IN VITRO AND IN VIVO EVALUATION OF ORAL (USP 1-May-2021) DOSAGE FORMS
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- PURPOSE
- SCOPE
- BACKGROUND INFORMATION
- IN VITRO CHARACTERIZATION
- Drug Substance Properties
- Drug Product Properties
- Design of In Vitro Characterization
- Experimental Variables
- Experimental Design
- Technical Requirements
- IN VIVO EVALUATION OF DOSAGE FORMS
- The Biopharmaceutics Classification System (BCS)
- CHARACTERIZATION OF DRUG SUBSTANCE
- CHARACTERIZATION OF THE ORAL (USP 1-May-2021) DOSAGE FORM
- IN VITRO–IN VIVO CORRELATIONS
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
DOWNLOAD PDF HERE
1 PURPOSE
This chapter focuses on the in vitro and in vivo performance of oral dosage forms and (USP 1-May-2021) provides an overview of the methodology for characterizing the (USP 1-May-2021) properties of a drug substance as well as its associated oral dosage form that are critical to establish the relationship to the pharmacokinetic (USP 1-May-2021) properties of the drug product. Results of in vitro methods are linked with information from in vivo evaluations through an in vitro–in vivo correlation (IVIVC), or, if an IVIVC is not possible, an in vitro–in vivo relationship (IVIVR). Establishing an IVIVC may enable a waiver of in vivo bioequivalence study requirements as well as support scale-up and post-approval changes. (USP 1-May-2021)
Change to read:
2 SCOPE
The ultimate goal of these characterization studies is an understanding of the relationship between the physicochemical (USP 1-May-2021) properties of the drug substance and the in vitro performance of the drug product to the pharmacokinetic profile of the drug product. (USP 1-May-2021) This chapter outlines the in vitro and in vivo testing that goes into the development of the body of data required for (USP 1-May-2021) decision making relating to the formulation, manufacturing, post-approval changes (see FDA guidance Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations; www.fda.gov/media/70939/download),
(USP 1-May-2021) and related regulatory activities of an oral dosage form. An IVIVC is linked to the in vitro test conditions and the product critical quality attributes (CQAs) controlling the in vitro and in vivo release characteristics. If either is changed, the relationship may be compromised. An IVIVC includes the verification of relevant in vivo differences predicted by a discriminating in vitro method. (USP 1-May-2021) This chapter complements the information in general chapters, Assessment of Solid Oral Drug Product Performance and Interchangeability, Bioavailability, Bioequivalence, and Dissolution 〈1090〉 and The Dissolution Procedure: Development and Validation 〈1092〉 by detailing the essential in vitro and in vivo data elements underlying an understanding of bioequivalence and bioavailability. This chapter's text recognizes that regulatory guidances and a wealth of text books are available to elaborate on the content provided, and it is not the purpose of this chapter to provide an exhaustive discussion (USP 1-May-2021) on the subjects presented but rather to provide a guide and listing of the issues of interest.
Change to read:
3 BACKGROUND INFORMATION
Establishing a meaningful relationship between dissolution behavior and in vivo drug performance (i.e., IVIVC) has long been sought from the perspectives of both bioavailability and bioequivalence and quality control considerations. In order to ascertain lot-to-lot equivalence to the biobatch, clinically relevant specifications should be established preferably with an IVIVC. If an IVIVC is not possible, the use of an IVIVR may be acceptable. (USP 1-May-2021)
The earliest achievable in vitro characteristic thought to predict an acceptable in vivo performance was tablet and capsule disintegration. A test for disintegration was adopted in USP XIV (1950). At that time, no quantitative work was done to attempt to demonstrate a relationship (USP 1-May-2021) to in vivo product performance. Advances in instrumental methods and analytical precision ultimately opened up prospects for this work. The USP–NF Joint Panel on Physiologic Availability recognized that the disintegration test was insufficiently sensitive and in 1968 directed the identification of candidate articles for the first 12 official dissolution tests that used Apparatus 1.
(USP 1-May-2021) The special sensitivity of the dissolution test to changes in composition or method of manufacturing that do not result in signi cant changes in performance in vivo is well recognized. An understanding of the full complement of information given by in vitro and in vivo evaluation of the drug substance and product is the starting point in the development of a meaningful in vitro performance test.
Change to read:
4 IN VITRO CHARACTERIZATION
For the purpose of an IVIVC, the goal of the in vitro characterization is to understand the performance of the formulation under different conditions and to understand the CQAs that influence the in vivo performance of the dosage form. In order to best characterize the performance of the dosage form, an understanding of the drug substance physicochemical properties is needed.
5 Drug Substance Properties
With the goal of achieving an IVIVC, the required intrinsic physicochemical information will include the pH-solubility profile, stability in the gastrointestinal environment, and other dissolution-controlling variables of the drug substance that are related to the extremes of the physiological environment experienced by the dosage form. Permeability, another drug substance property, can be determined in vitro by cellular permeability models such as Caco–2 cells.
6 Drug Product Properties
The purpose of in vitro performance testing is to challenge the dosage forms under different conditions to predict the in vivo performance.
The primary characterization is related to the rate-determining mechanism with different considerations for each release pattern. Immediate-release dissolution is a stochastic process but extended-release and delayed-release have intentional modifications that enable the performance characteristics. The variables of importance for immediate-release would include but not be limited to wetting, disintegration, disaggregation, erosion, and dissolution of particles in suspension. In addition to factors critical to the in vivo performance of immediate-release formulations, the key variables of importance for delayed-release are onset of the dissolution and pH dependency. The rate- controlling mechanisms of importance for extended-release include but are not limited to erosion, diffusion, swelling, and osmotic pressure.
Identification of the rate-limiting steps in the dissolution process that can be linked to in vivo performance is of great importance.
7 Design of In Vitro Characterization
The goals of the in vitro characterization studies are topographical characterization of the in vitro performance of the dosage and evaluation of dosage form robustness. A robustness evaluation would include such aspects as effect of the pH of the gastrointestinal environment, effect of mechanical factors (e.g., agitation), and effects of food components and alcohol.
8 Experimental Variables
Experimental variables of the dissolution method parameters to consider in characterizing the dosage form include the media pH value, volume and composition of the dissolution medium, and agitation (hydrodynamic). Also use of different dissolution apparatus should be considered as a potential experimental variable.
The media composition may contain surfactants, lipids, or enzymes. Organic solvents may be added to the dissolution medium with appropriate justification. There should be consideration of the osmotic pressure of the dissolution medium used (e.g., compendial buffers are usually hypoosmotic compared to physiological values). See Osmolality and Osmolarity 〈785〉 for additional information.
9 Experimental Design
Experimental design should be refiective of the physiological conditions prevailing within the gastrointestinal tract. The variables may be constant throughout each experiment or change along the time course of the experiment. The experimental setup could stipulate parallel or sequential design. One should consider the combinations of experimental variables in the gastrointestinal tract such as pH and compositions as they would occur in human (or veterinary) gastrointestinal physiology. The addition of surfactants and enzymes may also be considered. Conversely the setup may be simply run at a typical intestinal pH value. Other combinations of experimental parameters may be defined according to a factorial design or other statistical methods.
10 Technical Requirements
One major criterion for selecting the dissolution method should be its discriminatory power to show changes in the critical attributes of the release mechanism of the chosen formulation.
The in vitro characterization should include a sufficient number of data points to support pivotal phases of dissolution kinetics and data points that provide a full profile with nearly if not complete dissolution (100% release). Analytical validation of the method is required. Results should be based on an adequate number of samples for regulatory acceptance. For example, a sample of 12 is needed.
