NITROSAMINE IMPURITIES
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
The presence of nitrosamine impurities has been detected recently in several drug substances and drug products. In 2018, N-nitrosodimethylamine (NDMA) and N-nitrosodiethylamine (NDEA) were detected in some Valsartan drug substances and the drug products manufactured from drug substances using specific synthetic routes. This observation triggered extensive synthetic route assessments and development of analytical procedures to quantify these two nitrosamine impurities. As additional pharmaceuticals were evaluated and, in some cases tested, other nitrosamines beyond NDMA and NDEA were added as impurities of concern. Given the potentially broad implications of the presence of carcinogenic members of this class of chemicals, this chapter has been developed to provide a science- and risk-based approach for the control of nitrosamine impurities to ensure that the potential presence of nitrosamines in drug substances and drug products is identified, assessed, and controlled.
Recommendations are provided regarding: a) the establishment of controls of nitrosamine levels in order to ensure their elimination or reduction; and b) analytical procedure performance characteristics for procedures used to monitor nitrosamine levels.
2 NITROSAMINE IMPURITIES
Nitrosamines addressed in this general chapter are listed in Table 1 by their common names and chemical names. This list is a compilation of the information shared by multiple global health authorities. As additional nitrosamines are identified as potential concerns, the principles described herein should be applied for the assessment of these nitrosamines. If a manufacturer finds a nitrosamine not listed in Table 1, the appropriate regulatory authority should be contacted for determining appropriate AI limits. The potential presence of any one or more of these impurities is dependent on the reaction chemistries and processes. The list of nitrosamines is not intended to be exhaustive but represents those that have been observed and communicated by regulators and manufacturers as being potentially present or observed.
N-nitroso compounds are among the structural groups of high potency mutagenic carcinogens in several animal species, and some are classified as probable or possible human carcinogens referred to as the “cohort of concern” in ICH M7: Assessment and Control of DNA Reactive (Mutagenic) Impurities in Pharmaceuticals to Limit Potential Carcinogenic Risk (1), a designation that carries with it a recommendation to control these impurities at or below the acceptable cancer risk. As a result of the potential toxicity associated with these impurities, it is recommended to take steps to control and limit their presence in pharmaceutical materials.
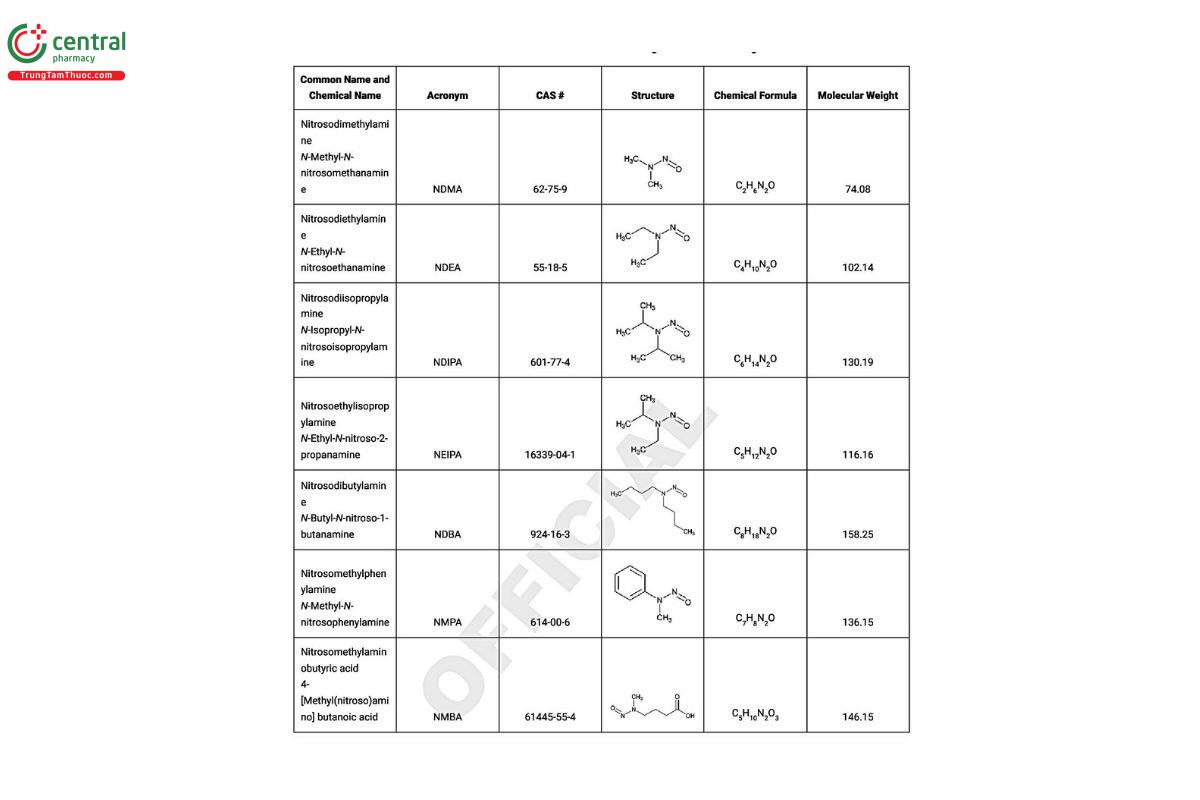
3 SOURCES OF NITROSAMINES
There are a number of pathways by which nitrosamines can be introduced into or generated as impurities in pharmaceutical drug products. Specifically, nitrosamines are formed by chemical reaction of secondary or tertiary amines with nitrites (the latter via intermediate degradation) under acidic conditions (see 3.1 Nitrosamine Formation Reaction). Some examples of the reported sources or pathways leading to the generation of nitrosamines identified empirically or reported in the literature (2–3) include (but are not limited to) the following:
• Drug substance processing under specific conditions and in the presence of certain reagents, solvents, raw materials, and processing aids. There is evidence that despite processing and purification steps, reactive species, whether intentionally added to or formed during the process/reaction sequence (e.g., nitrites and secondary amines in the presence of acidic conditions), can carry over to subsequent steps (see 3.1 Nitrosamine Formation Reaction). Special attention should be given to the formation of nitrogen-containing heterocycles by employing azide followed by quenching with nitrous acid to remove excess azide.
