Potency Assays to Evaluate Coagulation Factor VIII and Factor IX
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION AND SCOPE
Blood coagulation is one of the steps of hemostasis necessary to stop bleeding. The coagulation cascade proceeds through the activation of several coagulation factors in the blood which include factor VIII (FVIII) and factor IX (FIX). There are two predominant methodologies used to assess the potency of FVIII and FIX concentrates intended for the treatment of hemophilia A and B: the one-stage (OS) clotting method and the chromogenic substrate (CS) method. Both methodologies require the use of a reference or potency standard to enable the calculation of relative potency. Guidance and recommendations on the selection of which method and reference standard to use depend on several factors, including local laboratory practices and regulatory considerations.
Despite concerted global efforts toward standardization, considerable variability and discrepancies in assay performance and potency values are observed for both OS clotting and CS methods. Sources contributing to variabilities and discrepancies in assay performance and potency values include, but are not limited to, diversity in critical reagent composition, procedural and instrumentation differences, selection of reference standard, sample preparation, and data analysis. Assay discrepancies have been observed where statistically valid potency estimates differ not only by different methods but also within the method using different reagents when a test sample is assayed against the same reference standard. This issue is prevalent for recombinant and novel recombinant forms of FVIII and FIX when assayed against the current WHO International, pharmacopeial, or FDA/CBER standards. The potency labeling method for these products should be carefully considered and agreed with competent regulatory authorities. Similar considerations should also be applied when monitoring FVIII or FIX activity after treatment with a FVIII or FIX gene therapy product.
Given the number of factors that can impact method performance, obtaining precise and accurate potency values for FVIII and FIX concentrates is dependent on understanding the specific relationship in method behavior between the samples of interest and the selected reference standard.
2 METHOD TYPES
The OS method and CS method are the two method types used to determine the potency of FVIII and FIX concentrates. During verification and validation of the methods when the test sample is assayed against the primary International Standard (IS) or secondary pharmacopeial standard, pre-dilution of standard and test preparations in the appropriate factor deficient plasma should be carried out to minimize matrix effects. For FVIII methods, the content of von Willebrand Factor (vWF) in the FVIII deficient plasma should be within the normal range. Pre dilution in buffer containing 1% of high purity protease free bovine or human albumin is acceptable once the method has been validated locally to give similar results to pre-dilution in deficient plasma.
2.1 One-Stage Clotting Method
The OS method is based on the activated partial thromboplastin time (APTT or aPTT) clotting method. The OS method measures the ability of a sample containing the coagulation factor of interest to shorten the clotting time of the appropriate factor deficient plasma. The method relies on the sequential and linked macromolecule interactions that are part of the intrinsic and common pathways of coagulation. The method requires factor deficient plasma, a source of phospholipids containing an added contact activator, the sample containing the clotting factor to be measured by the method, and calcium chloride. The assay response is the formation of a plasma gel-like clot which can be detected by mechanical or photo-optical means. The time required for clot formation, the clotting time, is considered directly dependent on the clotting factor activity present in the sample being tested. The reaction has a limited linear range and data transformation is typically required to enable calculation of the potency as compared to a reference standard of known potency.
There are general considerations that are required to facilitate good method performance. The selection of appropriate reagents is critical to method performance; however, given the use of a reference standard, it is especially important to consider the source, composition, and method behavior of the reference standard in relation to the sample being tested.
There are several best practices that should be taken into consideration when performing an OS method. Only citrated anticoagulated factor deficient plasma should be used: the level of citrate should be in accordance with local and national best practices validated to be appropriate for use in clotting methods. The citrated deficient plasma being tested should only be deficient in the coagulation factor being tested and contain all other clotting factors and components of normal plasma (i.e., no other clotting factors, cofactors, or essential macromolecules are absent or compromised). It is especially important to verify FVIII immunodepleted plasma contains vWF. The contact activator and the Phospholipid reagent, contained in the APTT reagent, may be obtained from different sources (see Table 1). FVIII and FIX contain a phospholipid-binding domain and thus the interaction with the specific phospholipids present in the APTT reagent has the potential to greatly impact method performance. The choice of phospholipid reagent must be evaluated carefully as differential reagent interactions of the sample and reference standard can result in non-parallel behavior between the dose-response curves and lead to variable relative potency results. When choosing an APTT reagent, the overall method performance with respect to robustness and precision must be assessed, including evaluation of different lots of reagents.
