yes | Yes | Yes | Yes | Yes | |||
Receiving | Yes | Yes | Yes | Yes | No | Yes | |||
Picking | Yes | Yes | Yes | Yes | No | Yes | |||
Packing | Yes | Yes | Yes | Yes | No | No | |||
Sales | Yes | Yes | Yes | Yes | Yes | No | |||
Storage | Yes | Yes | Yes | Yes | No | Yes | |||
Transportation | Yes | Yes | Yes | Yesa | No | Yes | |||
Supplier qualification | Yes | Yes | Yes | Yes | Yes | Yes | |||
Customer qualification | Yes | Yes | No | No | Yes | No | |||
Quality agreements | Yes | Yes | Yes | Yes | Yes | Yes | |||
Licenses and authorizations | Yes | Yes | Yes | Yes | Yes | Yes | |||
Recall | Yes | Yes | Yes | Yes | Yes | Yes | |||
Return | Yes | Yes | Yes | Yes | No | No | |||
Temporary parking | Yes | Nob | Yes | No | No | Yes | |||
Excursion handling | Yes | Yes | Yes | Yes | No | Yes | |||
Disposal of expired and nonconforming drug products (e.g., suspect, expired, recalled, quarantined) | Yes | Yes | Yes | Yes | Yes | No | |||
Pest control and pallet conservation | Yes | Yes | Yes | Yes | No | Yes | |||
Training | |||||||||
Documentation (Manuals, Procedures, Protocols, Records) | |||||||||
Training | Yes | Yes | Yes | Yes | Yes | Yes | |||
Resources | |||||||||
Product segregation | Yes | Yes | Yes | Yes | No | Yes | |||
Storage are (layout/logical flow) | Yes | Yes | Yes | Yes | No | Yesc | |||
Maintenance | Yes | Yes | Yes | Yes | No | Yes | |||
Calibration | Yes | Yes | Yes | Yes | No | Yes | |||
Monitoring systems and alarms | Yes | Yes | Yes | Yes | No | Yes | |||
Appropriate number of personnel | Yes | Yes | Yes | Yes | Yes | Yes | |||
Organizational chart and job descriptions | Yes | Yes | Yes | Yes | Yes | Yes | |||
Qualification and Validation | |||||||||
Temperature mapping | Yes | Yes | Yes | Yes | No | Yes | |||
Shipping packaging qualification | Yes | Yes | Yes | Noc | No | No | |||
Software validation (automated checking systems, inventory management system) | Yes | Yes | Yes | Yes | Yes | Yes | |||
a Unless the healthcare entity ships between owned facilities or to the patient.
b If the ownership of the product has already transferred to the distributors, then temporary parking could be the responsibility of the distributors/wholesaler.
c Applied only if the logistics service provider has a temporary storage area.
4.1 Documentation and Procedures
Documentation provides written information that allows for consistency and traceability of actions. For this reason, documentation is a fundamental part of any quality system. In a risk-based approach, documentation is a category of risk mitigation. Some examples of documentation include quality manuals; standard operating procedures (SOPs); labeling; records pertaining to procurement, receiving, storage, and transportation; supplier qualification records; quality agreements; recalls; and excursion-handling records.
The sections that follow describe key types of documents that are fundamental for a QMS supply chain system. These documents can be used in a range of different organizations (see Table 2).
4.1.1 Quality manual
A quality manual is a top-level quality document for all areas of the business affected by the quality system. The quality manual contains the quality policy, quality objectives, quality system structure, and information related to the management of a specific organization. The content may also include information on inspection management, customer complaints, recalls, withdrawals and holds, corrective and preventive actions, nonconformance and change control, information about regulatory issues, and performance evaluation through quality indicators.
4.1.2 Standard operating procedures
SOPs are controlled documents, with document owners and approvers, effective dates, revision management, and scheduled reviews. Procedures should cover areas governed by the quality manual and should cover all aspects of the operation that may affect product quality, including handling, distribution, and all regulated activities in relation to the specific business (e.g., national and international laws). SOPs should also address actions that are performed to identify and mitigate risks.