• The drug substance itself, which may degrade under some conditions resulting in the formation of nitrosamines (e.g., Ranitidine).
• Degradation of solvents (e.g., dimethylformamide [DMF]) leading to the formation of dialkyl amines.
• Impurities in raw materials, solvents (including recycled solvents), reagents, or catalysts.
• Impurities in materials and intermediates, reagents, and solvents used to prepare the starting materials or intermediates.
• Impurities in water, excipients, or processing aids used in the production of the finished drug product.
• During drug product manufacture under certain reaction conditions and in the presence of requisite precursors necessary for the formation of nitrosamines.
• Impurities in the container–closure system for the finished drug product, which may include impurities capable of forming nitrosamines, especially if associated with materials containing amines and potential sources of a nitrosating agent (e.g., nitrite, nitrocellulose).
A risk assessment should be conducted to determine the materials that contribute to the potential for inclusion of nitrosamines in the drug product. All potential sources for the introduction of nitrosamines should be considered in the risk assessment including, for example, the drug substance, excipients, water, solvents, the manufacturing process, packaging components, and formation on stability. See Figure 1 for a diagram of some potential sources to be considered.
Ongoing assessments and evaluations have identified risks associated with several of the potential sources of nitrosamines. Some of the examples identified are summarized in Table 2.
Table 2
| Potential Source of Nitrosamines | Observed Risk |
|---|---|
| Solvents | Presence of residual dialkyl amines or tri-substituted amines that can degrade to form intermediates that can further react with nitrosating agents |
| Presence of nitrites or other nitrosating agents | |
| Presence of acid | |
| Limited controls/specification limits for recycled solvents | |
| Poor quality solvents | |
| Water | Presence of residual dialkyl amines or impurities that can degrade to form dialkyl amines |
| Presence of acid and nitrosating agents | |
| Excipients | Presence of nitrites or other nitrosating agents and/or nitrosamine impurities (if applicable) |
| Drug substance | Use of sodium azide in the synthesis followed by use of nitrites in acidic medium (nitrous acid) for quenching excess azides |
| Use of di- or tri-alkylamines and amides (e.g., DMF, DMA, TEA, NMP) in the presence of nitrites and acid media | |
| Use of recycled solvents that may contain nitrosamines or their precursors | |
| Use of sanitized water (e.g., chloramine) | |
| Insufficient purification | |
| Manufacturing process | Degradation of drug substances containing functional groups that can participate in nitrosation reactions |
| Contamination | |
| Use of poor quality or recycled solvents | |
| Presence of nitrous oxides in air used to dry the drug substance or drug product | |
| Carryover of reactive species into subsequent steps | |
| Drug product (including stability) | Secondary, tertiary, or quaternary amine group in molecule of drug substance |
| Presence of nitrate counter ions | |
| Potential reactions within formulation matrix during stability | |
| Container–closures | Packaging materials containing vulnerable amines reacting with nitrosating agents |
ᵃ General chemical reactions leading to formation of nitrosamines can be found in 3.1 Nitrosamine Formation Reaction.
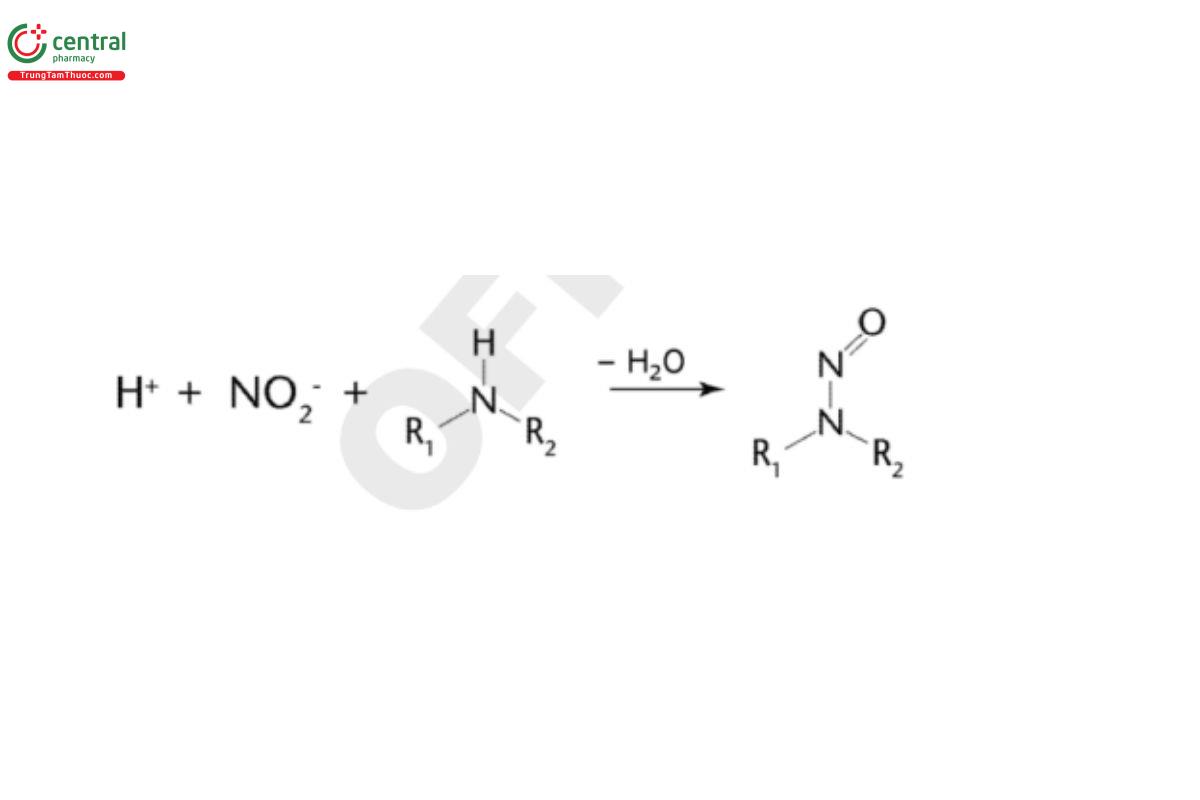
3.1 Nitrosamine Formation Reaction
The general schematic representation of the chemical reaction responsible for the formation of nitrosamines from secondary amines is described in Figure 2. Examples of representative reactions are described in the scientific literature (2–3).