Table 1. Common OS Method Reagents
Factor Deficient Plasma | APTT Reagent | Buffer | Trigger | |
Phospholipid | Activator | |||
Congenital Immunodepleted | Mammalian tissue derived Synthetic Plant-based | Silica Kaolin Ellagic acid | Albumin containing | Calcium chloride |
Plasma-derived FIX products may contain heparin, an additive that lowers thrombogenicity risk. As Heparin can interfere with an accurate determination of the FIX potency, the amount of heparin must be determined and neutralized before potency measurement. This can be done by adding Protamine sulfate or hexadimethrine bromide (e.g., polybrene) to the sample [10 µg of protamine sulfate and 100 µg of hexadimethrine bromide neutralize 1 International Unit (IU) of heparin].
2.2 Chromogenic Substrate Method
The CS method is also referred to as an amidolytic method. The CS method is performed using reagents that represent only a portion of the clotting cascade and therefore may be performed without the use of plasma, although factor deficient plasma may be used as a sample diluent. The CS method is considered a biochemically defined method which means the composition of the reagents are better understood and less complex than those used in the OS method. There are specific CS methods for measuring FVIII and FIX that differ in reagent composition; however, the basic operating principles are the same for both. The CS method is performed in two sequential steps; the first step enables the generation of activated factor X (FXa) and in the second step the amount of FXa formed is determined using an FXa chromogenic substrate. Under appropriate method conditions, the amount of FXa formed is directly dependent on the FVIII or FIX activity present in the sample and is quantified using a reference standard.
CS methods are typically performed using commercially available kits that can be adapted to different assay platforms (e.g., automated analyzers, microtiter plates, or manual test tubes). In-house or local methods are possible if careful attention to reagent composition and method conditions are taken into consideration.
The components of the CS method reagents should be sufficiently purified to not contain interfering substances. The purified coagulation factor proteins used in CS methods are usually human or bovine derived. Alternative sources for purified coagulation factor proteins are possible but should be demonstrated to not impact method performance. Activators that are required for CS methods, typically thrombin for the FVIII CS method and human factor XIa (FXIa) for the FIX CS method, should be of appropriate quality and quantity to facilitate efficient activation of FVIII or FIX. The source of the activator, i.e., human or bovine, may impact the kinetics of activation and should be evaluated when selecting an activator. A phospholipid reagent from natural or synthetic sources is also required and should be of suitable composition to enable robust generation of FXa. The method conditions selected should enable an adequate amount of FXa to be generated so that detection is sensitive, robust, and most importantly, consistent between sample and standard to facilitate consistent method performance. The amount of FXa generated should be less than the maximal amount possible (using 50% of maximal generation as a target). The chromogenic substrate used should be highly selective for FXa and contain an inhibitor to prevent further FXa generation (e.g., chelating reagent such as EDTA) and preferably also a thrombin inhibitor to eliminate substrate hydrolysis by thrombin. The amount of chromogenic substrate cleaved by FXa should be directly dependent on the FVIII or FIX activity present in the sample and be amenable to spectrophotometric quantification.
Further dilution of samples to reach suitable activities for the method are made with method buffer containing 1% albumin. As for the OS method, heparin, if present in the sample, needs to be determined and neutralized before assay.
2.3 Points to Consider in Method Performance
For both OS and CS method types, it is crucial to have strict control of method performance variables and to handle samples and reference standards in an equivalent manner. Regarding FVIII, the time frame from reconstitution/thawing to start of an assay should be limited due to the inherent instability of FVIII although this instability may partially be alleviated using various constituents in product formulation.
The potency determination for plasma-derived FVIII and FIX products are not usually problematic when potency is assigned using plasma derived reference FVIII/FIX standards. However, with the advent of recombinant and extended half-lives (EHL) FVIII and FIX products, assay discrepancies (e.g., over-estimation or under-estimation of potency) are common between OS and CS methods and within OS methods when using different APTT reagents or variations in APTT reagent lots. Discrepancies with CS methods are less common, but have been observed, mainly due to deviation from the bioassay ”like-vs-like” principle, a foundation in potency assessment of biologics.
Any change to a new lot of APTT reagent or FVIII/FIX deficient plasma in an OS method should always be accompanied by careful evaluation against qualified reagent lots using a bridging approach before replacement. Regarding FVIII deficient plasma, the vWF content should be within the normal range.