The selected dosage form then will be varied—such as by composition, by critical material attributes (e.g., drug substance particle size), and by manufacturing process—so there are different formulations for in vitro and in vivo testing that represent different levels of drug release in vitro and in vivo. These different levels should show rank order at a minimum, that is rank order of the fastest in vitro result corresponds to the dosage form that shows the highest rate of absorption, and so forth with the slowest in vitro results to the slowest absorption rate (see Figure 1). The IVIVC is for a particular formulation and cannot be applied to other formulations. Ideally, the dissolution method used to establish the IVIVC should be identical to the regulatory dissolution method used for routine quality control testing. In general, a quality control method that is not based on an IVIVC is sensitive to differences of in vitro performance, such as changes in manufacturing, which may not have in vivo significance. An IVIVC method is based on differences in in vivo performance. To prove the discriminatory power and to set a meaningful specification, the IVIVC needs to be valid for critical formulation batches.
As the method development evolves, the guidance in 〈1092〉 is essential. In early drug product development, the dissolution method may be complex attempting to explain the in vivo performance. As the understanding of the product evolves, the method should be robust and eventually suitable for the regulatory dissolution test.
(USP 1-May-2021)
Change to read:
11 IN VIVO EVALUATION OF DOSAGE FORMS
In evaluating a drug product's performance, analysts fundamentally must ask what type of study should be performed to give reasonable assurance of bioequivalence (USP 1-May-2021) to the clinical trial product that demonstrated safety and efficacy. 1 (USP 1-May-2021)
Although they provide important information concerning the release characteristics of the drug from the dosage form, in vitro dissolution studies at present are used primarily for setting or supporting specifications for drug products (e.g., shelf life) and manufacturing process control (e.g., scale-up or postapproval changes).
(USP 1-May-2021)
The following discussions are intended to provide guidance for drug substance evaluation and the design, conduct, and evaluation of studies involving dosage forms. Although these guidelines focus on oral drug delivery systems, the principles may be applicable to other routes of drug administration (e.g., transdermal, subcutaneous, intramuscular).
Animal data can be used only for veterinary products and not for establishing an IVIVC for human drug products based on animal models.
12 The Biopharmaceutics Classification System (BCS)
FDA has issued a guidance document titled Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System (see www.fda.gov/regulatory-information/search-fda-guidance-documents/waiver-vivo-bioavailability-and-bioequivalence-studies-immediate-release-solid-oral-dosage-forms). An additional guidance document from FDA is M9 Biopharmaceutics Classification System-Based Biowaivers (see www.fda.gov/media/117974/download).
Pharmacokinetic Profiling
Analysts should thoroughly characterize the input absorption profile of the drug substance from a formulation that shows a rapid absorption rate (an oral solution, or a well-characterized immediate-release drug product). An intravenous solution provides instantaneous and complete absorption and is required by the Loo-Riegelman model for level A correlations. In turn, this formulation serves as a reference to evaluate the input profile of the modied-release dosage form. This information, together with the pharmacokinetics of the drug, can characterize drug absorption and can predict changes in drug absorption rate when input is modified as in modified-release dosage forms.
For example, if the drug exhibits saturable first-pass hepatic metabolism, a change in systemic availability could result after oral administration if the input rate is altered.
The information required may include the following.
1. Pharmacokinetic parameters–clearance, area under the time–plasma concentration curve (AUC), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), volume of distribution, half-life, mean residence time, or model-dependent parameters
2. Linearity or characterization of nonlinearity over the dose or concentration range that could be encountered
3. Drug/active metabolite accumulation
4. Metabolic profile and excretory pathway, with special attention to the active metabolites and active enantiomers of racemic mixtures
5. Enterohepatic circulation
6. Protein-binding parameters and effect of dialysis
7. The effects of age, gender, race, and relevant disease states
8. Plasma/blood ratios
9. Information on chronopharmacokinetics (USP 1-May-2021)
Delete the following:
13 CHARACTERIZATION OF DRUG SUBSTANCE
13.1 The Biopharmaceutics Classification System (BCS)
FDA has issued a guidance titled “Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System”
(www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070246.pdf). A key assumption in the approach is that drug release and dissolution is suficiently rapid so that an in vitro–in vivo correlation is not possible and/or useful. When applicable, the BCS allows dissolution rate data in lieu of BA or BE studies for product approval.
Pharmacokinetic Properties
Analysts should thoroughly characterize the input absorption profile of the active drug entity from a formulation that shows rapid BA (an intravenous solution, oral solution, or a well-characterized immediate-release drug product). In turn, this formulation serves as a reference to evaluate the input profile of the modified-release dosage form. This information, together with the pharmacokinetics of the active drug entity, can characterize drug absorption and can predict changes in drug BA when input is modified as in modified-release dosage forms. For example, if the active drug entity exhibits saturable first-pass hepatic metabolism, a reduction in systemic availability could result after oral administration if the input rate is decreased.
In designing an oral modified-release dosage form, analysts may find it useful to determine the absorption of the active drug entity in various segments of the gastrointestinal tract, particularly in the lower gastrointestinal tract (colon) for delayed-release dosage forms that release drug in this region. Food effects also may be important and should be investigated.
13.2 Drug Disposition
The information required to characterize drug disposition may include the following.
1. Disposition parameters—clearance, area under the time—plasma concentration curve (AUC), maximum plasma concentration (Cmax), time to maximum plasma concentration (Tmax), volume of distribution, half-life, mean residence time, or model-dependent parameters.
2. Linearity or characterization of nonlinearity over the dose or concentration range that could be encountered.
3. Drug/metabolite accumulation.
4. Metabolic profile and excretory pathway, with special attention to the active metabolites and active enantiomers of racemic mixtures.
5. Enterohepatic circulation.
6. Protein-binding parameters and effect of dialysis.
7. The effects of age, gender, race, and relevant disease states.
8. Plasma: blood ratios.
9. A narrow therapeutic index or a clinical response that varies significantly as a function of the time of day (chronopharmacokinetics).
13.3 Pharmacodynamic Properties
Before developing a dosage form, analysts should obtain concentration–response relationships over a dose range sufficiently wide to encompass important therapeutic and adverse responses. In addition, the equilibration-time2
characteristics between plasma concentration and effect should be evaluated. For modified-release products that typically have larger drug doses in the dosage form, these concentration–esponse relationships should be suficiently characterized so that a reasonable prediction of the safety margin can be made if dose dumping should occur. If there is a well-defined relationship between the plasma concentration of the active drug substance or active metabolites and the clinical response (therapeutic and adverse), the clinical performance of a new modified-release dosage form could be characterized by plasma concentration–time data. If such data are not available, clinical trials of the modified-release dosage form should be carried out with concurrent pharmacokinetic and pharmacodynamic measurements.(USP 1-May-2021)
Change to read:
14 CHARACTERIZATION OF THE ORAL (USP 1-May-2021) DOSAGE FORM
14.1 Pharmacokinetic Properties: Immediate-Release Products
The types of pharmacokinetic studies that should be conducted are based on how much is known about the drug, (USP 1-May-2021) its clinical pharmacokinetics, and its BCS class (USP 1-May-2021) when an FDA-approved drug product undergoes changes in the manufacturing of the product after the product has been approved. Such changes are common and can be caused by expansion in the size of the lots manufactured, new manufacturing locations, or the introduction of new technology. Necessary in vitro dissolution tests and/or in vivo bioequivalence tests are described in the FDA guidance SUPAC-IR: Immediate-Release Solid Oral Dosage Forms: Scale-Up and Post-Approval Changes: Chemistry, Manufacturing and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation (see www.fda.gov/regulatory-information/search-fda-guidance-documents/supac-ir-immediate-release-solid-oral-dosage-forms-scale-and-post-approval-changes-chemistry).(USP 1-May-2021)
14.2 Pharmacokinetic Properties: Modified-Release Products
Similar to immediate-release products, (USP 1-May-2021) the types of pharmacokinetic studies that should be conducted for modied-release products are based on how much is known about the drug, (USP 1-May-2021) its pharmacokinetics, and biopharmaceutics, and whether pharmacokinetic studies are intended to be the sole basis for product approval. At a minimum, two studies are required to characterize the product when no reference modified-release product exists: (1) a single-dose crossover study for each strength of a modified-release dosage form and (2) a multiple-dose, steady-state study using the highest strength of a modified-release dosage form. A
(USP 1-May-2021) study to evaluate the potential for food effects including robustness of the dosage form (including dose dumping) from extended-release dosage forms also is required as a separate study or is included as an arm of a crossover study. Oral solutions, or well-characterized immediate-release drug products are possible reference products to evaluate a modified-release formulation. If the drug exhibits saturable first-pass hepatic metabolism from the small intestine, a reduction in systemic availability could result after oral administration if the input rate is decreased. An increase in systemic availability could be observed if a drug is absorbed from the colon from a delayed-release dosage form that targets the colon, thus avoiding a first-pass effect.