Some key components are, but not limited to, the following:
- Corrective action/preventive action (CAPA)
- Documentation
- Record keeping
- Inventory management
- Licensing
- Management reviews
- Nonconforming product (for example, but not limited to, damaged or adulterated, expired, recall, suspect or illegitimate product, and temperature deviation)
- Order processing Purchasing
- Picking
- Packing and shipping
- Receiving
- Returns
- Storage
- Training
Procurement: One of the risks in procurement is buying and selling for transport with an unlicensed trading partner, leading to legal sanctions. A procurement procedure ensures that a product is purchased according to product specifications and that purchases are made from qualified partners that are licensed as appropriate.
Receiving: One potential risk when moving a product forward and backward along the supply chain is the introduction of substandard, illegal, or counterfeit products into the legitimate supply chain. Receiving is the operation related to the entrance of cargo into the operation’s facility, starting with unloading the cargo from vehicles and then receiving, checking, and stocking the operation’s facility. Each organization should have a receiving procedure that determines the appropriate checks for this operation. A checklist can be used as a reminder of what to inspect and what to record. Where appropriate, the transport vehicle can be inspected before unloading to verify that adequate protection from contamination was maintained during transit. To avoid the risk of receiving a product that was not ordered, all deliveries should be verified at the time of receipt in order to check that containers were not damaged and that the consignment corresponds to the order.
All incoming products should be segregated from salable inventory and identified. Products should be transferred to their respective storage areas according to their classification and storage specification. When products arrive at warehouse loading docks and other arrival areas, they should be transferred as quickly as possible to a designated storage area within a time period that is consistent with the organization's receiving SOP.
Inclement weather, natural disasters, and traffic disruption can cause receiving delays and potential temperature excursions. Rescheduling the delivery, temporarily parking while waiting for an opportunity to unload, rescue services, or even reconditioning materials to ensure temperature maintenance during delays are all mitigation strategies that can be implemented. Procedures to follow for these contingencies should be written, and personnel should be trained on them.
Storage and transportation: During storage and transportation, two approaches can be used to keep the product within its required labeled specifications:
1. Controlling the environmental conditions within equipment, storage rooms, and transportation vehicles; and when applicable, using thermostatically controlled devices such as a heating, ventilation, and air conditioning (HVAC) system or refrigerators
2. Using packaging materials that allow for the control of environmental conditions (e.g., passive/thermal packaging, thermal blankets, temperature stabilizers, desiccants, and light-resistant material)
The organization should have written procedures for qualification of storage, shipping containers, and transportation (in-transit storage) of drug products, taking into account at a minimum:
- Product category (e.g., narcotics, medical devices, temperature-sensitive or hazardous products)
- Layout of the area [e.g., floor-standing pallets, pallet racking, and boxes inside refrigerators where practical and applicable (not feasible for air freight)]
- Volume of stored product (including peaks of storage)
- Air circulation and environmental conditions (e.g., temperature, relative humidity, pressure, shock, and vibration)
- Contingency plan for outages and employee breaks
The procedure should be written based on a risk assessment of factors that can impact product quality during storage and transportation. This procedure will be a measure to mitigate this risk.
If there are problems with vehicles during the transportation process (e.g., breakdowns, accidents, and loss of fuel), a product should be protected against environmental factors, theft, and diversion. The risk assessment and written procedures should take these situations into consideration. Depending on the probability of occurrence and the level of risk, an organization may consider backup systems or access to backup systems in the event of logistics disruption (e.g., severe weather).