If the potential for the presence of nitrosamines is identified, where appropriate, a control strategy should be developed. If nitrosamines are identified as impurities in ingredients, they may be controlled as appropriate in the ingredients (e.g., manufacture of the drug substance or controls placed on the drug substance). If nitrosamines are identified as degradation products (i.e., being formed during manufacturing of the drug product or formed during product storage), they should be controlled as appropriate in the drug product. In some cases, changes to the manufacturing process(es) or ingredients may be required to achieve acceptable levels or the elimination of nitrosamine impurities in the drug product.
4 NITROSAMINE RISK ASSESSMENTS—DEVELOPMENT OF A CONTROL STRATEGY
In order to determine the level of control, if any, which may be required for ensuring that levels of nitrosamines are at or below the acceptable intake (AI) if their presence cannot be avoided, the components of drug products should be assessed by the drug product manufacturer for the potential to form nitrosamines or to be contaminated with nitrosamines. Although one of the sources with the highest potential for nitrosamines is the drug substance synthetic route, the drug substance manufacturing process, drug product manufacturing process, and excipients and raw materials should also be included in a risk assessment to establish if controls or additional controls are needed. An example of high-level process flow for evaluating materials is shown in Figure 3.
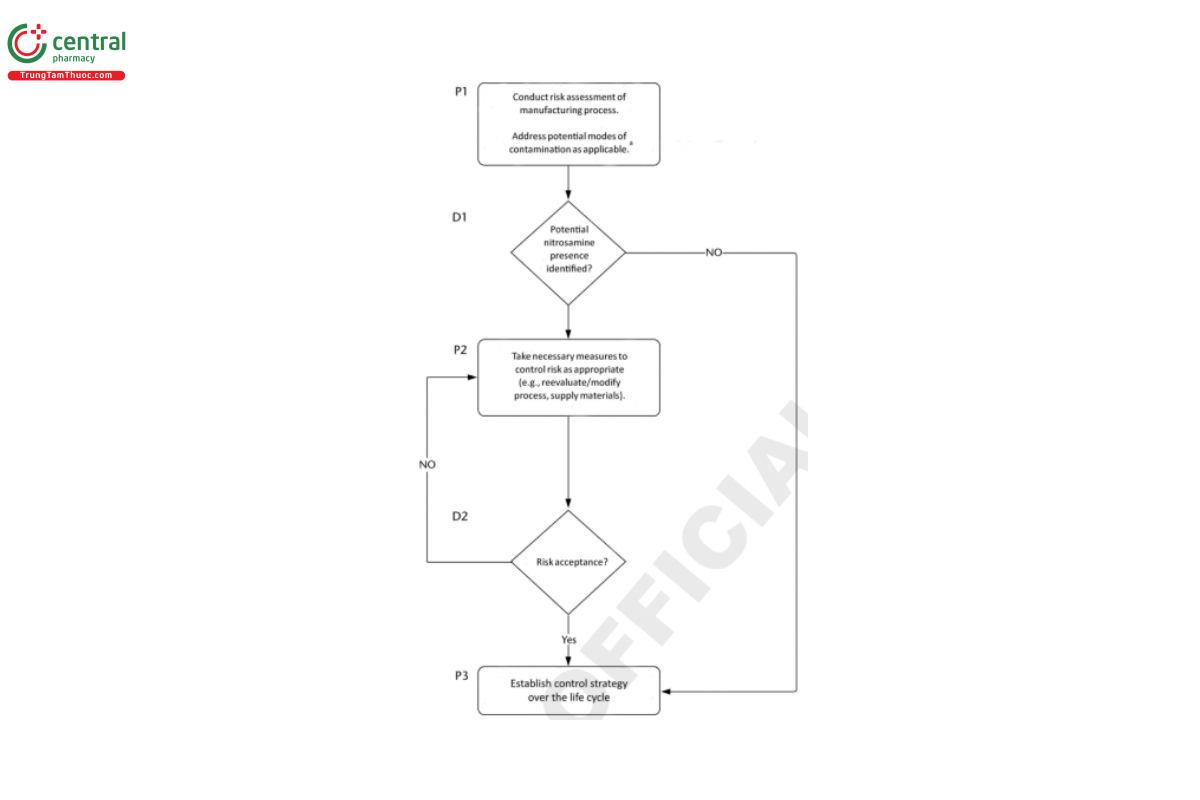
In all cases, if nitrosamines are predicted by the risk assessment or confirmed to be present through testing of the drug substance, drug product, or other materials, a control strategy should define an approach to ensure that the nitrosamine levels comply with the established AIs. The control strategy should be aligned with the current regulatory requirements in place.
5 LIMITS OF NITROSAMINES
Nitrosamine impurities identified in this chapter have potential and established toxicity with no therapeutic value. Because nitrosamines are among the structural groups of high potency mutagenic carcinogens of the “cohort of concern” in ICH M7 (1), the threshold of toxicological concern (TTC) does not apply. Instead, the available safety data should be used to establish a material-specific AI on a case-by-case basis. The AI is defined as an intake level that poses a negligible health risk.
5.1 Derivation of AI Limits
There are a number of methodologies that toxicologists have applied in establishing AIs. In this case, the median tumorigenic dose (TD₅₀) of NDMA, NDEA, and other nitrosamines was used as representative data in a linear extrapolation to establish an acceptable risk level. The limits have been published in the FDA Guidance for Industry: Control of Nitrosamine Impurities in Human Drugs (4).