PK studies are required for IVIVC of modified-release dosage forms. The pharmacokinetic studies may serve as the basis for characterization of the dosage form. Techniques to develop IVIVC assume linear pharmacokinetics in the dosing range. Manufacturers seeking regulatory approval without conducting clinical trials should consult the authorities to ensure that an adequate database of safety and efficacy information exists. These studies also apply to an FDA-approved product that has undergone scale-up and postapproval changes (SUPAC). Necessary in vitro dissolution tests and/or in vivo bioequivalence tests are described in the FDA guidance SUPAC-MR:
Modified Release Solid Oral Dosage Forms: Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution
Testing and In Vivo Bioequivalence Documentation (www.fda.gov/regulatory-information/search-fda-guidance-documents/supac-mr-modified-release-solid-oral-dosage-forms-scale-and-postapproval-changes-chemistry). (USP 1-May-2021)
Change to read:
15 IN VITRO–IN VIVO CORRELATIONS
The term "IVIVC" first appeared in the pharmaceutical literature as a result of the importance of identifying the relationship between bioavailability parameters and in vitro dissolution of a dosage form. (USP 1-May-2021) IVIVC refers to the establishment of a mathematical (USP 1-May-2021) relationship between (USP 1-May-2021) a parameter derived from drug plasma concentrations produced by a dosage form, and a dissolution (USP 1-May-2021) property or characteristic of the same dosage form. The bioavailability parameters (USP 1-May-2021) most commonly used are one or more pharmacokinetic parameters such as C or AUC, obtained following the administration of the dosage form. The in vitro (USP 1-May-2021) property most commonly used is a dosage form's in vitro dissolution profile (USP 1-May-2021) (e.g., percent of drug released under a given set of conditions versus time).
In vitro–in vivo correlation (IVIVC): A predictive mathematical model describing the relationship between an in vitro property of an extended-release dosage form (usually the rate or extent of drug dissolution or release) and a relevant in vivo response (e.g., plasma drug concentration or amount of drug absorbed). (USP 1-May-2021)
In many cases the actual drug plasma concentration profile can be predicted from in vitro dissolution data.
IVIVC analysis is most successful for oral drug products, where compared to in vivo absorption, in vivo dissolution is the rate-limiting step for bioavailability. For extended-release products the dissolution rate is intentionally designed so that in vivo dissolution is the rate-filimiting step for bioavailability. Some correlations with immediate-release products can be expected where in vivo dissolution is the rate-filimiting step. As described in the BCS, solubility and permeability classification are helpful in assessing feasibility of IVIVC with immediate-release products. Taking into account the changes in the absorption rate along the gastrointestinal transit, non-linear regression analysis may be useful.(USP 1-May-2021)
15.1 General Considerations
In an ideal case, correlations should be generated by using the entire profiles of both in vitro dissolution testing and plasma concentrations from bioavailability studies. A point-to-point data analysis allows establishment of a functional relationship, which does not imply a full correlation. One formulation of the product under investigation suffices for that determination. Data analysis performed with in vitro and in vivo parameters require at least three formulations to establish a correlation. Three correlation levels are available. Each correlation level displays important differences in the quality of the correlation. This chapter provides a discussion of each type of correlation as to when each may be the most appropriate type of relationship to establish and then to describe its potential utility within the framework of product development, the development of release specifications, and its regulatory utility in supporting biowaivers. Typically, one target formulation and one or two alternative formulations with expected higher and/or lower drug release are included in the IVIVC study protocol.
If feasible an oral solution is administered to a selected number of subjects in order to compute the relative oral bioavailability and to perform a model-free numerical deconvolution procedure. The target formulation will be used for the correlation development, and it should be near a commercial form, although further modifications may be planned. This allows the task of developing an IVIVC to occur early enough in product development so that it helps in predicting the in vivo linkage and minimizes bio studies, and it also has a role in specification selection. If the IVIVC is established too early in the drug product development process, the formulation used in the study may not be representative of the final dosage form.(USP 1-May-2021)
15.2 Correlation Levels
Three correlation levels have been defined and categorized in descending order of quality. The concept of correlation level is based on the ability of the correlation to reflect the entire plasma drug concentration–time curve that results from administration of the given dosage form. The relationship of the entire in vitro dissolution curve to the entire plasma concentration–time profile defines the strength of the correlation and, therefore, the predictability. If permeability rather than dissolution is rate limiting, or if there is solubility-limited absorption, the development of an IVIVC/R should not be attempted.(USP 1-May-2021)
Level A
This level is the highest category of correlation. It represents a point-to-point relationship between in vitro dissolution and the in vivo input rate (e.g., absorption rate of the drug from the dosage form). For a Level A correlation, a product's in vitro dissolution curve is compared to its in vivo input curve (i.e., the curve produced by deconvolution of the plasma profile). Deconvolution can be accomplished using mass balance model-dependent methods, such as the Wagner-Nelson or Loo-Riegelman methods, or by model-independent, mathematical deconvolution (linear system analysis). The model-dependent mass balance approaches do not allow for prediction of oral bioavailability, whereas the numerical deconvolution requires linear pharmacokinetics and is based on the assumption of absorption and elimination processes being invariant over time.
In an ideal correlation, the in vitro dissolution and in vivo absorption rate curves are superimposable or can be made superimposed by the use of a constant offset value or a multiplier of the time scale. The equations describing each curve are the same or stand in a linear relationship. Superimposition is not an absolute requirement for a Level A correlation. If the dissolution and absorption curves are different and a mathematical relationship can be developed to relate the two, the plasma level profile is predictable from the in vitro dissolution data.
This is called an internal validation. If the same correlation is used to predict the plasma profile of a lot not used to establish the IVIVIC; this is an external validation. This relationship must be true not only at that single input rate but also over the entire quality control dissolution range for the product, provided that the in vitro dissolution profiles are similar in shape (i.e., they can be made superimposable by multiplication).(USP 1-May-2021)
The advantages of a Level A correlation are as follows.