Recalls: All supply chain partners are responsible for maintaining the quality and integrity of products under their control. Anytime a deviation is found that likely affects the safety or efficacy of a marketed product, an appropriate action should be taken, including a potential recall. A challenge involved in recalling a product is ensuring that the amount of product that was initially distributed is returned. Thus, sharing information on product recall increases the efficacy of the recall procedure, provides transparency to supply chain partners, and mitigates the risk of reintroducing a recalled product into the supply chain. The organization should have a written procedure that establishes the steps for recalling products and the control of recalled products, such as product identification and segregation. The extent of the recall needs to correspond to the level of risk. The extent of the recall may change if the risk is re-evaluated. Returns: Accepting a returned product for restocking poses the risk that the product may not be authentic or its quality may have decreased. A risk-based evaluation should be performed to determine if the product will be acceptable for restocking and resale or if it needs to be destroyed. During the evaluations, returned products should be kept in a segregated area designated specifically for returns until nal disposition. A written procedure for handling returns should be in place, taking into account:
- Reasons for return
- Appearance and integrity of the original packaging
- Evidence of conditions in which the cargo was transported and stored throughout the entire time span
- Duration of time between the original shipment and its return
- Authenticity of the product, to include product identifier verification covered by applicable traceability and serialization laws and evidence of proper storage while in possession of the entity returning the product (e.g., ongoing assurance) Representative sampling for quality control analysis (if applicable and following regulation)
- Expiry date and batch number
- Batch trace history of excursion
- Information from any track-and-trace system in place
Supplier qualification (logistics service provider, third party logistics provider, material suppliers, maintenance providers, etc.): Supplier qualification is a process in which the organization assesses its suppliers regarding their licenses, authorizations, and compliance with regulatory requirements for the distribution of drug products. The organization should establish a written procedure for how suppliers are selected and evaluated, including the criteria for qualification and the period for requalification on a risk-based approach.
4.1.3 Labels
Labels are fundamental to material identification. For this reason, any label change should be communicated to downstream supply chain partners. Labels applied, even to small containers, should be clear, indelible, unambiguous, and permanently fixed in the format established by the manufacturer, packager, or repackager. The shipping label should include wording or icons to emphasize storage and transportation conditions, handling requirements, and hazards. The use of symbols that are recognized by international organizations is strongly recommended. ▲▲ (USP 1-Dec-2023)
4.1.4 Quality agreements
Written agreements (e.g., quality agreement, technical agreement, service level agreement) should be in place between applicable organizations involved in the supply chain. Each supply chain partner should ensure that its respective service level agreements and supporting documents cover delivery and receiving responsibilities. The use of written agreements ensures clarity and transparency, while delineating the responsibilities of each organization in the supply chain.
4.1.5 Excursion handling
Short-term temperature excursions can occur during distribution, storage, and transportation (see Packaging and Storage Requirements 〈659〉). Each excursion should be documented and handled with a deviation or appropriate risk assessment. Product disposition should be established on the basis of an assessment of the excursion (i.e., the temperature to which the material or product was exposed, and for how long), the stability data obtained from traditional stability studies (under accelerated, intermediate, if appropriate, and long-term conditions and performed in accordance with ICH guidelines), and distribution stability studies (e.g., extremes of temperature, thermal cycling, and freeze–thaw studies, as appropriate). Combining stability data from long-term and accelerated studies with mean kinetic temperature (MKT), temperature-excursion, and thermal-cycling studies should provide the information necessary to evaluate the effects of excursions. Excursions out of temperature range defined by thermostability data or 〈659〉 should be addressed/corrected in order to prevent recurrence using a risk-based approach. Downstream handlers of finished drug products may rely on the manufacturer's product disposition instructions. See 〈659〉 for excursion allowances and MKT limits and Mean Kinetic Temperature in the Evaluation of Temperature Excursions During Storage and Transportation of Drug Products 〈1079.2〉 for MKT. MKT should be calculated for the period of time that a drug is in residence at a warehouse and/or in transit on a truck to avoid the problem of diluting the impact of excursions by calculating annual MKT values.1,2
4.2 Training
Training is a teaching-learning process used in the workplace to provide knowledge and develop skills and behaviors. The objective of training is to reduce the gap between the existing competencies and those required for performing the work. Training comprises knowledge of the topic to be taught and the types of training practices such as class-like, on-the-job training, web-based, blind sample analysis, mocks, and simulations. The type of training can influence the effectiveness of that training.