Potency determinations based on the CS method are usually performed with commercially available kits using the protocol described on the product package insert. It must be verified that the kit diluent buffer working solution contains 1% albumin to avoid compromising the precision of the CS method. The stability of new reagents lots when stored reconstituted or used on an automated instrument should be verified to demonstrate they are suitable throughout the timeframe of the method run.
When switching kit lots, potential impact to method performance should be evaluated using a system suitability sample or control sample or other established method performance indicators. To overcome these difficulties, it is highly recommended to restore the "like-vs-like" principle in routine sample testing of recombinant and EHL products by establishing product-specic potency standards.
2.4 Points to Consider with Statistical Analysis of Assay Results
Activity assays for coagulation factors are relative potency biological assays (bioassays) that require a reference standard with known activity for value assignment. Therefore, it is assumed that the test sample and reference standard contain the same active principle, have the same mechanism of action, and behave the same at similar activities. To ascertain the validity of this assumption, the dose-response relationship of the test sample and the reference material should be assessed using an appropriate statistical model. Non-similar dose response relationships for the test sample and the reference standard suggest the presence of interfering substances or other process related components in the test sample and that a different assay design or protocol may need to be considered to ensure validity of comparison.
Dose-response relationships of coagulation factors and inhibitors can usually be described using parallel line or slope ratio statistical models. The design of the method needs to consider criteria required by these statistical models: 1) Independence of doses, which requires randomization of assay design, with true replicates of doses; 2) Normality of responses, the responses at each dose should be normally distributed; and 3) Constant variance, the variability of responses should be similar across the dilution range (i.e., the variability of responses for high dilutions should be similar to those for low dilutions). These three conditions should be assessed during method development and validation stages to select the most appropriate model for routine analysis. For each individual assay, a significant regression (i.e., slope of dose response curve) and linearity (for parallel line model: untransformed or transformed response against logarithm of dose; for slope-ratio model: untransformed or transformed response against dose) should be confirmed. Additionally, for the parallel line model, the dose response of the test sample should be parallel to the dose response of the reference standard and, for the slope-ratio model, the dose response of the test sample should intercept the y-axis (i.e., zero dose) at the same point as the dose response of the reference standard. To fulfill these requirements of the parallel line and slope ratio models, at least three, and preferably four adjacent concentrations for each test sample and reference standard should be tested, with replicate responses at each dilution.
A parallel line model usually gives the best t for results from OS clotting methods and a slope ratio model gives the best t for data from CS. Some method results may t both models, but the laboratory should determine which provides better agreement with the model assumptions described above by performing multiple assays during the development and/or validation of their own method procedures. Once the best model has been determined for their data, the laboratory should continue to use that model for all routine analysis. Details on development, design, analysis, and validation of bioassays are available in Design and Development of Biological Assays 〈1032〉, Biological Assay Validation 〈1033〉, and Analysis of Biological Assays 〈1034〉. European Pharmacopoeia Chapter 5.3 (1) also provides helpful information on analysis of bioassays.
Validated software packages for analysis are commercially available and may be preferable to perform manual calculation of the test results. To determine the assay validity or sample suitability, the analysis of variance (ANOVA) can be used to assess the statistical similarity between the standard and test sample (or assay control) slopes in the parallel line model or the Y-intercept in the slope ratio model. However, the ANOVA approach may "punish" the assays with high precision and result in a high assay invalid rate due to the high closeness of the sample replicate responses. An equivalence test (also called two one-sided t-test), with allowable difference established using historical data from assays of the same sample (or assay control) or relevant locally defined assay validity or sample suitability criteria, may be applied to minimize the number of invalid assays.
Multiple independent assays should be carried out for each lot or batch of test sample to obtain a reportable estimate together with condence limits that refiect the uncertainty of measurement. Methods for the combination of potencies from multiple assays are described in Design and Analysis of Biological Assays 〈111〉 which should be followed to obtain a nal potency estimate.
2.5 Points to Consider with Method Validation
Coagulation factor methods are typically based on an enzymatic reaction and categorized as bioassays. General chapter 〈1033〉 provides guidance regarding the validation of FVIII and FIX potency assays. In addition, the FDA guidance Analytical Procedures and Methods Validation for Drugs and Biologics (2) and ICH Q2(R1) Validation of Analytical Procedures: Text and Methodology (3) should be followed.