1. It develops a point-to-point correlation. This is not found with any other correlation level. It is developed using every plasma level and dissolution point collected at different time intervals, so it reflects the complete plasma level curve. A Level A correlation is reversible and allows computation of in vivo profiles from in vitro profiles and vice versa.(USP 1-May-2021) As a result, in the case of a Level A correlation, an in vitro dissolution test (USP 1-May-2021) can serve as a surrogate for in vivo performance studies. (USP 1-May-2021) A change in manufacturing site, method of manufacture, raw material supplies, minor formulation modifications, and even product strength using the same formulation can be justified without the need for additional bioavailability–bioequivalence (BA–BE) studies.2,3
2. A truly meaningful quality control procedure that indicates in vivo performance and is predictive of a dosage form's performance is fide fined for the dosage form because specifications may be set in a way to assure bioequivalence. (USP 1-May-2021)
3. The extremes of the in vitro quality control standards can be justified (USP 1-May-2021) by deconvolution (using the upper and lower confidence interval limits). Interpolation of specifications from the so-called side batches is a commonly accepted way, whereas extrapolation requires further efforts.(USP 1-May-2021)
Level B
This correlation uses the principles of statistical moment analysis. The mean in vitro dissolution time is compared to either the mean residence time or the mean in vivo dissolution time. As with a Level A correlation, Level B uses all of the in vitro and in vivo data but is not considered a point-to-point correlation. It does not correlate the actual in vivo plasma profiles but rather parameters that result (USP 1-May-2021) from statistical moment analysis of a plasma profile, (USP 1-May-2021) such as mean residence time. Because a number of different plasma profiles can produce similar mean residence time values, one cannot rely on a Level B correlation alone to predict a plasma profile from in vitro dissolution data. The advantage of a Level B correlation is its applicability for non-linear pharmacokinetics.(USP 1-May-2021) In addition, in vitro data from such a correlation can (USP 1-May-2021) be used to justify values at the extremes of quality control standards, if all in vitro profiles are similar in shape and the in vivo performance of the side batches are fully included in the confidence intervals defining bioequivalence.(USP 1-May-2021)
Level C
This category relates one dissolution time point (e.g., t50%, t90%) to one pharmacokinetic parameter such as AUC, Cmax, or Tmax. It represents a single-point correlation and does not reflect the complete shape of the plasma profile. Level C correlations do not reflect the shape of the profiles; therefore, Level C correlations cannot be used to predict plasma profiles.(USP 1-May-2021) Because this type of correlation is not predictive of the full rate and extent of in vivo product dissolution characteristics, it generally serves primarily as a guide in formulation development or as a production quality control procedure.(USP 1-May-2021) Because of its obvious limitations, a Level C correlation has limited usefulness in predicting in vivo drug performance and is subject to the same caveats as a Level B correlation in its ability to support product and site changes as well as justification of the extreme values in quality control standards. The advantage of a Level C correlation is that it selects parameters from the in vivo pro files, which are of a therapeutic relevance.(USP 1-May-2021) The FDA guidance Extended Release Solid Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations (www.fda.gov/regulatory-information/search-fda-guidance-documents/extended-release-oral-dosage-forms-development-evaluation-and-
application-vitroin-vivo-correlations(USP 1-May-2021) states that manufacturers can obtain biowaivers based on multiple Level C correlations. The guidance shows how manufacturers can achieve this correlation. (USP 1-May-2021)
15.3 Developing a Correlation
This chapter does not define the only procedures for developing an IVIVC, and any well-designed and scientifically valid approach is acceptable. To assist the pharmaceutical scientist, one possible procedure for developing a Level A correlation follows:
1. In order to perform deconvolution properly, analysts should be familiar with the pharmacokinetics of the drug substance.(USP 1-May-2021)
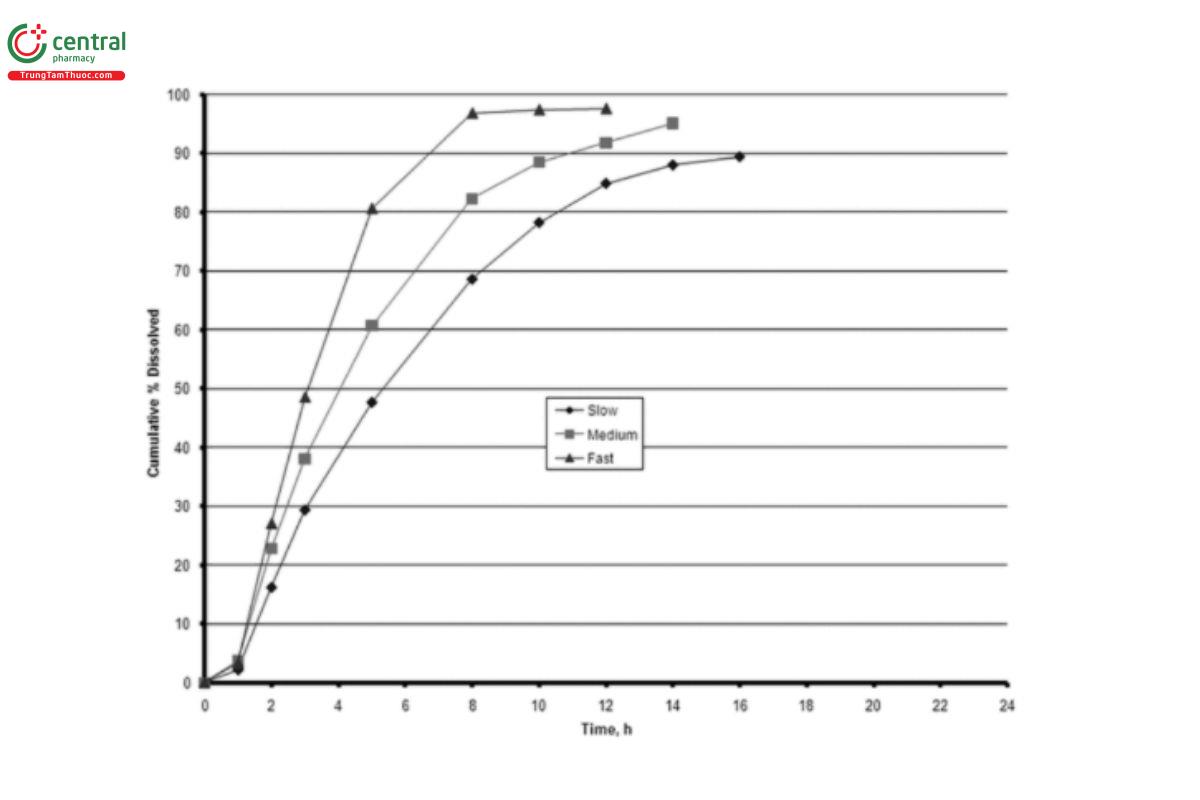
2. The formulation should be modified to produce different dissolution profiles so that the formulation has the same excipients in all the lots that will be tested. The products under investigation should be pharmaceutically equivalent. The formulation modifications used should be based on factors that would be expected to influence the product's modified-release rate and could occur during normal product manufacture or represent stability effects. In vitro drug release is performed on the formulation that will be used in the bioavailability study. Figure 2 illustrates an example of dissolution profiles in which the formulations were tested in an acid buffer. (USP 1-May-2021)
rpm, 900 mL, (USP 1-May-2021) pH 4.5 buffer, 37°).