A written training procedure is necessary to establish who can be the trainer, how training needs are identified, and how training effectiveness will be evaluated. Selection of trainers must adhere to applicable laws as appropriate. Training needs could be identified and linked to job descriptions, complexity of duties, types of product handled, management reviews, and any kind of human resources program for competence development.
SOPs are the foundation of a quality system, and training must be provided for each SOP to the appropriate job roles responsible for executing the processes outlined in each SOP. Frequency of training and when training or retraining should occur should also be outlined in the overarching training SOP. Training records should be maintained either as hard copies or electronically.
The effectiveness of training should be considered and evaluated to determine its impact on task execution and quality. Evaluation of training effectiveness is not necessarily a separate document, but it may include a review of written or performance tests, observation, error rates, non-conformance or CAPAs, customer complaints, and internal and external audits. Any identified training effectiveness gaps should be corrected and evaluated. This may include retraining or evaluation and modification of SOPs, training materials, training method, or the instructor.
As a risk mitigation strategy, training should be performed before the SOP becomes effective. All employees whose actions have an impact on product quality and security should have initial and ongoing training based on an approved schedule. Basic training should be provided regarding risks and mitigation for storage and transportation of drug products so that employees will better understand how individual and collective actions impact product quality, and so they will develop awareness and a risk-based mindset.
4.3 Resources for Storage, Transportation, and Personnel
In the context of this chapter, resources are warehouses, vehicles, and organizational personnel. Facilities, equipment, and transportation vehicles are emphasized as systems that function to control environmental conditions in accordance with product specifications. Evaluation of facilities, equipment, packaging, and transportation systems should be part of the quality system of the organization. Sampling family or type of equipment/vehicles (e.g., shipping containers or trailers) should be part of the risk assessment, when evaluation of individual systems is not feasible. Additionally, trading partner facilities/vehicles can be evaluated by risk by family or type, which may include sample lane mapping to evaluate the exposure of product through the transportation system.
4.3.1 Storage
The facility should be designed to maintain the quality and integrity of the stored drug product. Buildings should be constructed in such a way that they are appropriate for the intended operations, taking into account:
- Security
- Product characteristics (e.g., narcotics, radiopharmaceuticals, re/explosion risk)
- Product status (e.g., approved, recalled, returned, rejected, quarantined, falsified)
- Product required storage temperature
- Ease of cleaning and maintenance
- Logical ow of personnel and materials
- Means of preventing mix-ups and cross-contamination
- Ergonomic measures
- Product demand in order to prevent capacity constraints
- Any local, national, or international requirements
- Necessary environmental controls
Product segregation and proper identification can reduce the risk of mixing up products with different statuses, such as quarantined (rejected, expired, recalled, returned, or falsified) and released/salable. Products with special handling authorization, such as narcotics, should be segregated and locked in a secure area per applicable regulations. Radiopharmaceuticals should be contained in dedicated, locked storage areas. Hazardous products should be managed per applicable regulations for each supply chain partner. Other products that require special storage conditions such as hazardous products (see Hazardous Drugs—Handling in Healthcare Settings 〈800〉) should also be segregated per applicable safety standards. Any system used to replace physical segregation should offer the same level of security and protection.
Buildings and facilities used for the warehousing, storage, and/or holding of drug products should be of adequate size for their intended use to prevent product overcrowding. The building and facility should be designed to control environmental conditions, where necessary, and should be made of materials that are readily or easily cleaned. Sanitation and pest control procedures should be written, indicating the frequency of cleaning and the materials and methods to be used. The pest control program should prevent contamination and ensure the safe use of pesticides. Records of all cleaning and pest control activities should be maintained.
