POSITRON EMISSION TOMOGRAPHY DRUGS-INFORMATION
If you find any inaccurate information, please let us know by providing your feedback here

Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
Positron emission tomography (PET) drugs contain radionuclides that undergo nuclear transformation, or radioactive decay, predominantly by the emission of a positron. Positrons undergo annihilation upon interaction with electrons to produce two photons that are emitted in nearly opposite directions to each other. Each photon possesses an energy of 511 keV, which lies in the gamma ray portion of the electromagnetic spectrum. These radionuclides are used in a wide range of PET imaging studies, including research, investigational, and clinical applications.
The radionuclides used in PET imaging studies typically possess short physical half-lives (denoted as T₁⁄₂). Some common examples of PET radionuclides and their associated half-lives are included in Table 1. Note that Table 1 includes radionuclides currently in predominant use and is not intended to illustrate all positron-emitting radionuclides used in PET.
Table 1
| PET Radionuclide | Half-life, T₁⁄₂ |
| Fluorine-18 | 109.8 min |
| Carbon-11 | 20.4 min |
| Nitrogen-13 | 10.0 min |
| Oxygen-15 | 2.0 min |
| Copper-64 | 12.7 h |
| Gallium-68 | 68 min |
| Rubidium-82 | 75 s |
Because certain of these radionuclides such as Carbon-11 and Nitrogen-13 when found in biological systems have the same chemical and physical properties as their stable counterparts found in biological systems, PET offers a unique platform for in vivo imaging studies of complex biochemical pathways. As a result, PET radionuclides have found widespread use in cardiology, oncology, and neurology applications. PET drugs have also attracted interest as potential tools to accelerate and reduce the cost of therapeutic drug discovery efforts.
Most PET radionuclides are produced at the point of use by a particle accelerator (e.g., a cyclotron) or a radionuclide generator. A cyclotron accelerates charged particles such as protons or deuterons to velocities sufficient to induce a nuclear transformation of the target nucleus into a different element. High-energy particles and/or radiation are emitted from the target nucleus during the transformation process. An example of a transformation is the bombardment of stable Oxygen-18 nuclei with accelerated protons to produce Fluorine-18 nuclei along with the concomitant emission of a neutron. This process may be summarized according to the following shorthand notation:
18O(p,n)18F
The chemical form and physical state of the bombarded nuclei vary, depending on the target and the subsequent usage of the PET radionuclide. This allows the production of PET radionuclides in gaseous, solid, and solution phases.
Radionuclide generators offer an alternative method for point-of-use access to some PET radionuclides. A radionuclide generator contains an immobilized parent radionuclide that undergoes radioactive decay to a daughter radionuclide, which may be eluted from the generator. An example of a PET radionuclide generator is the Germanium-68/Gallium-68 generator. The parent Germanium-68 (T₁⁄₂ = 271 days) is adsorbed onto the column packing material. The Germanium-68 undergoes radioactive decay by electron capture into Gallium-68, which is not adsorbed onto the column packing material and may be eluted from the generator.
Regardless of the production method, other radionuclides may be present in addition to the desired PET radionuclide. In the case of cyclotron-produced PET radionuclides, other radionuclides may result from the bombardment of the target holder and/or other nuclei present in the target material. For generator-produced PET radionuclides, break-through of the parent radionuclide may occur. Safeguards are readily available to avoid unnecessary contamination with radionuclidic impurities of the final PET drug.
Once the PET radionuclide has been produced, it should be processed into a suitable form of a PET drug. The simplest form of processing involves the purification and/or formulation of the PET radionuclide. An example is the purification of the [18F]fluoride ion. After production of the [18F]fluoride ion by cyclotron bombardment, ion exchange procedures may be used to remove impurities and yield a formulation suitable for PET imaging studies. Another example is the purification of gaseous [15O]oxygen through various solid supports before administration of the finished PET drug by inhalation.
