IMPURITIES IN DRUG SUBSTANCES AND DRUG PRODUCTS
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
DOWNLOAD PDF HERE
1 INTRODUCTION
Impurities are critical quality attributes of drug substances and drug products because they have the potential to affect the safety and efficacy of the product. This general information chapter provides guidance on the control of impurities in drug substances (process-related impurities and degradation products) and drug products (degradation products). (Definitions of key terms used in this chapter can be found in the Glossary).
This chapter covers drug substances and drug products described in the USP–NF. This chapter addresses impurities arising during the manufacturing process and/or storage of the drug substance. Also the chapter covers those impurities in drug products classified as degradation products of the drug substance or reaction products of the drug substance with an excipient and/or immediate container–closure system (collectively referred to as “degradation products”).
This chapter does not cover veterinary products, biological/biotechnological products, peptides, oligonucleotides, fermentation products and semisynthetic products derived from them, polymorphic forms, radiopharmaceuticals, herbal products, or crude products of animal or plant origin. In addition, impurities in excipients and leachables from the container–closure system are out of the scope of this chapter.
The regulatory and compendial standards for the control of impurities continue to evolve due to advancements in analytical science, technology, and toxicology. Therefore, communications about impurities and degradation products in drug substances and drug products can be improved by including in this Pharmacopeia the definitions for terms and the contexts in which these terms are used (see the Glossary). See General Notices, 5.60. Impurities and Foreign Substances for additional information about impurities. Some other general chapters added over the years have also addressed topics of purity or impurity as these have come into focus or as analytical methodology has become available. Analytical aspects are enlarged upon in Chromatography 〈621〉, Validation of Compendial Procedures 〈1225〉, and Verification of Compendial Procedures 〈1226〉.
Impurity measurements for drug products using chromatographic methods could present a challenge to the Pharmacopeial standards setting due to low concentration of the impurity and complexity of matrix. As a consequence, many monographs for Pharmacopeial preparations rely on chromatographic assays. Where more significant impurities are known, some monographs set forth specific tests for these impurities. In general, however, this Pharmacopeia does not quantify in drug products impurities that are evaluated in drug substances and that are not expected to increase in the drug product during manufacturing or storage (i.e., impurities that are not degradation products).
It is assumed that adequate retention specimens are in storage for the exact batch of drug substances used in any specific lot of a drug product. Whenever analysis of a drug product raises a question of the quality attributes of any of the drug substances used, subsequent analysis of retention specimens is in order.
2 DRUG SUBSTANCE
Impurities in drug substances can be classified into the following categories:
1. Organic impurities
2. Inorganic impurities
3. Residual solvents
Organic impurities can arise during the manufacturing process and/or storage of the drug substance. They can be identified or unidentified, volatile or nonvolatile, and include the following:
1. Starting materials
2. Reaction byproducts
3. Intermediates
4. Degradation products
5. Reagents and ligands
Inorganic impurities can result from the manufacturing process. They are normally known and identified and include the following:
1. Reagents and catalysts
2. Elemental impurities
3. Inorganic salts
4. Other materials (e.g.,filter aids)
Elemental impurities can include catalysts and environmental contaminants that may be present in drug substances. These impurities may occur naturally, be added intentionally, or be introduced inadvertently (e.g., by interactions with processing equipment and the container–closure system). When elemental impurities are known to be present, have been added, or have the potential for introduction, assessment of the drug substance in the context of its use in the finished product is required (see Elemental Impurities—Limits 〈232〉).
Residual solvents originate from organic liquids used as vehicles in the synthesis of a drug substance, as starting material, or as the intermediates involved in the synthesis. Because these are generally of known toxicity, the selection of appropriate controls is easily accomplished (see Residual Solvents 〈467〉).
Concepts for setting impurity limits in drug substances are based on chemistry and safety concerns. As such, limits for organic and inorganic impurities and residual solvents should be established according to the applicable guidances. Elemental impurity limits for a drug substance may be required depending on the outcome of the risk assessment for the finished product. The basic tenet for setting limits is that levels of impurities in a drug substance must be controlled throughout its development to ensure its safety and quality for use in a drug product.
Documented evidence that the analytical procedure used to evaluate impurities is validated and suitable for the detection and quantitation of impurities should be established.
