Immunological Test Methods—General Considerations
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
Change to read:
1 INTRODUCTION
This general information chapter provides a high-level description of principles for immunological test methods (ITMs) that can be used in specified monograph tests, along with information and approaches to analytical development and validation for ITMs. The scope of this chapter is to provide general information that is applicable to all ITMs. The chapter provides a foundation for specific chapters about different types of ITMs, e.g., Immunological Test Methods-Enzyme-Linked Immunosorbent Assay (ELISA) (1103), Immunological Test Methods -Immunoblot Analysis (1104) (USP 1-May-2021), and Immunological Test Methods-Surface Plasmon Resonance (1105). This suite of general information chapters is related to the bioassay general information chapters. Use of ITMs for process monitoring, diagnosis, and evaluation of clinical response, assessment of pharmacokinetics/pharmacodynamics/absorption, distribution, metabolism, and excretion (PK/PD/ADME), and other product characterization (nonrelease testing) is outside the scope of this chapter.
The basis of all ITMs used to measure a quality attribute of a biologic drug substance or drug product is the highly specific noncovalent binding interaction between an antibody and antigen. The antigen typically is an analyte of interest (e.g., protein, carbohydrate, virus, or cell), and the binder is usually an antibody (e.g., monoclonal antibody or polyclonal antiserum). ITMs are applicable to molecules that are either directly antigenic (immunogens) or can be rendered indirectly antigenic (haptens). The measurand in ITM is directly related to a quality attribute of the product under test.
ITMs are valuable because they exhibit high sensitivity and specificity for an analyte in complex matrices. They typically are used for qualitative and quantitative assessment of both an antibody and antigen, but their application also extends to the measurement of hapten, complement, antigen-antibody complexes, and other protein-protein interactions. These properties of ITMs allow their use for assessing identity, potency (strength), purity, impurities, stability, and other quality attributes of biological drug substances and drug products.
ITMs are useful for many applications because they can measure molecules over a wide range of sizes and binding types. In general, antibodies are stable during various chemical modifications that do not have a significant adverse influence on interactions with an antigen. Antibody molecules tend to withstand moderate acidic and alkaline pH changes better than other proteins do. Because of this characteristic, a variety of ITMs with high degrees of sensitivity and specificity are possible. The ability to accelerate contact between an antigen and antibody enables ITM formats that provide rapid or real-time results.
Generally, ITMs have higher precision and shorter turnaround time than do traditional biologically-based (i.e., cell-based and animal) assays. Although in some cases these advantages can support the replacement of a biological assay with an immunoassay, such changes should be approached systematically and with caution. Often it is challenging to prove the equivalence, or comparability, of results from bioassays and immunoassays because the interaction between antigen and antibody may not reflect the functional attributes observed in bioassays.
One major limitation of ITMs compared to physicochemical methods (such as liquid or gas chromatography) is that the latter generally are more precise and can simultaneously identify a set of impurities or unexpected substance(s). Another major limitation is that generally ITMs operate at high molar dilutions at which they are sensitive to disturbances caused by environmental factors in the sample matrix (i.e., matrix effects). Matrix effects can depend on ITM format and are not fully understood. Their specificity, a hallmark of ITMs, is sometimes compromised by structural or sequence similarities between the analyte and a closely related molecular impurity (cross-reactivity).
Most ITMs reflect physical interaction (binding) between an antigen and antibody and not the analyte's functional properties. Therefore, analysts must pay attention in the selection and execution of ITM format. Cell-based ITMs that can provide functional information about the analyte are beyond the scope of this chapter.
2 GENERAL CHARACTERISTICS OF ITMS
ITMs are based on the principle of specific, noncovalent, and reversible interactions between an antigen and antibody. In general, the primary antigen-antibody reaction is brought about by complementarity, which creates macromolecular specificity. This noncovalent interaction determines the degree of intrinsic affinity. Intrinsic affinity contributes to functional and/or relative affinity that depends on factors like reaction phase and valency, which in turn determines the degree of reversibility of an interaction. A better understanding of factors that affect antigen-antibody interactions provides the rationale for the development of a suitable ITM format (e.g., solid or liquid phase, competitive or noncompetitive binding, etc.).
A defining characteristic of ITMs is that they employ an antigen (or hapten) and antibody. In addition, ITMs may contain companion molecules such as complement components. The components of ITMs are defined as follows:
- Antigens-Comprise a wide range of molecules that are capable of binding to the antibody in a specific interaction. Generally, part(s) of an antigen [the immunogenic epitope(s)] is/are capable of eliciting antibody response.
- Haptens-Small molecules that, by themselves, are not capable of eliciting an antibody response but are capable of eliciting an immune response when attached to a large carrier such as a protein. Antibodies produced to a hapten-carrier adduct also may bind to the small-molecule hapten in a specific interaction.
- Complements-Companion molecules that, under certain conditions, aid in the functionality of antigen-antibody complexes but are not required for antigen-antibody or hapten-antibody interaction.