Storage facilities themselves, unless thermostatically controlled, cannot be validated. However, they can be qualified via a mapping process, with appropriate attention to geography and seasons. The generator backup power supply should be qualified. Drug product storage areas are required to maintain the product temperature between the limits defined on the product label (see 〈659〉). Product storage areas/units should utilize recording systems to log and track temperatures. Alarm systems should be an integral part of the monitoring system for product temperatures. Although automated systems monitor units continuously, manual checks should be performed as appropriate to ensure functionality. When automated systems are not available, manual systems may be used. A risk-based approach should be applied when using a manual system.
Controlled access to warehouses and vehicles is a measure used to prevent unauthorized personnel from coming into contact with the product. Access control can be accomplished with automated systems or by procedure. Adequate precautions should be taken to prevent theft and diversion of products.
4.3.2 Transportation
All vehicles used in supply chain activities—such as trucks, vans, trains, airplanes, sea vessels, mail delivery vehicles, and emergency medical services vehicles—should be suitable for the intended purpose because they are providing in-transit storage and should prevent exposure to conditions that affect stability and package integrity. Thus, all of the precautions needed to maintain product quality, integrity, and security should be taken. Risk identification and mitigation strategies should be applied to determine whether the transportation method adequately protects the product from environmental exposures such as temperature and vibration without the need for additional packaging, or if additional packaging is necessary to mitigate the risk.
4.3.3 Personnel
Organizations should hire personnel and contractors who meet the requirements for handling drugs safely and securely per applicable laws and regulations. Job descriptions and individual proles should be reviewed to ensure that all experience and training requirements are met and maintained.
4.4 Qualification and Validation
Qualification and validation for storage and transportation of drugs focus on the assurance that the storage and transportation methods meet predetermined criteria and that processes and procedures produce the desired outcome. Calibration is necessary for instruments used in qualification studies. Organizations need to include appropriate qualification, validation, and calibration activities in their master plan and SOPs, as well as schedules and the use of approved protocols to conduct these activities and produce nal reports. The sections below provide brief explanations of calibration, qualification, and validation, focusing on storage and transportation. The scope for each stakeholder within the supply chain is described in Table 3.
4.4.1 Calibration of instrument or device
Calibration ensures that measurements such as temperature and humidity meet recognized standards. Calibration frequency may be determined by the device manufacturer, device workload, operational demands, and/or damage that require repairs. Instruments or devices used for qualification or monitoring of set criteria must be calibrated to recognized standards such as those from the National Institute of Standards and Technology (NIST) and those found in Monitoring Devices—Time, Temperature, and Humidity ▲〈1079.3〉▲ (CN 1-Dec-2023) .
Table 3. Calibration, Qualication, and Validation Activities of Organizations
Calibration, Qualification, and Validation Activities | Role of Organization within Supply Chain | |||||
Manufacturer | Wholesaler and Distributor | Pharmacy and Compounding Pharmacy | Hospital and Healthcare Provider | Broker | Logistics ServiceProvider (LSP) | |
Calibration | ||||||
Temperature recording devices used for temperature mapping or monitoring during storage and transportation | Yes | Yes | Yes | Yes | No | Yes |
| Qualification | ||||||
Calibration | ||||||
Temperature mapping of warehouse and equipment (freezers, refrigerators) | Yes | Yes | Yes | Yes | No | Yes |
Shipping packaging performance qualification | Yes | Yes | Yes | Yes | No | No |
Transportation vehicle performance qualification | Yes | Yes | Yes | Yes | Yes | Yes |
| Validation | ||||||
Software validation for systems that make quality decisions | Yes | Yes | Yes | LYes | Yes | Yes |
4.4.2 Qualification
For the purposes of this chapter, performance qualification is defined as all tests designed and executed to evaluate whether the storage rooms and areas, warehouse facilities, utilities, equipment, transport vehicles, and shipping containers are suitable for their intended purpose. Qualification studies should reflect actual load configurations and environmental conditions. Testing should be performed on both active and passive thermal packaging systems.