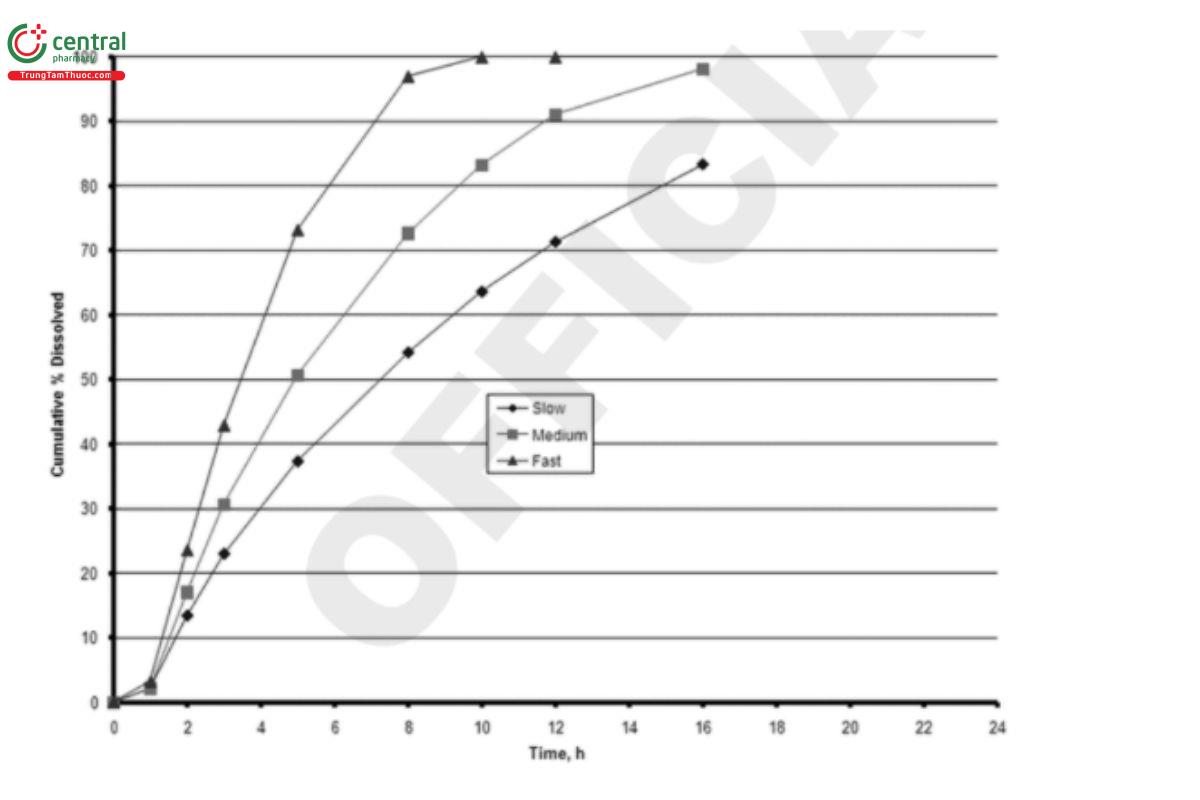
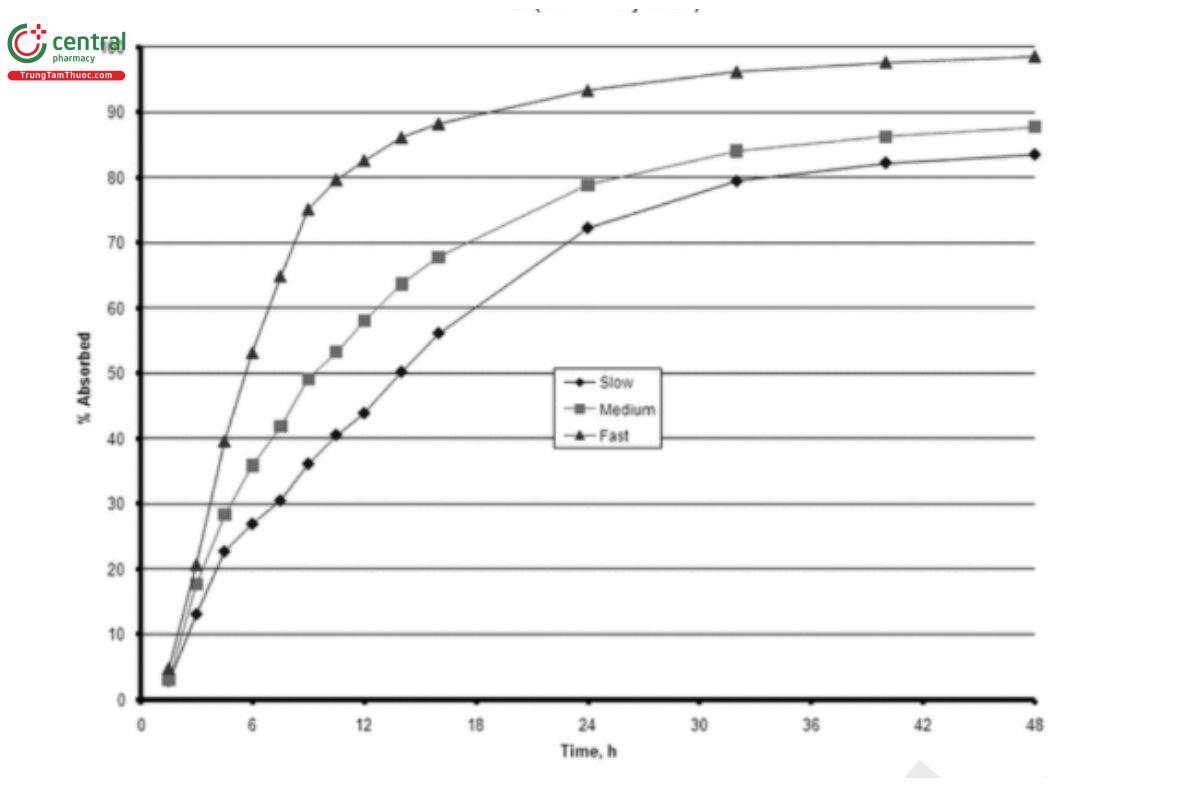
3. The plasma levels (USP 1-May-2021) obtained in the definitive bioavailability study of the modified-release dosage form are treated by a deconvolution procedure. The resulting data may represent the apparent (USP 1-May-2021) drug input rate of the dosage form. They also represent in vivo dissolution when the rate-controlling step of the dosage form is its dissolution rate (i.e., drug absorption after dissolution is considered to be instantaneous). Any deconvolution procedure (e.g., mass balance or mathematical deconvolution) will produce acceptable results. However, the results of mass balance algorithms versus numerical deconvolution are not comparable.(USP 1-May-2021) Figure 3 illustrates the results of numerical deconvolution of the plasma profiles (USP 1-May-2021) obtained for the formulation in (USP 1-May-2021) Figure 2. The plasma levels obtained in the definitive bioavailability study of the modified-release dosage form are analyzed by using a deconvolution procedure. The resulting data may represent the apparent drug input rate of the dosage form. The deconvolution represents in vivo dissolution when the rate-controlling step for oral drug absorption is its dissolution rate (i.e., drug absorption after dissolution is considered to be instantaneous). Any deconvolution procedure (e.g., mass balance or mathematical deconvolution) will produce acceptable results if properly validated. However, the results of mass balance algorithms versus numerical deconvolution are not comparable. Figure 3 illustrates the results of numerical deconvolution of the plasma profiles obtained for the formulation in Figure 2.(USP 1-May-2021)
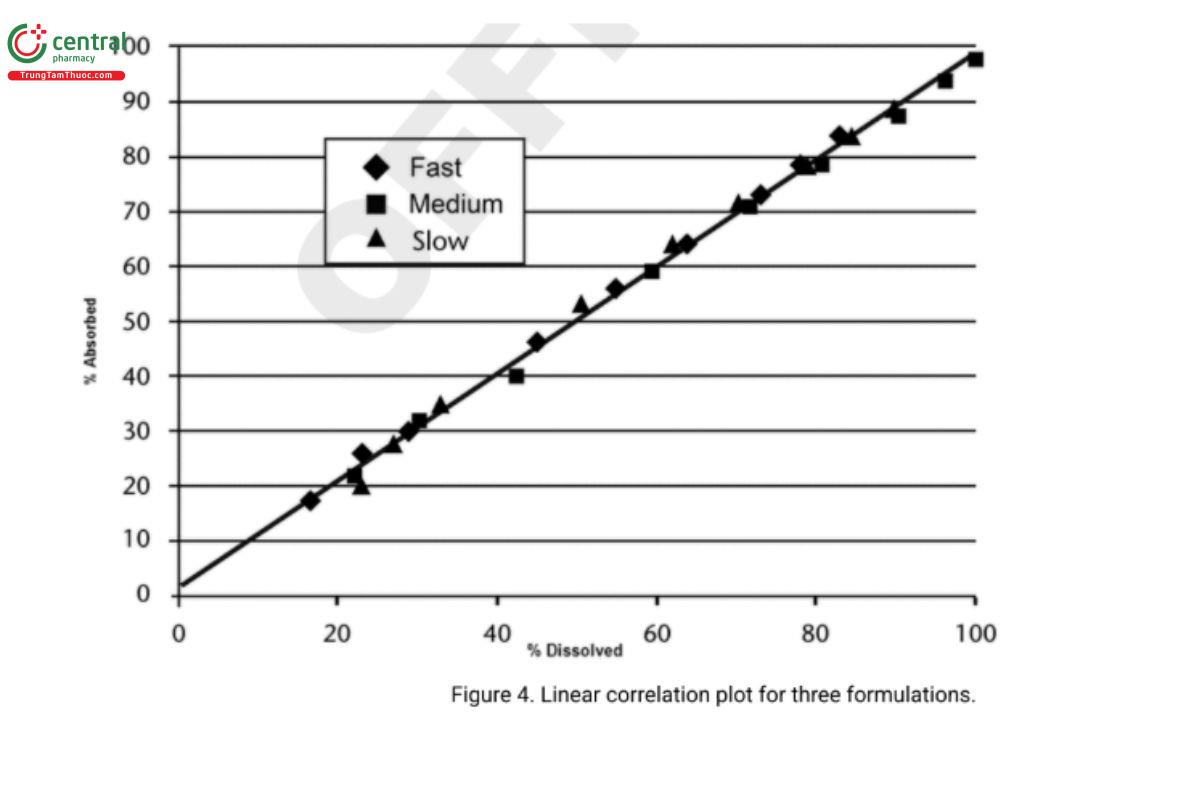