- Antibodies-Proteins with regions that impart a high degree of specific binding to antigens (and haptens). The structural elements of an Immunoglobulin G (IgG) antibody are shown in Figure 1.
In addition to these components, ITMs require some means to detect or monitor the binding reaction between the antigen and antibody.
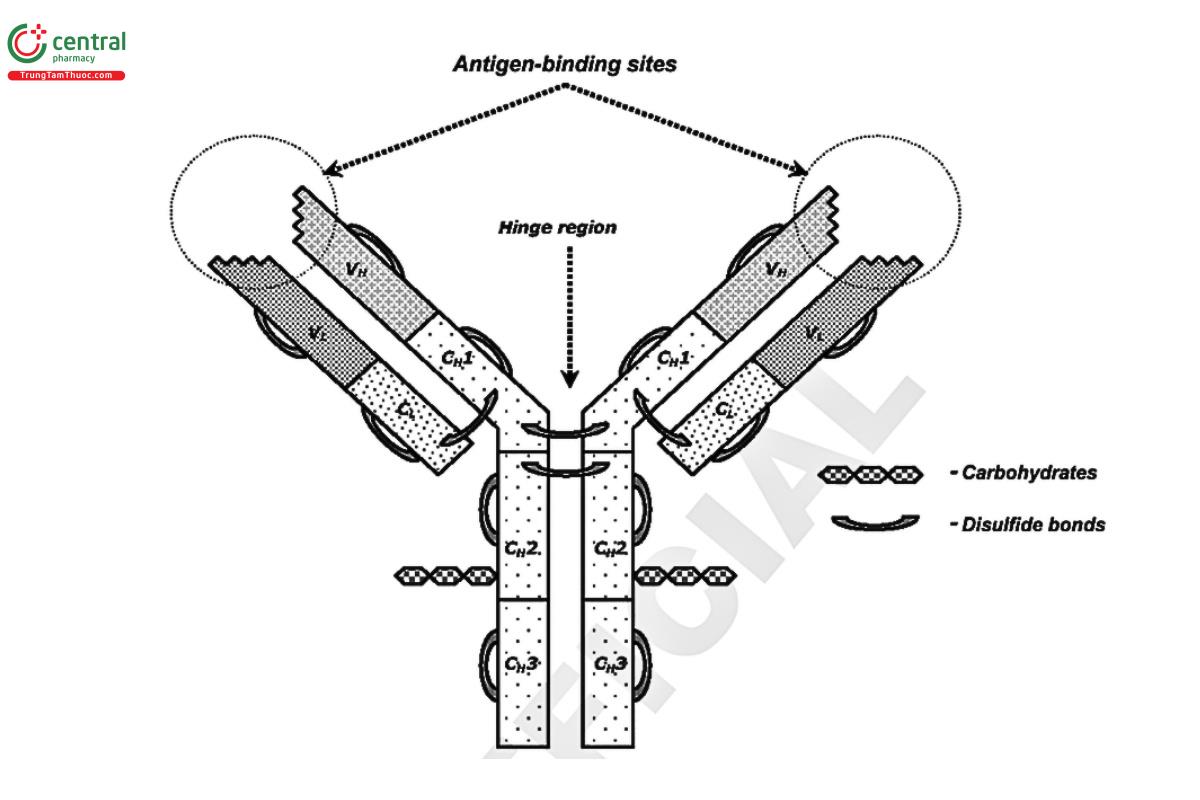
Figure 1. The structure of IgG. The IgG molecule is characterized by a distinctive domain structure of heavy (H) and light (L) chains, both of which are divided into variable and constant regions (V and C, respectively). Light chains consist of V and C domains, and heavy chains consist of a variable domain (VH) and three constant domains (CH1, CH2, and CH3). All domains are stabilized by disulfide bonds, and CH2 domains contain carbohydrates. The flexible hinge region between the CH1 and CH2 domains allows the independent behavior of two antigen-binding sites formed by variable domains.
3 TYPES OF ITMS
Measurement of antigen-antibody binding can be performed in a variety of assay types and formats: solid or liquid phase, manual or automated, labeled or nonlabeled, competitive or noncompetitive, qualitative or quantitative, homogeneous or heterogeneous, or combinations of some of these. The distinguishing characteristic of all these assays is the binding of an antibody or antigen to the analyte (which can be an antigen or antibody as well), followed by detection of the antigen-antibody complex. Although many different formats can be used for the binding reaction, along with different methods for detection, quantification of the analyte in the test article is always performed by comparison of the measurement to a reference standard. Thus a number of ITM technologies support investigations of product quality. Commonly used assay designs include enzyme-linked immunosorbent assay (ELISA), Western blotting, flow cytometry, competitive enzyme-linked immunosorbent assay, surface plasmon resonance (SPR), rate nephelometry, radioimmunoassay (RIA), radial immunodiffusion, precipitation, and agglutination. These methods are described below.