3 DRUG PRODUCT
The specification for organic impurities in a drug product should include a list of degradation products expected to occur during the manufacture of the commercial product and under the recommended storage conditions. Stability studies, forced degradation studies, knowledge of degradation pathways, product development studies, compatibility studies, and laboratory studies should be used to characterize the degradation profile. The selection of degradation products in the drug product specification should be based on the degradation products found in batches manufactured by the proposed commercial process.
The rationale for specifications should include a discussion of the degradation profiles observed in the safety and clinical development batches and in stability studies, together with a consideration of the degradation profile of batches manufactured by the proposed commercial process. For degradation products known to be unusually potent or to produce toxic or unexpected pharmacological effects, the quantitation/detection limit of the analytical procedures should be commensurate with the level at which the degradation products should be controlled.
For drug products, the concept for setting degradation product limits is based on sound scientific assessment as applied to available data on the safety and stability of the drug product, data that may include the degradation pathways of the drug substance, the manufacturing process, known excipient interactions, any safety assessment studies (e.g., predictive toxicology programs, mutagenicity assessments, and in vitro and in vivo toxicology studies), stability studies conducted under the recommended storage conditions, and ancillary studies that may provide additional information on the stability profile of the drug product. Impurities that are not degradation products (i.e., process-related impurities from the drug substance) are often not controlled in the drug product, as they are typically controlled in the drug substance and these impurities are not expected to increase over time. In cases where the studies show that impurities from the drug substance can increase over time (i.e., they are also drug product degradation products), they should be controlled in the drug substance and also in the drug product. Additional guidance for setting limits can be found in various International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) guidelines, such as ICH Q3A, Q3B, and M7; FDA guidance documents; and USP monograph submission guidelines.
Documented evidence that the analytical procedure used to evaluate impurities is validated and suitable for the detection and quantification of impurities should be established.
General chapter 〈232〉 specifies limits for the amounts of elemental impurities in drug products.
Drug products should contain no higher levels of residual solvents than can be supported by safety data (see 〈467〉).
4 ORGANIC IMPURITIES IN DRUG SUBSTANCES AND DRUG PRODUCTS
All drug substances and drug products are subject to control of organic impurities. A threshold-based approach described in the ICH Q3A and Q3B guidelines may be used for the control of organic impurities in drug substances or drug products generated during the manufacturing process or storage (see Control of Organic Impurities in Drug Substances and Drug Products 〈476〉 for additional information).
In this chapter, “impurity” can refer to process-related impurities and degradation products for a drug substance, and “degradation products” can refer to process-related impurities and degradation products for a drug product (see Glossary for additional information).
The organic impurities to be controlled in drug substances are the process-related impurities and degradation products. The organic impurities to be controlled in the drug product are those resulting from the degradation of the drug substance or from the interaction of the drug substance with excipients and/or the primary container–closure system. Drug substance process-related impurities need not be controlled in the drug product unless they are also degradation products. However, in some cases drug substance process-related impurities may be included in the drug product specifications, if appropriate, and limited by appropriate acceptance criteria.
As described in 〈476〉, unusually toxic impurities (e.g., mutagenic impurities) in drug substances and drug products require more stringent control compared to non-mutagenic impurities.
Manufacturers are responsible for controlling organic impurities in accordance with current regulatory standards. Manufacturers should consider the chemical characteristics and safety aspects of impurities when they identify and classify impurities in a drug substance or drug product. Analytical procedures for the detection and quantitation of impurities should be verified or validated. (For additional information, see〈1226〉 and 〈1225〉). For impurities that are known or suspected to be highly toxic (e.g., mutagenic) or that produce undesired pharmacological effects, the quantitation/detection limit of the analytical procedures should be commensurate with the acceptance criteria for the suspected highly toxic impurity and current applicable regulatory guidance to ensure patient safety. Impurities or degradation products that are also significant metabolites and present in animal and/or human studies are generally considered qualified.
If an individual monograph does not include a procedure for quantifying an impurity or acceptance criterion for an observed impurity, the manufacturer is responsible for developing and validating analytical procedures and establishing appropriate acceptance criterion. USP requests submission of the alternative/new procedure to evaluate for potential inclusion in the appropriate monograph(s).
In cases of complex impurity profiles, it may not be feasible to resolve each of the impurities individually or to detect them and quantify them using a single analytical procedure. In such cases, manufacturers should consider alternate approaches such as the use of multiple analytical procedures to test for impurities.