5.2 Example Calculations of Nitrosamine Limits
The AIs in nanograms per day and the maximum daily dose (MDD) of the drug substance (DS) from the drug label in milligrams per day can be used to calculate the maximum nitrosamine concentration limits, in ppm, for individual drug products using the following equation:
Concentration = AI / DS_dose
Since the exposure to nitrosamines is related to the MDD of the DS, different concentrations of nitrosamines (ng/g) may be acceptable for each material evaluated. The acceptable concentration in the material can be calculated using the following equation:
Acceptable nitrosamine content = AI / MDD
AI = acceptable intake of the nitrosamine (µg/day)
MDD = maximum daily dose of the drug substance (g/day)
Table 3. Example Using an AI of 96 ng/day for the Target Nitrosamine
| Name | Acceptable Concentration (µg/g) | ||||
|---|---|---|---|---|---|
| Nitrosamine 1 | 0.050 (50-mg dose) | 0.100 (100-mg dose) | 0.250 (250-mg dose) | 1.00 (1000-mg dose) | |
| 1.920 | 0.960 | 0.384 | 0.096 | ||
6 TESTING FOR THE PRESENCE OF NITROSAMINES
Upon completion of the risk assessment, exploratory testing may need to be performed to confirm the conclusions of the risk assessment and proposed control strategy. On the basis of the controls identified (e.g., incoming material testing or specification limit; drug substance or drug product specification limit), it may be necessary to implement routine testing for nitrosamines. If testing is applied to ensure that the nitrosamine(s) concentration(s) do not exceed the AI, methods should be established following the recommendations detailed in this chapter. Example analytical procedures can be found in 8. Analytical Procedures.
6.1 Presence of Two or More Nitrosamines
The current published AIs reflect limits for the presence of a single nitrosamine. If multiple nitrosamines are possible and are determined analytically to be present at levels exceeding the maximum amount permitted by the regulatory authority, the relevant health authority should be consulted to determine a specific path forward. Manufacturers should contact FDA for determining the AI limits if multiple nitrosamine impurities are detected in a drug substance or drug product in which the total nitrosamine level exceeds 26.5 ng/day based on MDD.
7 TEST METHOD PERFORMANCE CHARACTERISTICS OF NITROSAMINE METHODS
The AIs associated with nitrosamines require the application of sensitive analytical procedures. In many cases, the most reliable procedures take advantage of the sensitivity and selectivity of chromatographic separation techniques coupled with quantitation by mass spectrometry (e.g., HPLC–MS/MS, GC–MS/MS). For additional guidance on validation of alternative methods for nitrosamines, see Validation of Compendial Procedures 〈1225〉.
7.1 Considerations for Sample Preparation
Appropriate sample preparation is a critical step in trace impurity analyses such as those required to evaluate the levels of nitrosamines in drug substances and drug products. This is particularly critical to prevent the loss or generation of nitrosamines as artifacts of the analytical procedure itself, as in the following circumstances.
• The presence of dialkyl amine (dimethylamine) as a process impurity or counter ion of the salt form of the active ingredient in the presence of nitrite and acid can lead to in situ formation of nitrosamines as an artifact, especially in GC analyses.
• Total solubilization versus selective extraction: If the active ingredient contains a dimethylamino group, total dissolution of the drug substance should be avoided when applying GC techniques. High concentration of the active ingredient, when injected in the GC instrument, can generate nitrosamines in the injection port if a nitrosating agent is present. In these situations, sample extractions should be modified to prevent the solubilization of the active ingredient while maintaining the extraction efficiency for nitrosamines present in the material.
The recommended method performance characteristics that need to be evaluated for quantitative analysis of nitrosamines include range, accuracy, repeatability, intermediate precision, and limit of quantitation. If a limit test is intended for use, the recommended method performance characteristics to be assessed include specificity, recovery, detectability, and solution stability. The performance criteria for these parameters should be properly set and confirmed through validation to ensure that the method is suitable for its intended use based on the specific analytes, matrices, and required precision and accuracy of the analytical procedures. Precision and recovery depend highly on concentration and matrix complexity, and the final proposed acceptance criteria need to be justified in the procedure validation documentation. Higher variability may be tolerated or acceptable at concentrations approaching the limit of quantitation of the procedure while lower variability (higher precision) would be expected at higher concentrations. Examples of quantitative analytical procedures are included in 8. Analytical Procedures. Additional example methods may be provided as they become available for these additional nitrosamines.
8 ANALYTICAL PROCEDURES
The following procedures have been established as suitable for their intended (specified) purpose. Users should validate these methods while considering the effect of sample solubility and extraction efficiency on the test results for other materials for which they are intended to be applied (see 〈1225〉 and Verification of Compendial Procedures 〈1226〉).
8.1 Quantitative Procedures
Procedure 1: Quantitation of NDMA, NDEA, NDIPA, NEIPA, NMBA, NMPA, and NDBA in selected sartans (valsartan, Irbesartan, and Losartan potassium) by HPLC–HRMS
Diluent: Methanol
Solution A: 0.1% formic acid in water
Solution B: 0.1% formic acid in methanol
Mobile phase: See Table 4.
| Time (min) | Solution A (%) | Solution B (%) |
|---|---|---|
| 0 | 90 | 10 |
| 1.5 | 90 | 10 |
| 7.0 | 45 | 55 |
| 17.0 | 45 | 55 |
| 17.1 | 10 | 90 |
| 21.0 | 10 | 90 |
| 21.1 | 90 | 10 |
| 25.0 | 90 | 10 |
Chromatographic system
(See Chromatography 〈621〉, System Suitability.)
Mode: LC
Detector: High resolution mass spectrometer
MS conditions
Ionization: Electrospray ionization (ESI)
Scan settings: See Table 5.
| Nitrosamine Impurity | NDMA | NMBA | NDEA | NEIPA | NDIPA | NMPA | NDBA |
|---|---|---|---|---|---|---|---|
| Scan type | SIMᵃ | SIM | PRMᵇ | SIM | SIM | SIM | PRM |
| Polarity | positive | negative | positive | positive | positive | positive | positive |
| Scan start–end (min) | 1.0–3.5 | 3.5–5.5 | 5.5–7.0 | 7.0–8.5 | 8.5–10.0 | 8.5–10.0 | 13.0–15.5 |
| m/z isolated for PRM | N/A | N/A | 103.0866 | N/A | N/A | N/A | 159.1492 |
| NCEᵈ | N/A | N/A | 25 | N/A | N/A | N/A | 20 |
| Scan range (m/z) | 74.3–75.8 | 144.3–145.8 | 50.0–114.0 | 116.4–117.9 | 130.4–131.9 | 136.3–137.8 | 50.0–170.0 |
| Microscans | 3 | 3 | 3 | 3 | 3 (1)ᵉ | 3 (1)ᵉ | 3 |
| Resolution | 30,000 | 60,000 | 30,000 | 60,000 | 60,000 | 60,000 | 30,000 |
| AGC target value (%)ᶠ | 250 | 250 | 250 | 250 | 250 | 250 | 250 |
ᵃ SIM = selected ion monitoring.