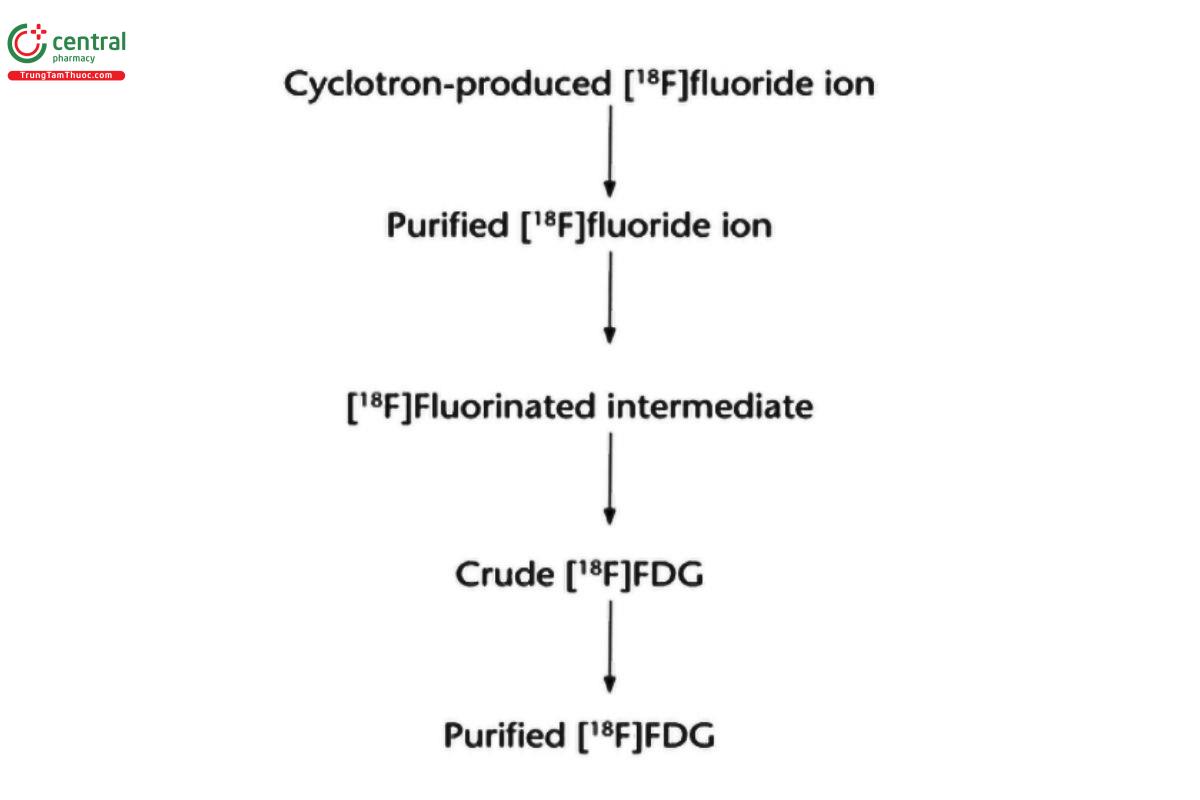
Simple forms of processing are rare, because PET drug products are typically synthesized from the appropriate radionuclide through multiple synthetic processing steps. The exact process depends on the desired PET drug. The synthesis of 2-[18F]fluoro-2-deoxyglucose ([18F]FDG) provides an illustration. The first step in this synthesis is the preparation of the anhydrous [18F]fluoride ion from cyclotron-produced Fluorine-18. The [18F]fluoride ion is activated with a phase transfer catalyst such as tetraethylammonium or [2.2.2]-cryptand to enhance its reactivity toward nucleophilic substitution. The resulting complex reacts with mannose triflate to yield a fluorinated 18F intermediate, which is deprotected and purified to yield [18F]FDG. This process is summarized in the following set of equations:
In addition to the chemical processing and/or purification steps used to generate the desired radiochemical of interest, other steps are typically required to formulate the PET drug product. For injectable PET drug products, formulation steps may include dilution, addition of a stabilizer, pH adjustment, and other steps. The final formulation of the PET drug product should take place in a manner that minimizes the presence of bacterial endotoxins and bioburden. Finally, the product should be sterilized (e.g., by passage through a membrane sterilizing filter) to provide a solution suitable for intravenous administration. In the above example, [18F]FDG becomes the drug product Fludeoxyglucose F 18 Injection.
Before use, the finished PET drug product should be tested to ensure that the product meets suitable standards of identity, strength, quality, and purity. Because of the short half-life of PET radionuclides, testing should be completed in a timely fashion; however, it is not possible to complete certain quality control (QC) tests within a suitable timeframe. In some cases, it may be necessary to adopt a “sub-batch” approach for QC testing, wherein a sub-batch of the PET drug product is prepared solely for purposes of QC testing. Examples of PET drug products with such short half-lives include [15O]water and [13N]ammonia.
Ultimately, the short half-lives of PET radionuclides create unique constraints for the production and testing of PET drug products and define how these products are used in research, investigational, and clinical settings. PET drugs are a unique class of products defined by the following characteristics:
The mass of the radioactive ingredient in a PET drug product usually ranges from nanogram to microgram quantities. This affects pharmacological and toxicological considerations for PET drugs, usually by creating large safety margins due to low mass of the active ingredient in the administered dose.
An entire batch of a PET drug product may be contained in a single vial. Samples withdrawn for QC testing are representative of the entire batch. PET drug products produced in this fashion undergo 100% QC testing.
PET drug products are produced and handled in environments with overlapping areas of regulatory authority. For example, a site may be subject to requirements of any combination of the following authorities: Nuclear Regulatory Commission or Agreement State Agency, Food and Drug Administration, Environmental Protection Agency, occupational safety entities, etc. These agencies may have different and conflicting requirements of the manufacturing facility, some through regulation, some through guidelines published by the authorities, and some through license commitments made by the facility.