The acceptance criteria for impurities shall be based on applicable guidances or other acceptable scientific means, with safety as the primary consideration, and not solely based on process capability. In cases of complex impurity profiles, acceptance limits may be established based on grouping of impurities, as appropriate.
Similar principles may be applied to set thresholds and acceptance criteria for degradation products in FDA over-the-counter (OTC) monograph drug products, which are not discussed in ICH guidelines or FDA guidances. Degradation products in these drugs should be reported, identified, and/or qualified.
Measurement of degradation products can be challenging for products containing multiple drug substances and complex formulations.
The use of Placebo products as controls in stability studies may aid in the deconvolution of chemical changes that could be related to excipients rather than the drug substance.
(Definitions for key terms used in this chapter can be found in the Glossary. Additional sources of guidance on impurities in drug substances and drug products may be found in Appendix 1: Additional Sources of Information and Guidance.)
5 ORGANIC IMPURITIES DECISION TREE
The decision tree shown in Figure 1 provides guidance for the control of organic impurities in drug substances and drug products.
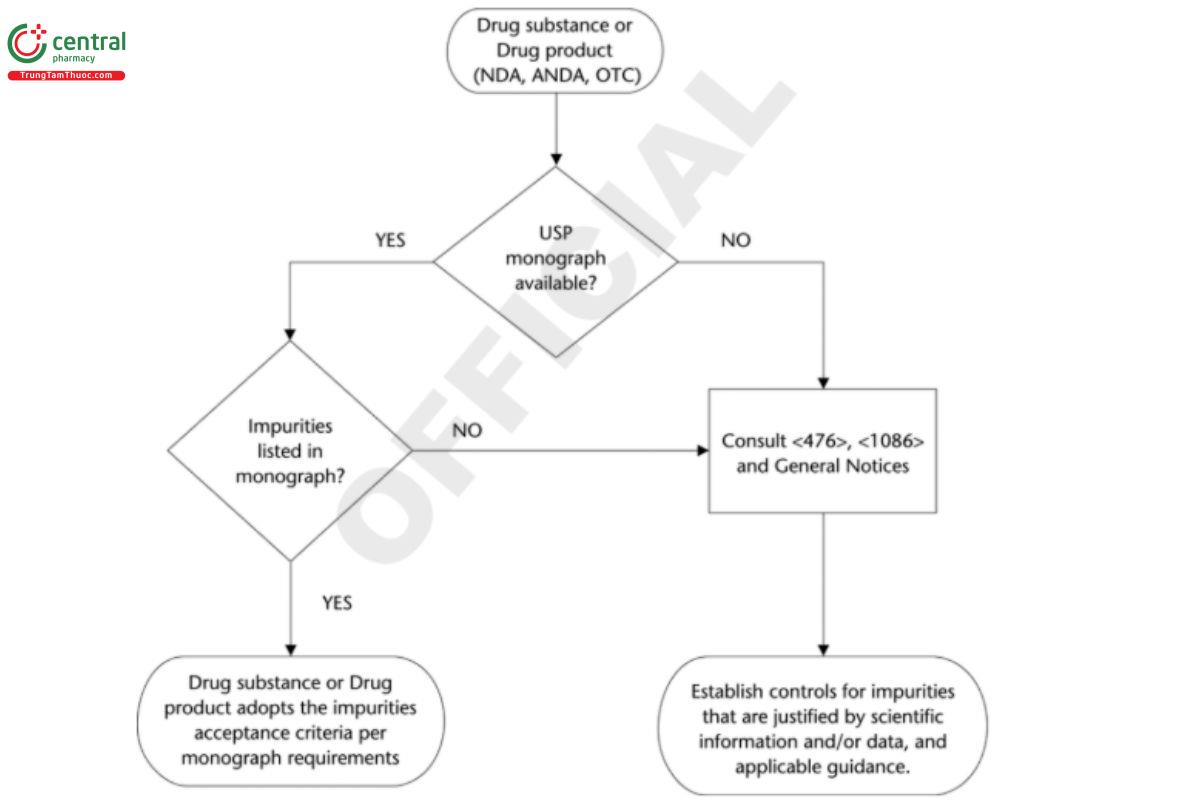
Abbreviated New Drug Application.