The in vitro dissolution curve is then compared to the drug absorption curve. This can be performed by various methods. Simply positioning one curve on the other often can indicate the existence of a correlation, which then may be quantified by defining the equation for each curve and comparing the corresponding constants. The simplest way to demonstrate a correlation is to plot the fraction absorbed in vivo versus the fraction released in vitro, as illustrated in Figure 4. With a Level A correlation, this relationship is often linear, as illustrated in Figure 4. Alternatively, as illustrated in Figure 5, a correlation may be curvilinear. The intercept may or may not be zero depending on whether there is a lag time before the system begins to release the drug in vivo, or the absorption rate is not instantaneous, resulting in the presence of some finite quantity of dissolved but unabsorbed drug. In either case, it is a point-to-point or a Level A correlation when the least-squares fit of the line approaches a coefficient of determination, r2, of 1.
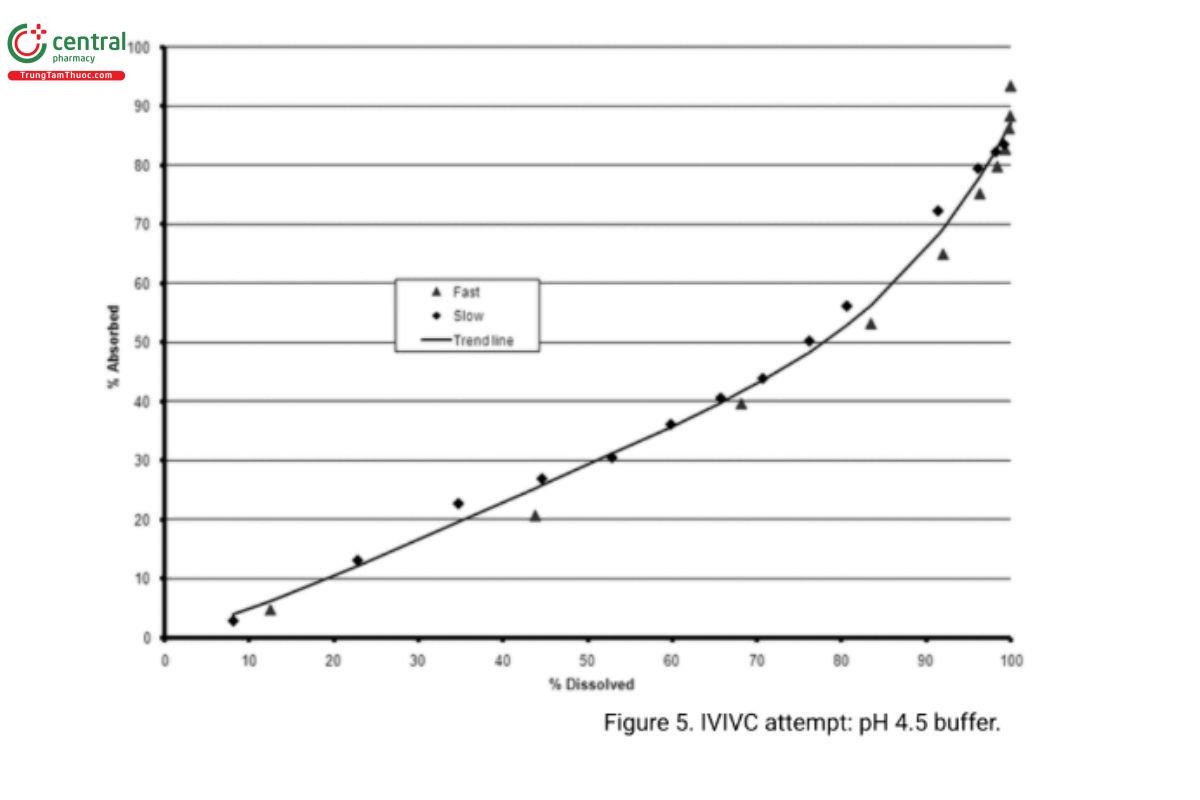
4. A discriminative in vitro method establishes that in vitro is predictive of in vivo absorption differences. This is achieved by preparing at least two formulations that have significantly different in vitro behavior. One should demonstrate a more rapid release while the other should be a slower release than the clinical bioavailability batch (or biobatch). A pilot BA–BE study should be performed with these formulations, and the previously established correlation should be demonstrated for both. The modifications of these formulations should be based on factors that are expected to influence the product's drug release rate.