ᵇ PRM = parallel reaction monitoring.
ᶜ m/z = mass to charge ratio.
ᵈ NCE = normalized collision energy.
ᵉ 1 microscan must be used only if both NDIPA and NMPA are present.
ᶠ AGC = automatic gain control.
[NOTE—Divert the drug substance from the MS source during the elution.]
Data processing: Peak areas in the extracted ion chromatograms (EIC) with an m/z extraction window of ±15 ppm are used for quantitation. The m/z values extracted are listed in Table 6.
| Nitrosamine Impurity | NDMA | NMBA | NDEA | NEIPA | NDIPA | NMPA | NDBA |
|---|---|---|---|---|---|---|---|
| m/z to be extracted | 75.0553 | 145.0619 | 75.0553, 103.0866 | 117.1022 | 131.1179 | 137.0709 | 57.0704, 103.0872, 159.1492 |
Column: 4.6-mm × 10-cm; 2.6-µm packing L43
Temperatures
Autosampler: 4°
Column: 40°
Flow rate: 0.6 mL/min
Flow rate to ion source: 0.6 mL/min
Injection volume: 3 µL
System suitability
Samples: Sensitivity solution and Standard solution
[NOTE—The relative retention times for NDMA, NMBA, NDEA, NEIPA, NDIPA, NMPA, and NDBA are 0.20, 0.31, 0.46, 0.57, 0.66, 0.67, and 1.00, respectively.]
[NOTE—NMBA and NEIPA exist as syn- and anti-conformers due to the restricted rotation of the N–N bond. These conformers are partially separated by the method’s chromatographic conditions. The NMBA peak is observed as a doublet. Integrate both of the NMBA peaks and use the combined peak areas for calculation of the NMBA concentration. The NEIPA peak may appear as a doublet or a single peak with a tailing shoulder. For NEIPA, if the conformers are resolved, integrate both peaks and combine the peak areas for the calculation of the NEIPA concentration. If the NEIPA conformers are not fully resolved (e.g., evidence of a shoulder is present), integrate the main peak and shoulder as a single peak and use the combined peak area to calculate the NEIPA concentration.]
Suitability requirements
Relative standard deviation: NMT 20.0% from 6 replicate injections, Standard solution
Signal-to-noise ratio: NLT 10, Sensitivity solution
Analysis
Samples: Standard solution and Sample solution
Calculate the concentration, in ppm, of each specified nitrosamine impurity in the portion of drug substance taken:
Result = (rᵤ / rₛ) × (Cₛ / Cᵤ) × 10⁶
rᵤ = peak response of the individual specified nitrosamine impurity from the Sample solution
rₛ = peak response of the corresponding nitrosamine impurity from the Standard solution
Cₛ = concentration of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, USP N-Nitrosodiisopropylamine RS, USP N-Nitrosodibutylamine RS, USP N-Nitrosomethylphenylamine RS, or USP N-Nitrosomethylaminobutyric Acid RS in the Standard solution (µg/mL)
Cᵤ = concentration of the drug substance in the Sample solution (µg/mL)
Report the nitrosamine impurity concentration in the drug substance in ppm (µg/g).
Procedure 2: Quantitation of NDMA, NDEA, NDIPA, and NEIPA in selected sartans (valsartan, irbesartan, losartan potassium, olmesartan medoxomil, Candesartan cilexetil, and Telmisartan) by headspace GC–MS
Diluent: Methanol
Internal standard stock solution: 0.4 µg/mL of NDMA-d6 in Diluent
Internal standard solution: 0.016 µg/mL of NDMA-d6 prepared as follows. Transfer 2.0 mL of Internal standard stock solution into a 50-mL volumetric flask, and dilute with Diluent to volume.
Nitrosamine RS stock solution: 0.4 µg/mL each of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, and USP N-Nitrosodiisopropylamine RS prepared as follows. Transfer an appropriate amount of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, and USP N-Nitrosodiisopropylamine RS into a suitable volumetric flask, and dilute with Diluent to the volume.
Standard stock solution: 0.016 µg/mL each of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, and USP N-Nitrosodiisopropylamine RS prepared as follows. Transfer 2.0 mL of Nitrosamine RS stock solution and 2.0 mL of Internal standard stock solution into a 50-mL volumetric flask, and dilute with Diluent to volume.
Standard solution: Transfer 1 mL of Standard stock solution to an appropriate headspace vial containing about 100 mg of imidazole and 1.0 mL of acetonitrile. Apply the stopper, cap, and crimp tightly.
Sensitivity stock solution: 0.004 µg/mL each of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, and USP N-Nitrosodiisopropylamine RS in Diluent prepared as follows. Transfer 0.5 mL of the Nitrosamine RS stock solution and 2.0 mL of Internal standard stock solution into a 50-mL volumetric flask and dilute with Diluent to volume.
Sensitivity solution: Transfer 1 mL of Sensitivity stock solution to an appropriate headspace vial containing about 100 mg of imidazole and 1.0 mL of acetonitrile. Apply the stopper, cap, and crimp tightly.
Sample solution: Transfer 200 ± 10 mg of drug substance and about 100 mg of imidazole into a headspace vial, and then add 1.0 mL of Internal standard solution and 1.0 mL of acetonitrile. Apply the stopper, cap, and crimp tightly.
Blank: Transfer about 100 mg of imidazole into a headspace vial, and then add 1.0 mL of Internal standard solution and 1.0 mL of acetonitrile. Apply the stopper, cap, and crimp tightly.
Chromatographic system
(See Chromatography ⟨621⟩, System Suitability.)