USP chapter 〈1033〉 provides validation goals pertaining to relative potency bioassays. For the coagulation assays where an IS is established, the potency results must be reported in IU assigned against the IS, rather than relative potency in percentage. A validation protocol should be prepared to instruct the validation study for the relevant validation characteristics with defined acceptance criteria; the validation protocol should be executed by trained analysts using qualified instruments. If automated instruments are used for the method, the automated system and its operational script/software should be qualified before the method validation to ensure data integrity.
Due to the inherent method variability, the best practice is to perform replicate testing when the method is used for routine testing with the average of the observed results presented as the reportable result. In this case, the reportable result should be used to assess the method performance during the validation.
FVIII and FIX products in aqueous solution may be unstable at ambient or refrigerated temperature while being kept in an automated test instrument. Sample stability should be investigated in detail during the validation.
If the potency method is used for in-process testing to report the process yield, the relevant in-process samples should be validated as appropriate in addition to drug substance (DS) or drug product (DP) samples for product release.
2.6 Points to Consider with the Analysis of New Generation Products
Potency determination of plasma-derived FVIII and FIX products is usually straightforward and with few issues. Plasma-derived FVIII/FIX reference standards are used and the "like-vs-like" principle is properly fulfilled, generally resulting in accurate potency assignments. This is observed with the noteworthy agreement between OS and CS methods during the calibrations of the WHO 8th IS FVIII Concentrate and the WHO 5th IS FIX Concentrate.
However, with the advent of FVIII and FIX products with extended half-lives (EHL-rFVIII/rFIX), assay discrepancy is common between OS and CS methods and also between OS methods using different APTT reagents. Large assay discrepancy has also been shown for FIX carrying the gain-of-function mutation R338L either present in family members or expressed via gene therapy. Similarly, OS and CS assay discrepancy have also been shown for recombinant B-domain deleted (BDD)-FVIII and BDD-FVIII expressed via gene therapy.
The frequent occurrence of assay discrepancy on potency assignment of new generations of FVIII/FIX products and gene therapy expressed FVIII/FIX brings much uncertainty in results from coagulation laboratories and this situation may worsen over time with increasing use of new products.
Before a new product is launched, a stringent evaluation of various OS and CS methods is recommended and supported by guidelines developed by regulatory authorities for manufacturers.
Either the CS or OS method can be used to evaluate the potency of FVIII and FIX concentrates. Ideally, the assays provide similar results and can be used interchangeably when a common non-product potency standard is used (i.e., WHO IS). However, discrepancies between OS and CS methods have been observed and documented across a number of FVIII and FIX materials. Comprehensive detailed technical investigations of all potential discrepancy scenarios are beyond the scope of this chapter, nevertheless, an investigational guide is provided below. The goal is to highlight some of the options available to users for investigating the sources of the discrepancy and aid in identifying potential solutions to enable aligning the results and performance of the OS and CS methods for a specific situation/product. This potential discrepant behavior should be taken into consideration when deciding on an appropriate method to use for product labeling and monitoring.
It is therefore important that manufacturers of new generation products perform a careful evaluation of OS and CS methods before selection of a method for potency assessment of the product. If assay discrepancy is obtained it is proposed to perform a comparison between a CS method and a modified OS method in which purified phospholipids with no contact activator are used in combination with human FXIa incorporated with the calcium chloride solution. With this method, denoted FXIa-OS method, the results are not skewed by discrepant contact activator interactions of product and a plasma-derived reference standard. It is suitable to use nal assay conditions of 40–100 μM phospholipids and 1–2 IU/mL human FXIa activity. The higher phospholipid concentration should preferably be used if the content of Phosphatidylserine in the phospholipid emulsion is below 15 mol%.
The evaluation of the OS and CS methods for new generation FVIII and FIX products may be performed in the following manner (see Figure 1).