PET drug products are generally produced at or near their point of use in small-scale facilities with limited personnel and resources. This requires:
- Allowance for multiple operations in one area with adequate controls
- Allowance for making and testing multiple PET drug products using shared equipment with appropriate cleaning between batches
- Appropriate requirements for aseptic operations
- Appropriate requirements for system suitability and other day-of-use activities
- Appropriate QC requirements for components, materials, and supplies
- Self-verification of significant steps in radionuclide production, PET drug production, compounding, and testing
- Single-person oversight of production or compounding, testing, review of batch records, and release authorization
PET drug products do not enter a traditional distribution chain. Instead, PET drug products require just-in-time deliveries typically performed by dedicated carriers with experience in handling radioactive materials.
It is not possible to complete sterility testing for PET drug products before their use. Therefore, considerations are made to provide for the assurance of sterility for injectable PET drugs intended for human use.
Procedures should be in place to notify the responsible individuals in a timely manner if a PET drug product is found to be in noncompliance after release for human use.
2 TECHNIQUES FOR PRODUCTION AND QUALITY CONTROL
The unique characteristics of PET drug products play a large role in the choice of instrumentation and techniques used in production and QC testing. Production techniques and analytical methods should be efficient and rapid. The selection of techniques and methods is also strongly influenced by the development stage of the PET drug. For example, early development efforts focus on radiolabeling, purification, QC methods development, and others. These efforts are designed to support the usage of the PET drug for in vitro studies, animal studies, and may even include first-in-human studies. At this stage, relatively small quantities of the PET drug are required at a limited number of institutions or geographic areas. Manual production techniques or semi-automated equipment may provide sufficient quantities to meet this demand. Analytical methods should also be suitable for the usage of the PET drug at the early stages of development. Typically, method development efforts focus on accuracy, precision, and linearity. Before the use of injectable PET drugs in human studies, provisions are made to ensure that bacterial endotoxins are controlled at suitable levels and that the product is sterile.
In later development stages, larger quantities of the PET drug may be required in more geographic areas. At this point, the synthesis, purification, and testing of the PET drug product should be well defined to support clinical trials. Development efforts at this stage typically focus on optimization and reliability of the production process. Manual production techniques are rarely used. Instead, semi-automated or fully automated techniques are typically required to provide sufficient quantities of the PET drug at multiple geographic locations. This approach simultaneously assures consistent quality attributes of the PET drug at multiple production facilities and minimizes radiation exposure of operators. Method development activities at this stage focus on specificity, ruggedness, robustness, and other characteristics required to support QC testing of the PET drug at multiple geographic locations.
Finally, at the commercial stage of production, semi-automated or fully automated techniques and equipment may be required to provide sufficient quantities of the PET drug in numerous geographic areas. Production techniques and analytical methods for commercial PET drug products should be well defined and adequately described in the appropriate marketing authorization.
Not all PET drugs are intended to advance through the various development stages from in vitro studies, to animal studies, to clinical trials, to commercial production. For example, a PET drug product may be produced at several institutions for research purposes without the intention of commercialization. In other instances, a PET drug product may be Food and Drug Administration (FDA) approved but is only available in a single geographic area. These factors often result in different strategies for production techniques, analytical methodologies, and the underlying studies and documentation to support these strategies.
3 QUALITY ASSURANCE
The goal of quality assurance (QA) is to ensure that the techniques and equipment used in production and testing result in a product that meets established standards for that specific PET drug product. The QA program should be sufficient to establish the reliability and suitability of the techniques and equipment as appropriate to the development stage of the PET drug product. The following topics should be considered in a QA program.
3.1 Reagents and Materials
Reagents and materials used in the synthesis and testing of a PET drug product should conform to established acceptance criteria. Procedures for procuring, receiving, testing, storage, and use of reagents and materials should also be considered. An audit trail may be established for the traceability of specific lots of reagents and materials to specific batches of product. These acceptance criteria, procedures, and the audit trail should be appropriate for the development stage of the PET drug product.
3.2 Change Control
Changes in the synthesis and test methods should be evaluated for their potential for altering the product quality and be approved in advance through established procedures. If the resultant PET drug product does not meet the criteria appropriate to the development stage, the process change is unacceptable. Acceptable changes should be appropriately documented.
3.3 Validation
Production and analytical test methods should be validated at a level that is appropriate for the development stage of the PET drug product. Validation of the production process should result in a process that is reliable and consistent. Analytical method validation demonstrates that a method can quantitatively measure a PET drug product reliably and reproducibly. It is not necessary to validate analytical methods that are described in USP compendia. The emphasis on validation typically increases as the PET drug product moves through the development stages toward commercialization. Validation of the computer software used to control automated production equipment should also be considered as appropriate.