During the qualification process, temperature mapping is performed to provide information relative to temperature consistency throughout a product area. This information may confirm a uniform temperature over the mapped area or it may identify areas that need mitigation. Such mitigation may include the movement or installation of insulation, HVAC units, or fans, or the need to modify the relevant SOPs. The identification, documentation, and rationale for the mapping procedure are the foundation of temperature mapping of any temperature-controlled space (e.g., facility, vehicle, shipping containers, refrigerator, and freezer). Temperature variability in mapped locations and the level of thermal risk to the product should both be defined, unless another process to ensure environmental control has been put in place (see 〈1079.3〉 (CN 1-Dec-2023) ).
Temperature mapping in facilities and equipment: The following factors, which may contribute to temperature variability in a facility, should be considered during the process of temperature mapping for storage locations: 1) size of the space; 2) location of HVAC equipment, space heaters, and air conditioners; 3) sun-facing walls; 4) low ceilings or roofs; 5) geographic location of the area being mapped; 6) airflow inside the storage location; 7) temperature variability outside the storage location; 8) workflow variation and movement of equipment (weekday vs.
weekend); 9) loading or storage patterns of product; 10) equipment capabilities (e.g., defrost mode, cycle mode); and 11) SOPs. The duration of temperature recordings during the thermal mapping of a warehouse or cold room should capture workflow variation that may impact airflow and the resulting temperature fluctuation; for example, this process could last from 1 day to 1 week, depending on the workflow cycle.
Temperature mapping for shipping packaging and vehicles: Pharmaceutical manufacturers should consider primary, secondary, and tertiary packaging that best protects the drug product during storage and distribution. Shipping package performance testing should be documented as part of a QMS. Several standard test procedures are available to evaluate package performance for factors such as shock, vibration, pressure, compression, and other transit events. (See Additional Sources of Information for standards for test methods.)
Packaging at the tertiary level (e.g., outer, external, or shipping package) or thereafter for the distribution of the drug product should be selected and tested to ensure that product quality is maintained and to protect the contents from the rigors of distribution, including environmental or physical damage. Active, passive, or semi-active shippers and transport systems are typically subjected to operation performance qualification by the manufacturers or suppliers of such equipment.
Thermal packaging and vehicles used for transporting the drug product for pharmaceutical manufacturers, wholesalers, and pharmacies should be evaluated based on the labeled storage or transport conditions of the product as well as anticipated environmental conditions. Special consideration should be made for seasonal temperature differences, transportation between hemispheres, and the routes and modes of transport.
Identification of risks and mitigation strategies to protect the product should be based on documented studies of specific distribution environments, including domestic and international lanes (as appropriate), mode(s) of transport, duration, temperature, and other potential environmental exposures or sensitivities that may impact product quality. If risks to product integrity have been identified by using historical data, observation of current practices, or operational changes, mitigation strategies may be employed to address the identified risks. Strategies include temperature-controlled vehicles, active or passive thermal containers or packaging, or ambient conditions based upon identified risks (product storage labeling, time, temperature, and geography).
Temperature monitors/indicators may include calibrated monitoring or recording devices, real-time monitors such as GPS, and chemical indicators of temperature. Monitoring devices may include an alert mechanism if the preset ranges are breached (see ▲〈1079.3〉▲ (CN 1-Dec 2023) ).
Thermal packaging and vehicles may be qualified based on historical data that are relevant to the process. If qualified thermal packaging is used without a temperature verification method (monitors or indicators), a plan should be in place to schedule transport within qualifying times and to mitigate and respond to exceptions. However, it may be acceptable to use product stability data from manufacturers and supply chain risk assessment to justify shipping without either continuous monitoring or qualification of the shipping system.
Operational and performance shipping studies should be part of a formal qualification protocol that may use controlled environments or actual eld testing, depending on the projected transport channel. These studies should reflect actual load configuration conditions and expected environmental extremes. Testing should be performed on both active and passive thermal packaging systems.