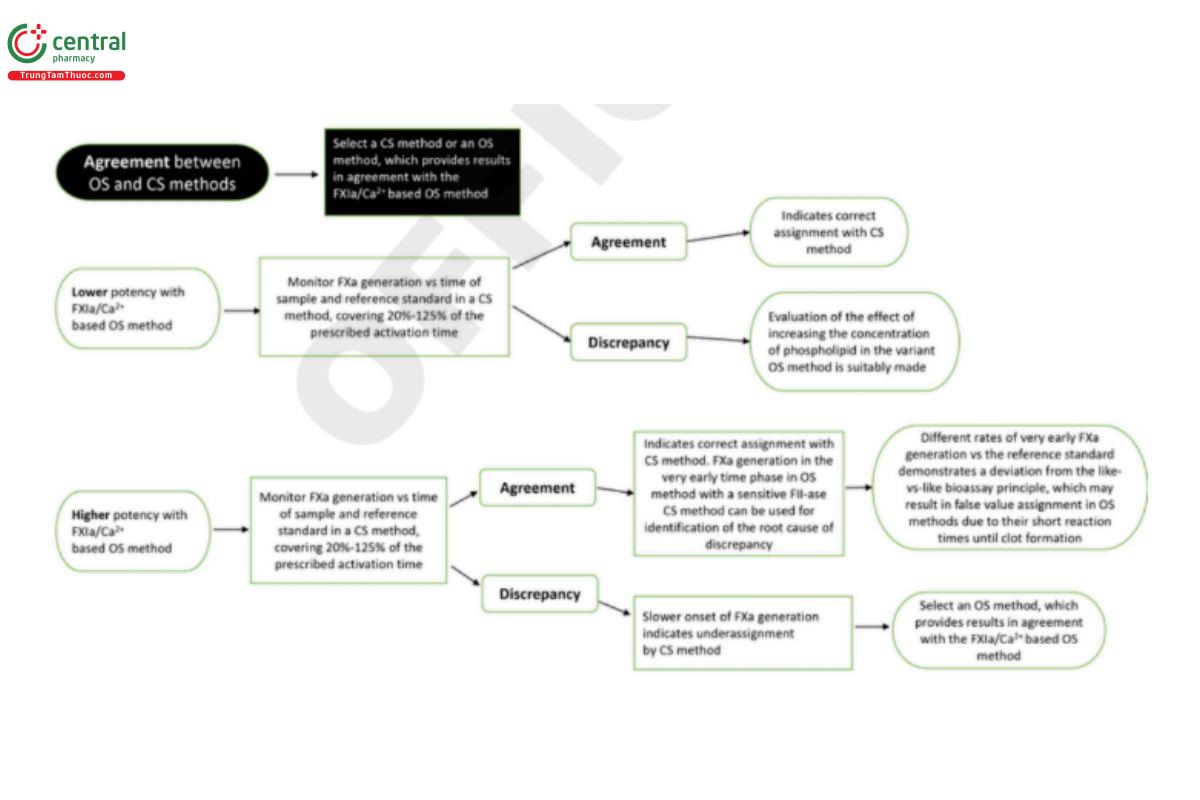
The comparability assessment of the OS and CS methods for new generation FVIII and FIX products has several points to consider. For this assessment, perform an initial product potency assignment with a CS method and the variant OS method. If the potency assignments with these methods agree, there is strong indication that both methods provide accurate results with the following caveats.
- If both the CS and the FXIa-OS method give similar results, a CS method or the FXIa-OS method may be selected for potency assignment. If an OS method is preferred, it should be selected after stringent evaluation to demonstrate similar results as the FXIa OS method.
- If there is a discrepancy in potency results between the FXIa-OS method and a CS method, this is likely related to an inherent property of the product. Suitably, FXa generation for the new FVIII/FIX product and the reference standard is then compared in a CS method, covering 20%–125% of the prescribed activation time. If similar FXa generation curves are obtained, it indicates that the CS method provides correct potency assignment.
- If the FXa generation for the product sample has a slower onset in the CS method than the reference standard, it indicates that the CS method may result in falsely low potency assignment of the product. This might be caused by a combination of low substrate afinity and high sample dilutions in CS methods.
- If the FXa generation curves are similar within the 20%–125% range of the prescribed activation time, a suitable next step is to determine FXa generation in the very early time phase of the FXIa-OS method and/or in a CS method where prothrombin and FV replaces thrombin for FVIII activation. Frequent subsampling is therewith made into a quenching medium for subsequent determination of generated FXa in which a chromogenic prothrombinase system is utilized to obtain a sufficiently high FXa sensitivity. Final assay conditions are suitably 30–50 nM human prothrombin, 10–15 μM phospholipids, and 1–2 nM FV. The FXa calibrator range is suitably 0–5 pM.