3.3.1 QUALIFICATION
Equipment used in the synthesis and testing of PET drug products should be qualified as appropriate for the development stage of the PET drug product. This may include qualification procedures (installation, operational, and performance), as well as procedures for periodic maintenance and equipment calibration. For commercially available equipment, the equipment vendor may be able to support qualification requirements.
3.3.2 STABILITY STUDIES
Stability studies should be performed on PET drug products to establish suitable storage conditions and the expiration time and date. Quality attributes such as radiochemical purity, appearance, pH, stabilizer, preservative effectiveness, and chemical purity should be evaluated. The frequency and extent of these studies should be appropriate for the development stage of the PET drug product. Significant process changes require stability studies to be repeated.
4 PRODUCTION
4.1 Equipment for Manual Synthesis
Equipment intended for the manual synthesis of PET drug products may be based on apparatus found in a typical chemistry laboratory such as glass reaction vessels, heating blocks, tubing, and other items. These items should be suitable for their intended use and selected according to the needs of the process. Items that are reused and come into contact with the reactants and/or final drug product of the synthesis should be cleaned and depyrogenated as appropriate for the use of the final product.
Written procedures for the manual syntheses of PET drug products should contain sufficiently detailed steps to ensure that a reproducible process is followed for each batch by the operator. Observations of critical process parameters should be documented as appropriate in the batch record.
4.2 Equipment for Automated Synthesis
Preparation of Fludeoxyglucose F 18 Injection and other common PET drug products can be adapted readily to automated synthesis. The use of programmable controllers and/or computers with switches, solenoids, and sensors allows the operator to control the sequence of steps in an automated fashion. Thus, an automated synthesis requires minimal operator intervention, and batch-to-batch consistency is maintained with each step executed in a reproducible manner. Software may provide feedback to the operator such as alarm conditions, status reports, or an end-of-synthesis report. Modules for automated synthesis may be commercially available. These modules may use individual reagents or cassettes required for the appropriate PET drug product synthesis. The cassettes provide preassembled collections of reagents, reaction vessels, tubing, filters, and other items necessary to produce a batch of a PET drug product.
Laboratory robots may be appropriate in situations where complex manipulations are required for the procedure, such as the physical movement of a reaction vessel from one station to another, or the dispensing of doses from a vial into individual syringes. Customized equipment may also be fabricated to perform a specific function in the PET drug product synthesis.
5 QUALITY CONTROL
QC tests should be designed and executed in a manner that is appropriate for PET drugs. The short half-life of PET radionuclides significantly limits the timeframe for the performance of QC tests, and it may not be possible to complete all QC tests before the use of the PET drug product.
5.1 Quality Control Sampling
In most cases, the entire batch of a PET drug is contained within a single vial. The relatively small batch volume of PET drugs limits the amount of material available for QC testing. This is different from traditional manufacturing practices, where a batch may consist of numerous vials and not all vials are sampled for purposes of QC testing. QC sampling of PET drug products use a larger proportion of the drug volume, and thereby offers a greater degree of assurance that quality-related problems will be identified in the QC testing process.
5.2 Reference Standards
Reference standards are necessary to identify the API and impurities in the PET drug product. Reference standards may be commercially available or prepared in-house. Commercial reference standards should include documentation from the supplier to ensure they are properly characterized. The characterization and qualification of reference standards should be appropriate for the development stage of the PET drug product.
5.3 Conditional Release
When a required QC test for a PET drug product cannot be completed because of a malfunction of test equipment, it may be appropriate to conditionally release the batch. It is not appropriate in the absence of critical QC tests to conditionally release batches (e.g., radiochemical identity and purity). Procedures should be developed to describe the conditional release process, including the role of historical data, other QC test results, sample retention, and other procedures.
5.4 Out-of-Specification Results
When the results of a QC test do not meet product specifications, an investigation should be conducted to determine if the result is due to an analytical error (false positive) or if the result is due to a product failure. Because of the short half-life of PET radionuclides, it may not be possible to conduct investigations in the same manner as in traditional drug testing laboratories.
6 ANALYTICAL METHODOLOGIES
Various analytical methodologies and chromatographic techniques may be used for the testing of PET drug products. The most commonly used methodologies are discussed in this section for purposes of illustration. Similar principles should apply to other methodologies not included in this section.
6.1 Thin-Layer Chromatography
Thin-layer chromatography (TLC) involves separation, identification, and quantification of analytes using a stationary phase on a support or plate and a developing solvent that may also be known as mobile phase. The stationary phase may be silica or alumina spread in a uniform layer onto a plate of glass, metal, or plastic. A small sample of the PET drug product is applied (spotted) onto the stationary phase, and the plate is developed in a chromatographic tank. The mobile phase moves through the stationary phase by capillary action, and the analytes are separated based on partition, ion exchange, and/or adsorption. The retardation factor (R,) and radiochemical purity of the PET drug on the developed plate can be determined by either counting the developed plate on a radio-TLC scanner, or cutting the plate into pieces and counting each piece in a radiation detector. The R, of an analyte, under specific TLC conditions, is considered an identifying characteristic of the analyte.