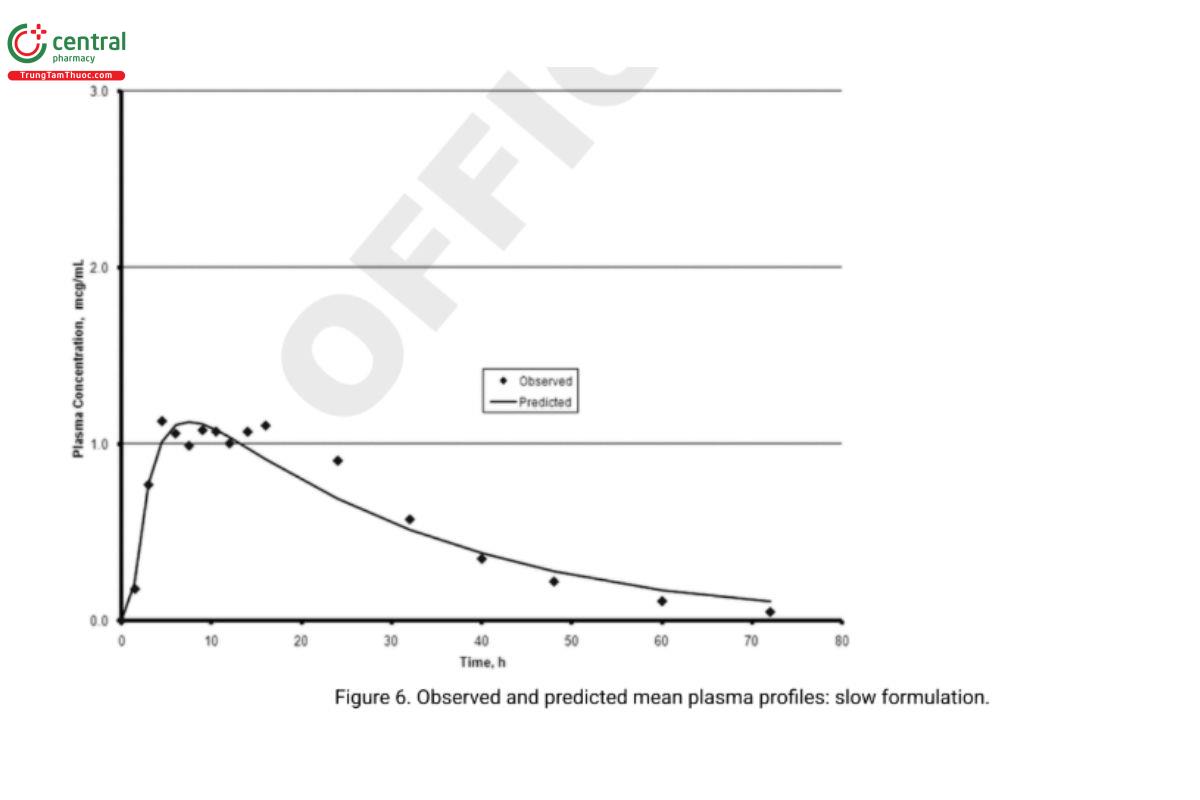
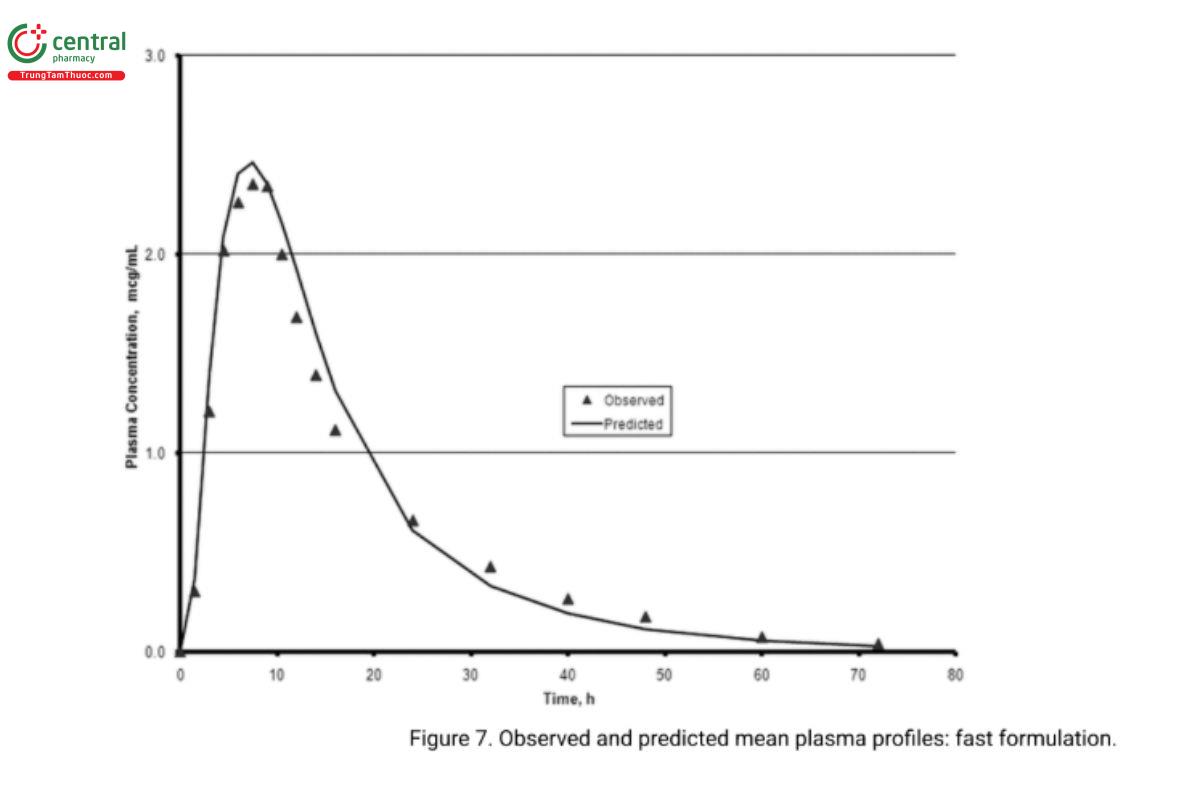
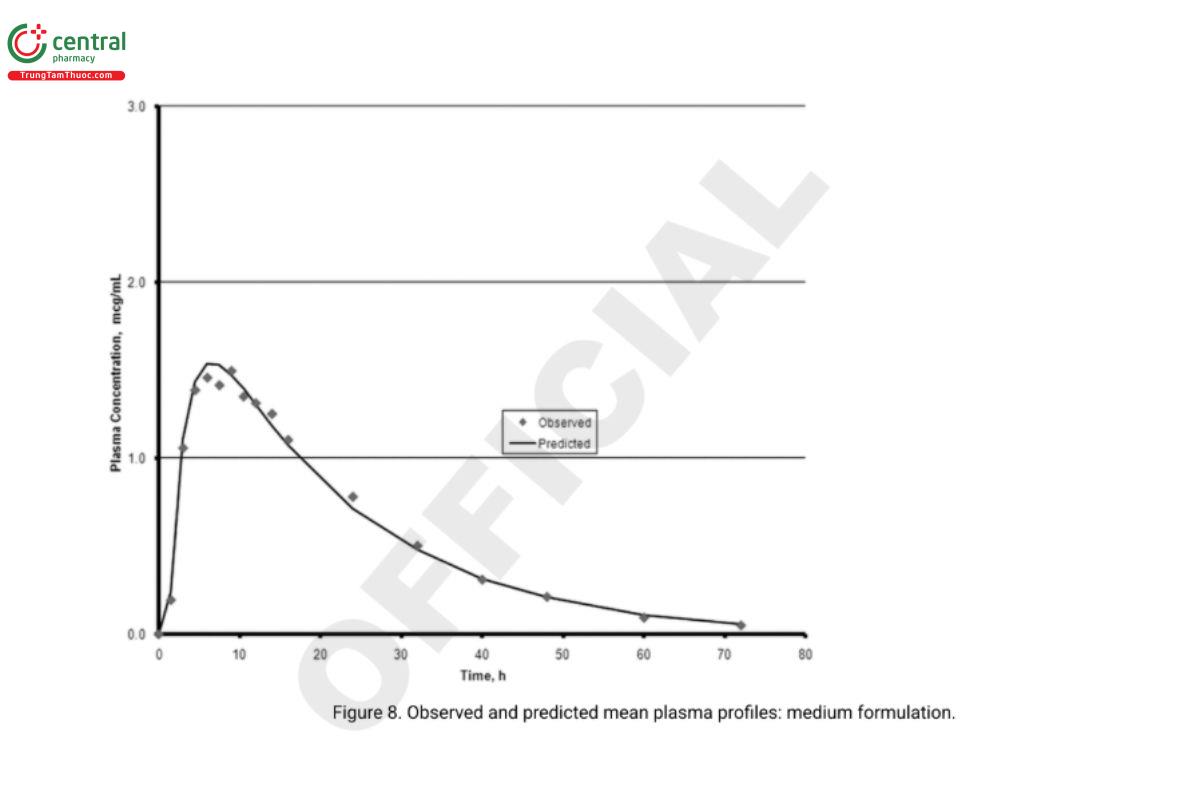
5. In vivo performance of the two biobatch formulations can be computed based on the correlation developed with the formulations that were used in the BA–BE study. Analysts can then compare the predicted and experimentally determined values (e.g., the prediction error). Figure 6 and Figure 7 illustrate an exercise that serves as an internal validation of the Level A correlation. An external validation would involve computing the drug plasma profile and comparing it with the observed plasma profiles for a formulation not included in the Level A correlation calculations. Figure 8 illustrates the results of such a validation using the in vivo data from the formulation of the lot ultimately selected. Once a Level A correlation is established, in vitro testing can be used to justify dissolution specifications and biowaivers.