- If a different rate of very early FXa generation is noticed for the product sample compared to the reference standard, observed as faster or delayed onset of FXa generation, it illustrates a deviation from the "like-vs-like" bioassay principle. It may then result in false potency assignment in OS methods due to their short reaction times until clot formation.
This evaluation may be used to obtain an in-depth understanding of the performance of a product in different methods and, in cases of assay discrepancy, specific approaches should allow identification of the root cause(s) of assay discrepancy. The compiled data and overall outcome from such investigations should then allow identification and selection of the most accurate method for product potency assignment. In this way any new product will be launched only after extensive evaluation and will benefit regulatory bodies and clinical coagulation laboratories.
3 DEVELOPMENT AND QUALIFICATION OF IN-HOUSE REFERENCE MATERIALS
Upon implementation of a new FVIII or FIX OS method or CS method for determination of potency it is recommended to establish product specific reference materials. A primary in-house reference material (PRM) and a secondary in-house reference material (SRM) are prepared as part of a two-tier reference material program that is a best practice in the industry. In the case where a large quantity of SRM can be obtained with sufficient stability, the SRM can be directly qualified against the IS, skipping the PRM. In-house reference materials should be formulated for optimal stability and practical use. It may be advantageous to prepare the PRM in a lyophilized form in high concentration to enhance long-term stability and the SRM, the working reference material for routine use, in liquid form with a lower concentration that would reduce the intra-assay variability. The nal choice of formulation may be different to that used for DS and DP and should consider the stability of the reference materials, the effect of stabilizing excipients on the behavior of the PRM and SRM in the methods relative to the IS, DS, and DP.
The in-house reference materials should be traceable to the related WHO IS. When a WHO IS is replaced, studies should be conducted to confirm the relationship between the new IS and the in-house reference materials. If different potency values are observed with the PRM, SRM, DS, and DP when a new IS is implemented this should be investigated. Studies should be conducted to confirm the traceability of the potency when there is a replacement IS and when new in-house reference materials are established. Likewise, the method and reagents used for the calibration need to be carefully selected and defined. As the method reagents can have significant impact on the labeled potency value when measuring modified EHL FVIII/FIX against the IS, consistent method performance must be routinely demonstrated by use of an appropriate system suitability assay control. Trending and tracking control data should be used to ensure reagent lot changes or reagent composition changes have little or no impact on the established control sample potency value. The use of a control sample can be used to assess potency drift over time and demonstrate continued alignment with the initial established product potency (reference) standard.
The in-house reference material development is closely linked to the chosen method, the production processes, and the product. It is essential that the relationship in method behavior between samples and the reference materials is well examined and understood. The samples and reference materials must be analyzed under the same conditions (e.g., identical incubation time, dilution buffer, reagents, and procedure).
The in-house reference materials may be derived from a DP or DS batch and may have added stabilizing excipients. Choice of which batch to use should be based on the release testing results to ensure that the batch is representative of the marketed product. The volume of the batch should be enough to produce reference material in filled containers to support the estimated use for several years to minimize product potency shifts or maintain potency assignment consistency over the product lifecycle. The target batch may be stored at a lower temperature to improve the stability. The target content of each vial of reference material should be for single use to avoid freeze-thaw degradation and re-sampling errors. The target potency of the reference material as well as any possible matrix effects must be evaluated to ensure the reference material composition does not interfere with the chosen method. The suitability of the candidate reference material should be verified by statistical evaluation based on its use in the potency analysis of multiple batches of DS and DP and comparison of estimated potencies relative to the IS.
4 REFERENCES
1. European Pharmacopoeia. 5.3. Statistical Analysis of Results of Biological Assays and Tests. 10.8.
2. US Food and Drug Administration. Guidance for industry. Analytical Procedures and Methods Validation for Drugs and Biologics. 2015. www.fda.gov/les/drugs/published/Analytical-Procedures-and-Methods-Validation-for-Drugs-and-Biologics.pdf.
3. International Council for Harmonisation (ICH). ICH Q2(R1), Validation of Analytical Procedures: Text and Methodology. 2005. https://database.ich.org/sites/default/les/Q2%28R1%29%20Guideline.pdf. (USP 1-Aug-2024)