Note that all impurities specific to a given drug product formulation may or may not be included in the USP monograph. If the impurity is listed in the monograph, follow monograph limits. If the impurity is not listed, consult this chapter and 〈476〉 for guidance. Nonmonograph tests and acceptance criteria suitable for detecting and controlling impurities that may result from a change in the processing methods or that may be introduced from external sources should be employed in addition to the tests provided in the individual monograph, where the presence of the impurity is inconsistent with applicable good manufacturing practices or good pharmaceutical practices.
6 TERMINOLOGY ASSOCIATED WITH ORGANIC IMPURITIES USED IN DRUG SUBSTANCE AND DRUG PRODUCT MONOGRAPHS
The Organic Impurities section in a monograph generally includes impurities/degradation products that are known to be detected by the tests prescribed in the monograph. There are several scenarios for citing specified organic impurities in USP–NF monographs.
For identified impurities/degradation products (i.e., the ones that have known and characterized chemical structure), the chemical name is usually provided (e.g., in an impurities table or as a table footnote or in the Reference Standard section). They can have reference material available or not, depending on the substance.
Unidentified impurities/degradation products (i.e., the ones for which a structural characterization has not been achieved) are named with a general designation (e.g., impurity 1, impurity 2) or by an analytical property (e.g., chromatographic retention time). The reference material is not available.
Specified impurities/degradation products can be either identified or unidentified. They are listed and limited with a specific acceptance criterion in monographs.
If an impurity/degradation product is not listed and is NMT the identification threshold, it is controlled as an individual unspecified impurity.
In all cases the terms “any individual impurity” or “any other individual impurity” correspond to unspecified impurities.
A USP monograph of a drug substance may contain acceptance criteria for: specified identified and unidentified organic impurities; “any individual impurity”, "any unspecified impurity", or “any other individual impurity”; and total impurities. Total impurities in the drug substance monographs are the sum of all specified and unspecified impurities above the reporting threshold.
A USP monograph of a drug product may contain acceptance criteria for: specified identified and unidentified degradation products; “any individual degradation product”, “any other individual degradation product”, or "any unspeci
fied degradation product"; and total degradation products. Total degradation products in the drug product monographs are the sum of all specified and unspecified degradation products above the reporting threshold.
The preferred terms applicable to organic impurities procedures are “impurity” and “total impurities” in the drug substance monographs and “degradation product” and “total degradation products” in the drug product monographs. Only if the drug substance process-related impurities are included in the drug product organic impurities procedure should the term “impurity” be used and the total be expressed as “total impurities”. Drug substance process-related impurities also detected in the drug product and included in its specification may include a note that certain drug substance process-related impurities are listed for information only and should not be included in the “total degradation products”. When this note is included, the total degradation products should only include all specified and unspecified impurities/degradation products above the reporting threshold, with the exception of these designated process-related impurities.
Some monographs may state “disregard any peak below [... certain value]” in chromatographic tests. Typically, the disregard limit for substances covered by a monograph is set in accordance with the reporting threshold given in 〈476〉. In this context, the disregard limit is the decision criterion (the numerical value) for the user whether a peak response of an impurity is to be included or excluded in the total impurities.
In chromatographic tests, during quantitation, peaks caused by solvents and reagents or arising from the mobile phase or the sample matrix shall be disregarded, as well as other peaks that the monograph explicitly states are to be disregarded. When the impurity test prescribes the total of impurities or there is a quantitative determination of an impurity, choice of an appropriate threshold setting and appropriate conditions for the integration of the peak areas is important. Principles from 〈621〉, 〈1225〉, and 〈1226〉, such as signal-to-noise ratio, quantification limit, and quantitation methods, should be observed. The quantitation limit for the analytical procedure should be NMT (≤) the reporting threshold.
7 GLOSSARY
Degradation product: An impurity resulting from a chemical change in the drug substance brought about during manufacturing and/or storage of the drug substance or drug product by the effect of, for example, light, temperature, pH, water, or by reaction with an excipient and/or the immediate container–closure system.
Disregard limit: See Reporting threshold. [Note—It may appear in a monograph as “disregard any peak below [.. certain value]”.]
Drug substance process-related impurity: An impurity generated during drug substance manufacturing. Process-related impurities may include starting materials, byproducts, intermediates, reagents, ligands, and catalysts. In this chapter, it is also called “process impurity”.