When developing a thermal package qualification protocol, the following factors and actions should be considered:
- Transportation temperature profile for the shipping lane(s)
- Delivery cycle time, accounting for reasonable delays due to weather, traffic, or customs
- Testing beyond qualification time to obtain worst-case data for excursion dispositioning
- Seasonal changes in the environment
- Use of seasonal (warm/summer and cool/winter) configurations versus universal configuration
- Testing with actual payload, worst case configuration, or a representative sample
- Variable order sizes, making it difficult to select a representative sample; testing for the minimum and maximum payload possible in each package may be necessary
- Probe placement should be inside or directly attached to the product (or representative samples) or in the most vulnerable temperature locations within the package; scientific justification should be given for the differences between monitoring for qualification and operation
- Perform at least 3 replicate tests on a representative package containing a representative thermal payload per season; tests may be performed at the same time in an environmental chamber. If a qualified environmental chamber is used for the test, it is only necessary to perform one test on a representative package containing a representative thermal payload per season.(USP 1-Dec-2023) Allow the product payload as well as coolant to condition before beginning testing per protocol
- Utilize a recognized standard to conduct thermal qualification (see Additional Sources of Information) or develop a written rationale for a qualification
4.4.3 Validation of information systems
In the context of this chapter, before an information system that can impact the quality of the product in storage or in transportation is brought into use, it should be demonstrated through appropriate validation or verification studies that the system is capable of achieving the desired results accurately, consistently, and reproducibly. (See Pharmaceutical Inspection Convention in Additional Sources of Information). The following are considerations for using validation as a mitigation strategy. These considerations do not suggest replacing or enforcing any existing regulatory standards in place for various members of the supply chain [e.g., current good manufacturing practices (cGMP) for manufacturers, wholesale distributors, or pharmacies].
Prior to their use, information systems should be tested according to approved protocols. The extent of the study depends on the risk or impact the software can have on product or service quality. Validation or verification is not applied to systems that have no impact on quality. An inventory of information systems should be done periodically and include at least:
- Information system identification (name, version)
- System supplier
- Processes where system is used
- Process owner
- Risk assessment
- Status (validated, not validated, validation in progress, verified, not verified, verification in progress, not applicable)
A multidisciplinary team with representatives from information technology, quality, and operations should be responsible for protocols and report approvals. Responsibilities for the tests should be assigned in the protocols. Validation or verification tests should cover the Products following:
- Security (e.g., access levels, proles, responsibilities inclusion, exclusion, or changing proles)
- Data validity (e.g., challenge the software with entries above and below specification and with entry value errors)
- Documentation (e.g., system design in accordance with user requirements and other documents)
- Functionality (e.g., calculations, operations) [Note— Most of the functionality tests for embedded software are covered during equipment qualification (installation, operation, and performance qualifications
- Data integrity (e.g., changes, traceability, backup, recovery, and protection)
After validation or verification studies, any modification to the system should be done according to change control procedures, and records of these changes should be kept.
5 GLOSSARY
Calibration: A process that typically focuses on instruments or devices to provide assurance that they produce results within specified limits. Organizations need to include the appropriate qualification, validation, and calibration activities in their SOPs and master schedules and should follow protocols to conduct these activities and nal reports.
Qualication: Qualication is the assurance that systems or equipment meet predetermined acceptance criteria. This process typically focuses on equipment and utilities such as refrigerators and HVAC systems, as well as packaging. There are several different types of qualification, and an organization should determine which to use and when. Some of these include design qualication, installation qualication, operational qualication, and performance qualication.
Service level agreement (SLA): An SLA or contract is a negotiated agreement between the customer and service provider that defines the common understanding about materials or service quality specifications, responsibilities, guarantees, and communication mechanisms. It can either be a legally binding document or an information agreement. The SLA may also specify the target and minimum-level performance, operation, or other service attributes.