6.2 Gas Chromatography
Gas chromatography (GC) involves separation, identification, and quantification of volatile analytes using a stationary phase and a gaseous mobile phase. A small sample of the solution is introduced into the GC instrument, which vaporizes the sample for passage over the stationary phase that is immobilized within a GC column. As the gaseous mobile phase passes over the stationary phase, the analytes are separated based on their partition between the gas and stationary phases. A detector located at the exit of the column provides an electronic signal to produce the gas chromatogram.
A GC instrument consists of a gas source, injection port, column, detector, and a data collection device. The injection port, column, and detector are temperature controlled and may be varied as part of the analysis. The gas source depends on the column and detector in use. The type of detector depends on the nature of the compounds analyzed. Typical detectors include flame ionization and thermal conductivity. In addition, radiation detectors may be used in GC analysis. Detector output is recorded over time, and the instrument response, measured as peak area or peak height, is a function of the amount present. The retention time of an analyte under specific GC conditions is considered an identifying characteristic. GC analysis is most typically used to quantify residual solvents in PET drug products but may also be used to determine radiochemical and chemical purity of the PET drug product formulation.
6.3 High-Performance Liquid Chromatography
High-performance liquid chromatography (HPLC) is an analytical technique used to separate, identify, and quantify the components of a solution. HPLC separations are based on the interaction of the analytes between the stationary and mobile phases, which in turn leads to the retention of the analytes. The mechanism of interaction between the analytes and stationary phase may be partition, size exclusion, adsorption, and/or ion exchange chromatography. The retention time of an analyte under specific HPLC conditions is considered an identifying characteristic.
HPLC instruments consist of a reservoir containing the mobile phase, a pump to force the mobile phase through the system at high pressure, an injector to introduce the sample into the mobile phase, a chromatographic column, a detector, and a data collection device. The type of detector used depends on the nature of the compounds analyzed. In addition to mass detectors such as refractive index, ultraviolet, and conductivity, radiation detectors are also used in HPLC analysis. An HPLC system with a radiation and mass detector allows the simultaneous determination of radiochemical and chemical purity, retention time, and the subsequent identity of the PET drug product. This configuration also allows the determination of specific activity. Elution methods include gradient or isocratic methods with aqueous and/or organic buffers.
The calibration of a GC or HPLC instrument may be achieved by different means. One approach involves the creation of a calibration curve from a range of standards with known concentrations. The calibration curve may be used over a specified period of time for product testing. A second approach for calibration involves the creation of a single-point calibration at the beginning of each testing cycle. The results may be averaged and used to provide a calibration factor for product testing. Regardless of the approach for calibration, the tailing factor and chromatographic resolution (or column efficiency as appropriate) should be determined routinely as a part of system suitability. Routine system suitability testing should include checks for these parameters.
6.4 Radiation Detectors Used in Chromatographic Techniques
Radiation detectors used in TLC, GC, and HPLC for quantitative measurements should be calibrated when possible with suitable NMI-traceable radionuclide standards at appropriate frequency. Routine system suitability testing should be performed on the radiation detection system to ensure proper operation. Because of the nature of TLC, system suitability for a radio-TLC scanner should address uniformity, positional accuracy, detector linearity, and resolution. The calibration of radiation detectors should be repeated as appropriate.
6.5 Multichannel Analyzer
A multichannel analyzer (MCA) is an instrument used to obtain a spectrum of gamma rays emitted by a PET radionuclide. The key component of a MCA is an energy-sensitive detector. On the basis of the gamma spectrum of a sample, radionuclides present in the sample may be identified and quantified. Calibration of the system is typically performed with certified standards and may involve one or more radionuclides with gamma energies and quantities that span the range of typical analyses at appropriate frequency.
6.6 lonization Chamber
An ionization chamber, often referred to as a dose calibrator, is an instrument used to measure the quantity of radioactivity in a PET drug product. The key component of an ionization chamber is an argon-filled chamber with an applied electrical potential that allows the detection of ions produced by the passage of gamma rays through the chamber. Calibration of the system is typically performed with certified standards at appropriate frequencies and may involve one or more radionuclides with gamma energies and quantities that span the range of typical analyses.
7 QUALITY ATTRIBUTES
Quality attributes should be defined and assessed for each PET drug. These attributes should be suitable for the intended use of the PET drug and should also reflect the dosage form. Because most PET drug products are administered by intravenous injection, this section focuses on quality attributes associated with injectable PET drug products. Different attributes may be appropriate for other dosage forms. In addition, the definition and assessment of quality attributes may vary with the development phase of the PET drug product in comparison to the final drug phase.