Identification threshold: A limit above which an impurity should be identified (i.e., its structural characterization).
Identified impurity/degradation product: An impurity or degradation product for which the structural characterization has been achieved.
Impurity: For a drug substance, any component of the drug substance that is not the chemical entity that is defined as the drug substance; for a drug product, any component of a drug product that is not the drug substance or an excipient in the drug product.
Qualification: The process of acquiring and evaluating data that establish the biological safety of an individual impurity or a given impurity profile at the level(s) specified.
Qualification threshold: A limit above which an impurity should be qualified.
Reporting threshold: In chromatographic tests, the limit above which an impurity should be reported and should be taken into account for calculating total impurities/total degradation products. Synonyms include “reporting level” and “reporting limit”. The older term “disregard limit” still appears in some USP–NF monographs; however, the ICH Q3 term “reporting threshold” is preferred. Peak responses must be corrected by the relative response factor when the information is provided in the individual monograph.
Specified impurity/degradation product: An impurity or degradation product that is individually listed and limited with an acceptance criterion in the drug substance or drug product monograph. A specified impurity or specified degradation product can be either identified or unidentified.
Total impurities/total degradation products: In a drug substance monograph, “total impurities” are the sum of all specified and unspecified impurities above the reporting threshold. Unless otherwise indicated, “total degradation products” in a drug product monograph are the sum of all specified and unspecified degradation products above the reporting threshold. Drug substance process-related impurities also detected in the drug product and included in its specification may include a note that certain drug substance process-related impurities are listed only for information and should not be included in the total degradation products. When this note is included, the total degradation products should only include all specified and unspecified impurities/degradation products above the reporting threshold, with the exception of these designated process-related impurities. For drug product monographs, the term “total degradation products” is considered a synonym to “total impurities”, with “total degradation products” being a preferred term.
Unidentified impurity/unidentified degradation product: An impurity or degradation product for which structural characterizations has not been achieved and that is identified solely by qualitative analytical properties (e.g., chromatographic retention times).
Unspecified impurity/degradation product: An impurity or degradation product that is not individually listed with its own specific acceptance criterion in the drug substance or drug product monograph. In Pharmacopeial monographs, any impurity/degradation product that is not individually listed is considered “unspecified” and is limited by a general acceptance criterion. Synonyms for “unspecified impurity/degradation product” include “other impurity/degradation product” or “other individual impurity/degradation product”, with “unspecified impurity/degradation product” being preferred terms.
8 APPENDIX
Appendix 1: Additional Sources of Information and Guidance
1. International Council for Harmonisation. Q3A(R2) Impurities in new drug substances. 2006. https://database.ich.org/sites/default/files/Q3A%28R2%29%20Guideline.pdf. Accessed 6 September 2017.
2. International Council for Harmonisation. Q3B(R2) Impurities in new drug products. 2006. https://database.ich.org/sites/default/files/Q3B%28R2%29%20Guideline.pdf. Accessed 6 September 2017.
3. International Council for Harmonisation. Q6A Specifications: test procedures and acceptance criteria for new drug substances and new drug products: chemical substances. 1999. https://database.ich.org/sites/default/files/Q6A%20Guideline.pdf. Accessed 6 September 2017.
4. International Council for Harmonisation. M7 Assessment and control of DNA reactive (mutagenic) impurities in pharmaceuticals to limit potential carcinogenic risk. 2017. https://database.ich.org/sites/default/files/M7_R1_Guideline.pdf . Accessed 6 September 2017.
5. U.S. Food and Drug Administration. Guidance for industry. NDAs: impurities in drug substances. 2000. http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM070577.pdf. Accessed 6 September 2017.
6. U.S. Food and Drug Administration. Guidance for industry. ANDAs: impurities in drug products. 2010.
http://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/UCM072861.pdf. Accessed 6 September 2017.
7. Organic Impurities in Drug Substances and Drug Products 〈476〉. USP. In: Pharm Forum 43(6) [Nov.–Dec. 2017]. Rockville (MD): United States Pharmacopeial Convention; 2017. http://www.usppf.com.
8. Consumer Healthcare Products Association. Your Health at Hand Book: Guide to OTC Active Ingredients in the United States. 2010.
http://www.yourhealthathand.org/images/uploads/Your_Health_at_Hand_Book.pdf. Accessed 6 September 2017. (USP 1-May-2021)