Mode: GC
Injector: Headspace (see Table 7 for parameters)
| Parameter | Value |
|---|---|
| Equilibration temperature | 95°–110° |
| Loop temperature | 150° |
| Rate 1 | 10°/min |
| Transfer line temperature | 160° |
| Pressurizing gas pressure | 20.00 psi |
| Equilibration time | 10.00 min |
| Pressurizing time | 2.00 min |
| Load time | 2.00 min |
| Injection time | 1.00 min |
| Vial size | 20 mL |
Injection type: (Split, Split ratio 1:1 or 1:3)
[NOTE—Split ratio can be modified to optimize sensitivity.]
Detector: Mass spectrometer
Column: 0.32-mm × 30-m fused-silica, coated with a 1.0-µm layer of phase G16
Column temperature: See Table 8.
| Initial Temperature (°C) | Temperature Ramp (°C/min) | Final Temperature (°C) | Hold Time at Final Temperature (min) |
|---|---|---|---|
| 45 | 0 | 45 | 3 |
| 45 | 10 | 130 | 3 |
| 130 | 15 | 190 | — |
| 190 | 40 | 240 | 10 |
Carrier gas: Helium
Flow rates
Gas: Constant flow at 1.8 mL/min (adjustment and verification are necessary for other carrier gases)
Purge: 3.0 mL/min or default value
MS conditions: See Table 9.
Event 1
| Parameter | Value |
|---|---|
| Name | NDMA |
| Start time | 10.0 min |
| End time | 12.0 min |
| Acquisition mode | multiple reaction mode (MRM) |
| Ch 1 m/z | 74.00 > 44.00 |
| CH 1 collision energy | 4.00 V |
| Ch 2 m/z | 74.00 > 42.00 |
| CH 2 collision energy | 15.00 V |
Event 2
| Parameter | Value |
|---|---|
| Name | NDMA-d6 |
| Start time | 10.0 min |
| End time | 12.0 min |
| Acquisition mode | MRM |
| Ch 1 m/z | 80.00 > 50.00 |
| CH 1 collision energy | 5.00 V |
Event 3
| Parameter | Value |
|---|---|
| Name | NDEA |
| Start time | 12.00 min |
| End time | 12.75 min |
| Acquisition mode | MRM |
| Ch 1 m/z | 102.00 > 85.1 |
| CH 1 collision energy | 6.00 V |
| Ch 2 m/z | 102.00 > 56.1 |
| CH 2 collision energy | 15.00 V |
Event 4
| Parameter | Value |
|---|---|
| Name | NEIPA |
| Start time | 12.75 min |
| End time | 13.35 min |
| Acquisition mode | MRM |
| Ch 1 m/z | 116.00 > 99.10 |
| CH 1 collision energy | 6.00 V |
| Ch 2 m/z | 99.00 > 44.10 |
| CH 2 collision energy | 9.00 V |
Event 5
| Parameter | Value |
|---|---|
| Name | NDIPA |
| Start time | 13.35 min |
| End time | 14.00 min |
| Acquisition mode | MRM |
| Ch 1 m/z | 130.00 > 42.00 |
| CH 1 collision energy | 10.00 V |
| Ch 2 m/z | 130.00 > 43.10 |
| CH 2 collision energy | 18.00 V |
[NOTE—Ch 1 m/z in multiple reaction mode (MRM) is used for quantitation.]
System suitability
Samples: Standard solution, Sensitivity solution, and Blank
[NOTE—The relative retention times for NDMA, NDMA-d6, NDEA, NEIPA, and NDIPA are 0.80, 0.80, 0.90, 0.96, and 1.00, respectively.]
Suitability requirements
Relative standard deviation: NMT 20.0% for the ratio of the impurity standard peak response to the internal standard peak response from 6 replicate injections, Standard solution
Signal-to-noise ratio: NLT 10 for the impurity peak, Sensitivity solution
Interference: There should be no interfering peak in the Blank.
Analysis
Samples: Standard solution and Sample solution
Calculate the concentration, in ppm, of each specified nitrosamine impurity in the portion of drug substance taken:
Result = 1 / W × (Rᵤ / Rₛ) × Cₛ
W = weight of the drug substance in the Sample solution (g)
Rᵤ = peak response ratio of the specified nitrosamine impurity to that of the internal standard from the Sample solution
Rₛ = peak response ratio of the specified nitrosamine impurity to that of the internal standard from the Standard solution
Cₛ = concentration of USP N-Nitrosodimethylamine RS, USP N-Nitrosodiethylamine RS, USP N-Nitrosoethylisopropylamine RS, and USP N-Nitrosodiisopropylamine RS in the Standard stock solution (µg/mL)
Report the nitrosamine impurity concentration in the drug substance in ppm (µg/g).
Procedure 3: Quantitation of NDMA, NDEA, NDIPA, NEIPA, NMBA, and NDBA in selected sartans (valsartan, losartan potassium, olmesartan medoxomil, candesartan cilexetil, and telmisartan) by HPLC–MS/MS
Diluent: 1% formic acid in water
Solution A: 0.1% formic acid in water
Solution B: 0.1% formic acid in methanol
Mobile phase: See Table 10.
| Time (min) | Solution A (%) | Solution B (%) |
|---|---|---|
| 0 | 97 | 3 |
| 1.5 | 97 | 3 |
| 4.0 | 50 | 50 |
| 7.0 | 25 | 75 |
| 8.1 | 15 | 85 |
| 9.2 | 5 | 95 |
| 12.0 | 5 | 95 |
| 12.1 | 97 | 3 |
Internal standard solution: 10 µg/mL each of NDMA-d6 and NMBA-d3, 1 µg/mL each of NDEA-d10 and NDBA-d18 in water
Nitrosamine standards stock solution mixture: Prepare a mixture of 200 ng/mL each of N-nitrosodimethylamine, N-nitrosoethylisopropylamine, N-nitrosodiisopropylamine, N-nitrosodibutylamine, and N-nitrosomethylaminobutyric acid by mixing appropriate volumes of the respective USP Reference Standards and dilute with water.
[CAUTION—Prepare Nitrosamine standards stock solution mixture in amber vials and store at −18° to −20°.]
NDEA standard stock solution: Prepare a solution of 132 ng/mL of N-nitrosodiethylamine by diluting USP N-Nitrosodiethylamine RS with water.