7.1 Appearance
Injectable PET drug products should be free of visible particulate matter. Because of the radioactive nature of PET drug products, the test for appearance should be a visual inspection that meets radiation safety requirements. The use of a visual standard should be considered to ensure the accuracy of appearance determinations.
7.2 pH
Injectable PET drug products should be in the pH range suitable for intravenous administration. Because of the limited volume of a PET drug product, pH measurement is typically performed using narrow-range paper strips. The use of pH standards should be considered to ensure the accuracy of pH determinations.
7.3 Total Radioactivity and Strength
The total radioactivity of a PET drug product may be determined by an ionization chamber and should be stated at a given date and time. From the total radioactivity and the volume of the PET drug product, the strength may be determined. The strength of the PET drug product should be stated, along with the date and time of the determination of total radioactivity.
7.4 Radionuclidic Identity
Half-life (also referred to as approximate half-life) is a characteristic of the radionuclide that may be used for its identification. To adequately confirm the identity of a PET radionuclide, the half-life should be measured in a suitable counting device over a period of time appropriate to the half-life of the radionuclide.
7.5 Radionuclidic Purity
By definition, all positron-emitting radionuclides emit 511 keV gamma rays but may also emit gamma rays with different energies. Therefore, it is generally not possible to determine the radionuclidic purity of PET drug products with a MCA. One suitable solution to this problem is the use of validation studies to ensure the removal and/or decay of accelerator-produced radionuclidic impurities during the production process. Other approaches may be appropriate depending on the characteristics and source of the radionuclidic impurities. In addition, periodic analysis of decayed samples in routine production with a MCA may be used to quantify radionuclidic impurities developed.
7.6 Radiochemical Identity and Purity
Depending on the physical and chemical properties of the PET drug product, the radiochemical identity and purity may be determined by TLC, HPLC, or GC. The identity of the active pharmaceutical ingredient (API), and possibly other analytes, should be based on the known retention time of the analyte. The simultaneous use of reference standards during sample analysis should be considered for purposes of radiochemical identification. The radiochemical purity of the PET drug product should be determined based on the sum of all chemical forms of the radionuclide of interest. As appropriate, it should be established during validation that all radiochemical analytes are eluting from the chromatographic system and that the radioactive detector is operating within its linear range.
7.7 Chemical Purity
The chemical purity of the PET drug product should be assessed for the presence of volatile impurities, residual reagents and/or precursors, and by-products. For example, residual solvents, such as acetonitrile and ethyl alcohol, may assessed by GC. [2.2.2]-Cryptand, a common reagent used in the preparation of PET drug products, may be assessed by colorimetric techniques.
7.8 Total Mass of the Active Pharmaceutical Ingredient and Specific Activity
Potential toxicity issues and pharmacological effects may be associated with the API in a PET drug product (e.g., the mass-dependent localization of neurotransmitters or other more generalized forms of toxicity). In these instances, the total mass of the API should be determined before use of the PET drug product. The total mass of the API contained in a patient dose should be defined for PET drug products where mass-related localization or toxicity concerns require such assessment, which may be determined by HPLC or GC. On the basis of the mass of the API, the specific activity may be calculated. To determine the appropriate quality of the injected drug product in the patient dose, any two of three measured parameters (total mass, total activity, or specific activity) are sufficient. Specific activity should be stated along with the date and time of determination.
7.9 Bacterial Endotoxin
The quantity of bacterial endotoxin in an injectable PET drug product should comply with USP standards (<175 USP Endotoxin Units/patient dose). The test for bacterial endotoxin uses a solution of limulus amebocyte lysate (LAL). Various methods exist for this test, including gel-clot, chromogenic, and others. Test samples should be obtained and handled in a manner that minimizes contamination before testing. In addition, control measures should be used to ensure accurate test results without interferences from the test solution, such as certain formulations that enhance or inhibit the interaction of LAL with bacterial endotoxin. This effect may be corrected by dilution of the PET drug product before testing. The necessity of dilution in this test is determined in the validation of the specific formulation of the PET drug product.
7.10 Sterility
Injectable PET drug products should be sterile. For reasons described in 1. Introduction, it is not possible to complete the sterility test before the use of PET drug products. The test for sterility should consist of inoculating a test sample into media capable of supporting the growth of aerobic and anaerobic microbes, which may be established with an appropriate certificate of analysis. The test samples should be obtained and handled in a manner that minimizes contamination before testing. To reduce radiation exposure to operators, the test samples may be allowed to decay before inoculation. The inoculation should use techniques that minimize the potential for false positives during the sterility test. In addition, control measures should be used to ensure accurate test results without interferences from the test solution. After inoculation, the sterility test samples should be incubated for the appropriate period of time at the prescribed temperature. If visible growth is detected during the incubation period, the investigator or physician should be informed in a timely manner, and an investigation should be initiated to determine whether the growth was due to accidental contamination during the sample handling process or to a true product failure. The investigation should include identification of the microbe(s) in the sterility test sample to determine the source of the contamination. This aspect of the investigation may be limited if production and sterility test sampling occur in the same laboratory.