Temperature excursion: An event in which a pharmaceutical product is exposed to temperatures outside of the range(s) prescribed for storage and/or transport. Temperature ranges for storage and transport may be the same or different; they are determined by the product manufacturer, based on stability data.
Validation: Validation typically focuses on processes and procedures, to provide assurance that the processes or equipment produce the desired outcome.
6 ADDITIONAL SOURCES OF INFORMATION
American Society for Testing and Materials International. ASTM D3103-14. Standard Test Method for Thermal Insulation Performance of Distribution Packages.
American Society for Testing and Materials International. ASTM D4169-16. Standard Practice for Performance Testing of Shipping Containers and Systems.
American Society for Testing and Materials International. ASTM D4332-14. Standard Practice for Conditioning Containers, Packages, or Packaging Components for Testing.
Health Canada. National Guidelines for Vaccine Storage and Transportation. In: Canada Communicable Disease Report. June 15, 1995; Vol 21- 11:93–97.
European Commission. Guidelines of 5 November 2013 on Good Distribution Practice of medicinal products for human use (2013/C 343/01).
International Council for Harmonisation. Quality Risk Management (ICH Q9).
International Council for Harmonisation. Pharmaceutical Quality System (ICH Q10).
International Council for Harmonisation. Good manufacturing practice guide for active pharmaceutical ingredients (ICH Q7), Chapter 10— Storage and Distribution, p 22; November 10, 2000.
International Organization for Standardization. Quality management systems—Fundamentals and vocabulary (ISO 9000:2015). International Organization for Standardization. Risk management (ISO 31000).
International Organization for Standardization. Quality management systems—Requirements (ISO 9001:2015).
International Safe Transit Association. Testing Standard for Thermal Transport Packaging Used in Parcel Delivery System Shipment (ISTA STD-7E).
Parenteral Drug Association. Guidance for Temperature-Controlled Medicinal Products: Maintaining the Quality of Temperature-Sensitive Medicinal Products through the Transportation Environment (PDA Technical Report 39); 2007.
Parenteral Drug Association. Last Mile: Guidance for Good Distribution Practices for Pharmaceutical Products to the End User (PDA Technical Report 46); 2009.
Parenteral Drug Association. Active Temperature-Controlled Systems: Qualification Guidance (PDA Technical Report 64); 2013.
Parenteral Drug Association. Passive Thermal Protection Systems for Global Distribution: Qualification and Operational Guidance (PDA Technical Report 72); 2015.
Pharmaceutical Inspection Convention. PIC/S Guide to Good Distribution Practice (GDP) for Medicinal Products (PE- 011-1); June 1, 2014. International Air Transport Association. IATA Temperature Control Regulations, Chapter 17—Air Transport Logistics for Time and Temperature Sensitive Healthcare Products; July 1, 2009.
World Health Organization. Model Guidance for the storage and transport of time- and temperature-sensitive pharmaceutical products (WHO Technical Report Series, No. 961, Annex 9); 2011.
World Health Organization. Supplement 7—Qualication of temperature-controlled storage areas (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. Supplement 8—Temperature mapping of storage areas (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. Supplement 11—Qualication of refrigerated road vehicles (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. Supplement 12—Temperature-controlled transport operations by road and by air (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. Supplement 13—Qualication of shipping containers (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. Supplement 14—Transport route profiling calculation (Technical supplement to WHO Technical Report Series, No. 961, 2011); 2015.
World Health Organization. WHO good distribution practices for pharmaceutical products (WHO Technical Report Series, No. 957, Annex 5); 2010.
1 Seevers RH, Hofer J, Harber P, Ulrich DA, Bishara R. The use of mean kinetic temperature (MKT) in the handling, storage and distribution of temperature sensitive pharmaceuticals. Pharmaceutical Outsourcing. May/June 2009;12–17.
2 Anderson C, Seevers R, Hunt D. The use of mean kinetic temperature to aid evaluation of temperature excursions: proper and improper application. Pharm Forum. 2018;44(4).