Standard solutions: Depending on the targeted nitrosamine concentration in the sample, prepare a set of 5 consecutive linearity solutions from Table 11 from the Nitrosamine standards stock solution mixture and NDEA standard stock solution by mixing specified volumes of each solution as indicated. [NOTE—Table 11 represents an example for preparing solutions for constructing the calibration curve. Other dilution schemes may be used for preparing the set of 5 linearity solutions covering the range of interest. L1 is used only for NDEA when applicable. For others, linearity starts with L2.]
| Linearity Solution # | Concentration Level | NDMA/NMBA/NDBA/NEIPA/NDIPA (ng/mL) / NDEA (ng/mL) | Content (ppb) | Std mix (µL) | NDEA Std (µL) | Water (µL) | IS (µL) | Total (µL) |
|---|---|---|---|---|---|---|---|---|
| 1 | L1 | 1.33/0.66 | 19.95/10 | 8 | 6 | 1174 | 12 | 1200 |
| 2 | L2 | 2/0.88 | 30/13.5 | 12 | 8 | 1168 | 12 | 1200 |
| 3 | L3 | 5/3.3 | 75/49.5 | 30 | 30 | 1128 | 12 | 1200 |
| 4 | L4 | 7.5/4.95 | 112.5/74.2 | 45 | 45 | 1098 | 12 | 1200 |
| 5 | L5 | 10/6.6 | 150/99 | 60 | 60 | 1068 | 12 | 1200 |
| 6 | L6 | 15/9.9 | 225/148.5 | 90 | 90 | 1008 | 12 | 1200 |
| 7 | L7 | 30/19.8 | 450/297 | 180 | 180 | 828 | 12 | 1200 |
| 8 | L8 | 60/39.6 | 900/594 | 360 | 360 | 468 | 12 | 1200 |
| 9 | L9 | 90/59.4 | 1350/891 | 540 | 540 | 108 | 12 | 1200 |
Sample solution: Transfer about 80 mg of the drug substance into a 2-mL lidded centrifuge tube. Add 1188 µL of Diluent and 12 µL of the Internal standard solution. Vortex at 2500 rpm for 20 min (except for losartan potassium, which should be vortexed NMT 5 min). Centrifuge at about 10,000 rpm for 10 min, and filter into a vial using a hydrophilic polytetrafluoroethylene (PTFE) filter of 0.45-µm pore size.
Chromatographic system
(See Chromatography ⟨621⟩, System Suitability.)
Mode: LC
Detector: MS/MS (triple quadrupole mass spectrometer)
MS conditions
Ionization: Atmospheric pressure chemical ionization (APCI)
Scan settings: See Table 12.
| Nitrosamine Impurity | Acquisition Mode | Polarity | MRM-1 | MRM-2 |
|---|---|---|---|---|
| NDMA | MRM | Positive | +75.0 → +43.0 | +75.0 → +44.1 |
| NDMA-d6 | MRM | Positive | +81.2 → +46.0 | +81.2 → +64.1 |
| NDEA | MRM | Positive | +103.1 → +75.1 | +103.1 → +47.1 |
| NDEA-d10 | MRM | Positive | +113.2 → +34.2 | +113.2 → +49.1 |
| NMBA | MRM | Positive | +147.1 → +44.1 | +147.1 → +117.1 |
| NMBA-d3 | MRM | Positive | +150.1 → +47.1 | +150.1 → +120.2 |
ᵃ MRM-1 is used for quantitation.
ᵇ NDEA-d10 is used as internal standard for NEIPA and NDIPA.
Column: 3.0-mm × 15-cm; 2.7-µm packing L1
Temperatures
Autosampler: 18°
Column: 60°
Flow rate: 0.5 mL/min
Flow rate to ion source: 0.5 mL/min
Injection volume: 20 µL
System suitability
Samples: Standard solutions
Generate the peak response ratio of the specified impurity to that of the internal standard versus the concentration standard curve for each nitrosamine impurity under test using the corresponding selected Standard solutions and perform the linear regression analysis.
[NOTE—The relative retention times for NDMA, NMBA, NDEA, NEIPA, NDIPA, and NDBA are 0.20, 0.31, 0.46, 0.57, 0.66, and 1.00, respectively.]
Suitability requirements
Correlation coefficient: NLT 0.99
Y-intercept: NMT 25% of the response of the medium concentration solution used in standard curve generation
Analysis
Samples: Standard solutions and Sample solution
Calculate the concentration, in ppm, of each specified nitrosamine impurity in the Sample solution using the corresponding calibration curve:
Result = [(Rᵤ − yᵢₙₜ) / a] × (1 / Cᵤ) × 10³
Rᵤ = peak response ratio of the specified nitrosamine impurity to that of the internal standard from the Sample solution
yᵢₙₜ = y-intercept of the calibration curve for the specified nitrosamine impurity from the Standard solutions
a = slope of the calibration curve for the specified nitrosamine impurity from the Standard solutions [(µg/mL)⁻¹]
Cᵤ = concentration of the drug substance in the Sample solution (mg/mL)
Report the nitrosamine impurity concentration in the drug substance in ppm (µg/g).
Procedure 4: Quantitation of NDMA, NDEA, NDIPA, NEIPA, NMPA, and NDBA in selected sartans (valsartan, losartan potassium, and candesartan cilexetil) by GC–MS/MS (triple-quad)
Internal standard solution: 50 ng/mL of NDMA:C13-d6 in methylene chloride
Standard solution: Prepare a mixture of 0.1 µg/mL each of N-nitrosodimethylamine, N-nitrosodiethylamine, N-nitrosoethylisopropylamine, N-nitrosodiisopropylamine, N-nitrosomethylphenylamine and N-nitrosodibutylamine by mixing appropriate volumes of respective USP Reference Standards and diluting with Internal standard solution.