8 STERILITY ASSURANCE
A suitable sterility assurance program should be established for PET drug products that are intended for intravenous injection. This section describes appropriate measures that assure the routine production of a sterile product suitable for injection.
8.1 Sterile Membrane Filtration
Injectable PET drug products are typically sterilized by passage of the solution through a sterile membrane filter into a presterilized vial. To provide the greatest assurance of sterility, the PET drug product should pass through an appropriate sterilizing filter into a presterilized vial that has been assembled using aseptic techniques.
8.2 Aseptic Techniques
Critical steps that affect the sterility of the PET drug product should be identified and, where appropriate, aseptic techniques should be used to complete these steps. Aseptic techniques should include the use of a suitable gowning, proper handling of components, environmental controls, and others. Aseptic techniques should be described in written procedures.
8.3 Presterilized Components
PET drug production typically employs various components and containers that are presterilized and pyrogen free. Appropriate sterile components are generally assembled aseptically in a controlled environment before use in production. Presterilized vials and sterile diluents such as sterile water for injection or sodium chloride solution are typically used. Other sterile components are sometimes utilized in nonsterile applications in production where a commercially available product may be useful. An example is use of a sterile transfer line for fluid transfer prior to a sterilizing filter, where the diameter or length of a commercially available sterile line is appropriate to the application.
8.4 Environmental Controls
The environment where aseptic techniques are executed should be controlled to ensure appropriate aseptic conditions. These controls may include temperature and humidity, ventilation and air filtration, cleaning and disinfection, equipment maintenance, proper garb, and microbiological monitoring. Air filtration standards should conform to local and/or national standards relevant to environmental standards, for example, International Organization for Standardization (ISO) standards.
8.5 Media Fills
A media fill, also known as a "process simulation", is the performance of an aseptic procedure using a sterile microbiological growth medium in place of the PET drug product. The goal of a media fill is to test whether the aseptic procedure is adequate to prevent microbiological contamination during the actual process. Media fills may be used to evaluate aseptic techniques used in the assembly of presterilized components and to qualify operators for aseptic techniques. A media fill should be designed to ensure that the simulation is representative of the aseptic manipulations performed during the actual process, including personnel, components, gowning, locations, batch size, number of replicates, and other factors. After completing the media fill, components filled with media should be incubated appropriately to permit the growth of microbes. The use of negative controls should be considered during the media fill procedure,
8.6 Suitability of Media
Sterility test media should be tested before use to ensure that the media adequately support the growth of microbes. These tests are typically referred to as growth-promotion tests and may be conducted in-house, by the supplier, or by a contract laboratory. Media should be used within the manufacturer's expiration date.
8.7 Suitability of the Sterility Test Method
The sterility test should be tested to ensure that the PET drug product is not bacteriostatic or fungistatic. The suitability of the sterility test may be conducted in-house or by a contract laboratory.
8.8 Membrane Filter Integrity Test
To ensure the proper sterilization of the PET drug product by passage through a membrane sterilization filter, the bubble point of the filter should be determined after completing the filtration process. The bubble point of the filter, measured in pounds per square inch (psi), is the pressure required to force air through the pores of the wetted membrane in the device. Other suitably validated procedures may be used.
8.9 Operator Training and Qualification
Operators involved in aseptic techniques should be trained in proper gowning, environmental controls, handling sterile components, and other techniques. Operators should be qualified through successful completion of media fills. Media fills should be periodically repeated to ensure ongoing competency of the technique.
9 LABELING
The labeling associated with PET drug products may evolve as the drug progresses through the various development stages. For example, the labeling for PET drug products used for in vitro and animal studies may be very simple and designed to avoid mix-ups in the routine use of the PET drug product. In later development stages, the labeling may include information required for the investigational use of the PET drug product in humans. Finally, at the commercial stage of production, the labeling should include ingredients, warnings, approved indications, and other elements required by the appropriate regulatory agencies (e.g., FDA, Nuclear Regulatory Commission, and others).
10 GLOSSARY
The following definitions apply to words and phrases as they are used in this chapter.
Accuracy: The closeness of test results obtained by that method to the true value established across the range of the method.
Active pharmaceutical ingredient (API): A radioactive substance that exhibits spontaneous disintegration of unstable nuclei by the emission of positrons and is incorporated into a PET drug product to furnish a direct effect in the diagnosis or monitoring of a disease or a manifestation of a disease in humans, or monitoring treatment of disease or therapeutic procedures (e.g., tumor therapy). Both radioactive and nonradioactive forms of the PET drug are included in the API.
Batch: A quantity of PET drug product that is intended to have uniform character and quality, within specified limits, and that is made in a single, defined operational cycle.