15.4 Establishment of Dissolution Specification Ranges
When establishing (USP 1-May-2021) a multipoint dissolution specification for a modified-release dosage form, the dissolution behavior of the biobatch can be used to define the amount that will be released at each time point. The dificulty arises in the variation that will be allowed around each time point. In the case of a Level A correlation, this can be done in two ways, both of which use IVIVC: convolution and deconvolution. convolution
Reasonable upper and lower dissolution values are selected for each time point established from the biobatch. The dissolution curves defined by the upper and lower limits (USP 1-May-2021) are convoluted to project the anticipated plasma level curves that would result from administration of these formulations to the same patients to whom the biobatch was administered. If the resulting plasma level data fall within the 90% (USP 1-May-2021) confidence intervals obtained in the definitive BA–BE study, these ranges can be considered acceptable.
15.5 Deconvolution
An acceptable set of plasma-level data is established both for a formulation (USP 1-May-2021) demonstrating a more rapid release and for one demonstrating a slower release than that of the biobatch. These can be selected by using the extremes of the 90% (USP 1-May-2021) confidence intervals or ±1 standard deviation of the mean plasma level. These curves are then deconvoluted, and the resulting input rate curve is used to establish the upper and lower dissolution specifications at each time point. In the case of Level B and C correlations, formulations (USP 1-May-2021) must be made at the proposed upper and lower limits of the dissolution range, and it must be demonstrated that these formulations (USP 1-May-2021) are acceptable by a BA–BE study.
15.6 Considerations for (USP 1-May-2021) Immediate-Release Dosage Forms
Because the mechanisms for drug release from modified-release dosage forms are more complex and variable than those associated with immediate-release dosage forms, one would anticipate that an IVIVC would be easier to develop with the latter formulations. Unfortunately, most of the correlation efforts to date with immediate-release dosage forms have been based on the correlation Level C approach, although there also have been efforts employing statistical moment theory (Level B). Although it is conceivable that the same Level A correlation approach can be used with immediate-release dosage forms, until data have been gathered to support this concept, Level B and Level C are the best approaches that can be recommended with these dosage forms.
Add the following:
15.7 GLOSSARY
Biobatch:
The lot of drug product formulated for purposes of pharmacokinetic evaluation in a bioavailability/bioequivalency study. For modified-release solid oral products, this batch should be 10% or greater than the proposed commercial production batch or at least 100,000 units, whichever is greater.
Biopredictive dissolution method:
A set of testing conditions for which in vitro dissolution profiles are capable of predicting pharmacokinetic profiles. These are typically based on classical or mechanistic IVIVC.
Biorelevant dissolution method:
A set of testing conditions for monitoring in vitro dissolution designed to closely mimic a relevant biological fluid and physiological environment.
Clinically relevant dissolution specification:
A set of in vitro dissolution testing conditions and acceptance criterion(ia) that can identify and reject drug product batches that are not expected to be bioequivalent to clinical pivotal product batches.
Discriminating dissolution specifications:
A set of in vitro dissolution testing conditions that, along with the acceptance criterion(ia), are able to differentiate drug products manufactured under target conditions versus drug products that are intentionally manufactured with meaningful variations (i.e., formulation and manufacturing variants) for the relevant manufacturing variables (e.g., drug substance particle size, compression force, tablet hardness).
In vitro–in vivo correlation (IVIVC):
A predictive mathematical model describing the relationship between an in vitro property of an extended release dosage form (usually the rate or extent of drug dissolution or release) and a relevant in vivo response (e.g., plasma drug concentration or amount of drug absorbed).
In vitro–in vivo relationship (IVIVR):
An IVIVR is a relationship which simply states that an in vitro change will result in an in vivo change (in rank order), but the amount of change is not mathematically predictable.
Modified release:
A dosage form release pattern when the rate and/or time of release of the drug substance is altered as compared to what would be observed or anticipated for an immediate-release product. Two modified-release profiles, delayed-release and extended-release, are recognized.
Pharmaceutically equivalent:
Drug products containing the same active ingredient(s), having the same route of administration, and with identical strength or concentration of the same dosage form.
Percent prediction error: Calculated as [(observed value − predicted value)/observed value] × 100.
Predictability:
Verification of the model's ability to describe in vivo bioavailability results from a test set of in vitro data (external predictability) as well as from the data used to develop the correlation (internal predictability).
Prediction error:
A measure of the prediction by the IVIVC model of in vivo performance from in vitro dissolution data. (USP 1-May-2021)
1 21 CFR 320.22 Criteria for waiver of evidence of in vivo bioavailability or bioequivalence.
2 Equilibration time is a measure of the time-dependent discontinuity between measured plasma concentrations and measured effects. The discontinuity is more often characterized by the degree of hysteresis observed when the effect-concentration plot for increasing concentrations is compared with that for decreasing concentrations. Where the equilibration time is very short (i.e., rapid equilibration with no active metabolites generated), there will be little or no hysteresis. That is, the same effect will be observed for a given concentration independent of the interval between the time of dosing and the time that measurements are made.
2 FDA guidance SUPAC-MR: Modified Release Solid Oral Dosage Forms: Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation (1997).
3 FDA guidance Extended Release Solid Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations: “If an IVIVC is developed with the highest strength, waivers for changes made on the highest strength and any lower strengths may be granted if these strengths are compositionally proportional or qualitatively