Calibration solutions: Depending on the targeted nitrosamine concentration in the sample, prepare a set of 5 consecutive solutions from Table 13 that are used for generating the calibration curve by following the preparation scheme shown in the table. Volumes may be adjusted to prepare larger quantities of the calibration solutions as needed, maintaining final concentrations of the nitrosamines. For each calibration solution, transfer the designated aliquot of Standard solution to the designated volumetric flask, and adjust the volume with the Internal standard solution.
| Calibration Solution ID | Standard Solution Aliquot (µL) | Final Volume (mL) | Final Nitrosamine Concentration (µg/mL) | Equivalent Nitrosamine Concentration (µg/g) |
|---|---|---|---|---|
| Cal 1 | 50 | 10 | 0.0005 | 0.005 |
| Cal 2 | 100 | 10 | 0.001 | 0.010 |
| Cal 3 | 200 | 10 | 0.002 | 0.020 |
| Cal 4 | 300 | 10 | 0.003 | 0.030 |
| Cal 5 | 400 | 10 | 0.004 | 0.040 |
| Cal 6 | 500 | 10 | 0.005 | 0.050 |
| Cal 7 | 1000 | 10 | 0.010 | 0.100 |
| Cal 8 | 1500 | 10 | 0.015 | 0.150 |
Sample solution: Transfer 500 mg of the drug substance into a disposable 10- to 15-mL glass centrifuge tube. Add 5.0 mL of the Internal standard solution. Cap the tube. Vortex the sample for 1 min, and then place in the centrifuge. Centrifuge the sample at 4000 rpm for 2.5 min. Transfer 2 mL of the bottom methylene chloride layer to a 5-mL syringe fitted with a 0.45-µm nylon filter. Filter 1 mL of sample extract into a 2-mL GC autosampler vial and cap.
Chromatographic system
(See Chromatography ⟨621⟩, System Suitability.)
Mode: GC
Injector: Split/splitless
Injection type: Splitless with purge
Purge time: 0.5 min
Detector: MS/MS (triple quadrupole mass spectrometer)
MS conditions
Ionization: Electron impact
Scan settings: See Table 14.
| Nitrosamine Impurity | Acquisition Mode | Polarity | MRM-1 | MRM-2 |
|---|---|---|---|---|
| NDMA | MRM | Positive | 74 → 44 | 74 → 42 |
| NDMA:c13-d6 | MRM | Positive | 82 → 48 | — |
| NDEA | MRM | Positive | 102 → 85 | 102 → 56 |
| NEIPA | MRM | Positive | 116 → 99 | 71 → 56 |
| NDIPA | MRM | Positive | 130 → 88 | 130 → 42 |
| NMPA | MRM | Positive | 106 → 77 | 77 → 51 |
| NDBA | MRM | Positive | 158 → 99 | 84 → 56 |
ᵃ MRM-1 is used for quantitation.
MS1 and MS2 resolution: Q1: normal; Q3: wide (1.5)
Minimum window: 1 min
Emission current: 50 µA
Column: 0.25-mm × 30-m; fused-silica coated with a 1.0-µm layer of phase G16
Temperatures
Injector: 250°
Transfer line to MS detector: 250°
Ionization source: 250°
Column: See Table 15.
| Initial Temperature (°C) | Temperature Ramp (°C/min) | Final Temperature (°C) | Hold Time at Final Temperature (min) |
|---|---|---|---|
| 40 | 0 | 40 | 0.5 |
| 40 | 20 | 200 | 0 |
| 200 | 60 | 250 | 3 |
Carrier gas: Helium
Flow rate: Constant flow at 1.0 mL/min (adjustment and verification are necessary for other carrier gases)
Injection volume: 2 µL
System suitability
Samples: Calibration solutions
[NOTE—The relative retention times for NDMA, NDEA, NEIPA, NDIPA, NDBA, and NMPA are 0.73, 0.78, 0.81, 0.82, 0.99, and 1.00, respectively.]
Generate the peak response ratio of the specified impurity to that of the internal standard versus concentration standard curve for each nitrosamine impurity under test using the corresponding Calibration solutions and perform the linear regression analysis.
Suitability requirements
Correlation coefficient: NLT 0.98
Signal-to-noise: NLT 10 for the impurity peak of the lowest concentration Calibration solutions used in the calibration curve
Analysis
Samples: Calibration solutions and Sample solution
Calculate the concentration, in ppm, of each specified nitrosamine impurity in the Sample solution using the corresponding calibration curve:
Result = 5 × (1/W) × [(Rᵤ − yᵢₙₜ)/a]
W = weight of the drug substance in the Sample solution (g)
Rᵤ = peak response ratio of the specified nitrosamine impurity to that of the internal standard from the Sample solution
yᵢₙₜ = y-intercept of the calibration curve for the specified nitrosamine impurity from the corresponding Calibration solutions
a = slope from the calibration curve for the specified nitrosamine impurity from the corresponding Calibration solutions [(µg/mL)⁻¹]
Report the nitrosamine impurity concentration in the drug substance in ppm (µg/g).
8.2 Limit Test Procedures
While a limit test analytical procedure for nitrosamines content is not currently available, recommended sample preparation procedures are shown in Table 16.
| Solutions | Solution Preparation |
|---|---|
| Internal standard solution | Prepare a suitable Internal standard solution that, when added to the Sample solution, will have the resultant peak response at the highest appropriate target limit of the nitrosamines of interest in the sample. |
| Sample solution preparation | Prepare a solution of the article to be examined spiked with the internal standard and prepared as described in the sample preparation. |
| Spiking solution | A solution of target N-nitrosamine(s) of a concentration that, if added to the amount of article used for the preparation of the Sample solution, would result in the acceptance limit. |
| Spiked sample solution | Prepare a solution of the article to be examined spiked with appropriate Spiking solution(s) and Internal standard solution prepared as described in the Sample solution. |
Analysis
Samples: Spiked sample solutions and Sample solution
Determine the peak response ratio of the respective target N-nitrosamine to the internal standard from the Sample solution: Rᵤ
Determine the peak response ratio of the respective target N-nitrosamine to the internal standard from the Spiked sample solution: Rₛₜ(i)
Acceptance criteria: Rᵤ/Rₛₜ(i) is NMT 0.5
9 ADDITIONAL SOURCES OF INFORMATION
Several test procedures have been developed for the specific testing of nitrosamines in sartans and have been based on different scientific principles and are publicly available from many regulatory agencies. The hyperlinks in this section direct the user to the respective regulatory agencies’ procedures. These can be used as alternative procedures and must be validated under actual use for the respective performance characteristics recommended in 7. Test Method Performance Characteristics of Nitrosamine Methods.