Chemical purity: The purity of a PET drug product based on the nonradioactive components of the formulation, for example, residual solvents and/or volatile impurities, reagents, and or precursors used in the synthesis and purification, stabilizers, excipients, or by-products produced in the synthesis.
Compounding: The process of synthesis or formulation of a PET drug for use under the practice of pharmacy and medicine.
Conditional final release: A final release for patient administration before completion of a required test because of a breakdown of analytical equipment.
Limit of detection: The lowest amount of analyte in a sample that can be detected but not necessarily quantified under the stated method conditions.
Linearity: The ability to elicit test results that are, directly or by a well-defined mathematical transformation, proportional to the concentration of analyte in samples within a given range.
Lot: A quantity of materials (e.g., reagents, solvents, gases, purification columns, and other auxiliary materials) that have uniform character and quality within specified limits and are used to make a PET drug product.
Lower limit of quantification: The lowest amount of an analyte in a sample that can be determined with acceptable precision and accuracy under the stated method conditions.
National Metrology Institute (NMI): A measurement standards body that is a laboratory of metrology that establishes standards for a country or organization. The National Institute of Standards and Technology (NIST) is the NMI for the United States.
PET drug: A radioactive substance (active pharmaceutical ingredient) that exhibits spontaneous disintegration of unstable nuclei by the emission of positrons and is incorporated into a PET drug product to furnish direct effect in the diagnosis or monitoring of a disease or a manifestation of a disease in humans, or monitoring treatment of disease or therapeutic procedures (e.g., tumor therapy).
PET drug product: A finished dosage form that contains a PET drug, whether or not in association with one or more other ingredients.
Precision (as repeatability): The degree of agreement among individual test results when the method is applied repeatedly to multiple samplings of a homogenous sample within a lab over a short period of time using the same analyst with the same equipment.
Quality assurance (QA): A planned and systematic program to ensure that a PET drug product possesses the quality required for its intended purpose.
Quality control (QC): A system for testing the quality of components, materials, supplies, and PET drug products by procedures, tests, analytical methods, and acceptance criteria.
Radiochemical identity: The molecular structure of the intended active radiopharmaceutical ingredient that is present in the radiopharmaceutical preparation.
Radiochemical purity: The ratio, expressed as a percentage, of the radioactivity of the intended active radiopharmaceutical ingredient to the total radioactivity of all radioactive ingredients present in the radiopharmaceutical preparation.
Radionuclidic identity: The intended radionuclide in the radiopharmaceutical preparation.
Radionuclidic purity: The ratio, expressed as a percentage, of the radioactivity of the intended radionuclide to the total radioactivity of all radionuclides in the radiopharmaceutical preparation.
Range: The interval between the upper and lower levels of a quality attribute that can be determined with a suitable level of precision and accuracy.
Reagents and materials: A reagent is a chemical used in the synthesis and/or testing of a PET drug, whereas a material is an ancillary object, such as tubing, glassware, vials, and others.
Retardation factor (TLC): The ratio of the distance the analyte moved from the origin line divided by the distance the solvent moved from the origin line denoted by the variable R,
Retention time (HPLC or GC): The time required, after the injection, for the analyte to move through the column and reach the detector.
Robustness: The measure of the capacity of an analytical method to remain unaffected by small variations in method parameters.
Ruggedness (as reproducibility): The degree of reproducibility of test results obtained by the analysis of the same samples under a variety of conditions, such as different labs, different analysts, different instruments, different lots of reagents, different assays, and different days.
Specific activity: The radioactivity of a radionuclide per unit mass of the element or compound. The unit of specific activity is the amount of radioactivity on a mass basis [e.g., mCi/µg (MBq/µg) or Ci/mmol (GBq/mmol)].
Specificity: The ability to assess unequivocally the analyte in the presence of components that may be expected to be present such as impurities, degradation products, and matrix components.
Strength: The radioactivity concentration of the active pharmaceutical ingredient in a PET drug product at a given date and time. The unit of strength is the amount of radioactivity on a volume basis [e.g., mCi/mL (MBq/mL)].
Sub-batch: A quantity of PET drug product having uniform character and quality, within specified limits, that is produced during one succession of multiple irradiations using a given synthesis or purification operation. A group of sub-batches collectively forms a batch that is intended to have uniform character and quality, within specified limits. Sub-batches may be required for PET drug products with very short-lived radionuclides (e.g., 13N and 150), because QC tests cannot be completed before use.
System suitability: Requirements used to verify that the system performs according to established criteria.
Tailing factor (symmetry factor): A measure indicating the non-ideality of a chromatographic peak resulting from the distribution and the migration of the analyte through the chromatographic column.
Validation: Establishment of documented evidence that a method, process, or system meets its intended requirements.
Verification: Confirmation that an established method, process, or system meets predetermined acceptance criteria.