3.1 Enzyme-Linked Immunosorbent Assay
An ELISA is a quantitative, solid-phase immunological method for the measurement of an analyte following binding to an immunosorbent and its subsequent detection using enzymatic hydrolysis of a reporter substrate either directly (analyte has enzymatic properties) or indirectly (e.g., horseradish peroxidase- or alkaline phosphatase-linked antibody subsequently bound to the immunosorbed analyte). The analyte usually is quantitated by interpolation against a standard curve of a reference material. Chapter (1103), discusses ELISA in greater detail, including ELISA development for quantitative analysis.
3.2 Western Blotting
A Western blot is a semiquantitative or qualitative method for the measurement of a protein analyte that has been resolved by polyacrylamide gel electrophoresis and subsequently transferred to a solid membrane (e.g., nitrocellulose, nylon, or polyvinylidene difluoride). Detection can be achieved directly by reacting with a labeled primary antibody (antibody specific to the analyte of interest) or indirectly by reacting labeled secondary antibody (antibody against the primary antibody) to the primary antibody bound to the membrane-immobilized antigen. The label can be a radioisotope or an enzyme that uses the substrate to produce color, fluorescence, or luminescence. This method is semiquantitative, especially when proteins are present in low concentration and in very complex mixtures. It is commonly used in early process development (e.g., antibody screening, protein expression, protein purification, etc.). Western blotting is a powerful method for analyzing and identifying proteins in complex mixtures, particularly after separation using 2-dimensional gel electrophoresis, which separates proteins based on size and charge (pl).
3.3 Flow Cytometry
Flow cytometry is a laser-based semiquantitative technology that permits the measurement of fluorophore-conjugated probes as they interact with their respective ligands on cells or particles. More details for flow cytometry can be found in Flow Cytometry (1027).
3.4 Surface Plasmon Resonance
SPR is a quantitative method for measurement of an analyte in a sample where the antibody-antigen complex formation can be measured in real time at the interface of a liquid and solid (e.g., gold surfaces or particles). The measurement taken is the real-time change in refraction of a polarized light and occurs during the formation of the antibody-antigen complex, resulting in changes to the plasmon resonance minima (i.e., the sensorgram). The quantity of analyte is determined by comparison to the measurement of a reference standard curve determined in the same assay. More details for SPR can be found in (1105).
3.5 Rate Nephelometry
Rate nephelometry is a quantitative method for measurement of an analyte in a sample in solution by measuring the light scatter introduced by small aggregates formed by the antigen-antibody complex. The quantity of analyte is determined by comparison to the measurement of a reference standard curve determined in the same assay.
3.6 Radioimmunoassay
RIA, a sensitive ITM first developed in the 1950s, is a quantitative method for measurement of an analyte in a sample. RIA usually uses a competitive antibody-antigen binding reaction, but it also can be used in sandwich immunoassay format, including immunoprecipitation. In competitive RIAs the analyte competes for binding with a radiolabeled (e.g., using 125I or 3H) reference antigen that is identical to the analyte; therefore, the analyte and the antigen both compete for binding to a fixed and limiting dilution of a specific (often polyclonal) antibody. The radiolabeled antigen is present in excess. The same unlabeled antigen in the test sample competes in binding to the same site on the antibody, which is present in a fixed quantity. Binding of the unlabeled antigen to the antibody leads to the displacement of the labeled antigen, resulting in a decrease in the radioactivity of the antigen-antibody complex fraction. To separate the antigen-antibody complex from the excess unbound antigen, the complex generally is either precipitated with a secondary antibody (or Protein G) immobilized on a solid matrix (e.g., glass or resin beads) or with an already immobilized primary antibody. The quantity of analyte usually is determined by interpolation against a standard curve of a reference material, where a fixed amount of antibody and radiolabeled antigen is mixed with an increasing amount of unlabeled antigen. Hence, even a small quantity of unlabeled antigen will result in a relative quantitative decrease in total bound radioactivity.
3.7 Single Radial Immunodiffusion
Single radial immunodiffusion (SRID or SRD) is a quantitative method for measurement of an analyte in a sample by measuring the diameter of the ring of precipitin formed by the antigen-antibody complex. Antigen is applied to a well in a gel infused with a constant level of antibody. Solutions with higher concentrations of antigen diffuse farther before being saturated with antibody and then precipitated. The quantity of analyte is determined by comparison to a reference standard curve measured by the same assay.
3.8 Precipitation
The underlying principle for this method is that the interaction of a multivalent antibody and antigen leads to the formation of a complex. In some cases a visible precipitate is formed. Other immunoprecipitation techniques involve the use of Protein A or Protein G beads to capture the antigen-antibody complex and facilitate the separation of the antigen-antibody complexes from the other antigens in the solution. Precipitation is not commonly used for quantitative analytical purposes because of the time required (days to complete), lack of sensitivity, and requirement for large quantities of antigen and antibodies.
3.9 Agglutination
Agglutination and inhibition of agglutination, respectively, provide qualitative and quantitative measures of certain antigens and antibodies. Inhibition of agglutination is a modication of the agglutination reaction that provides higher sensitivity to detect small quantities of proteins, chemicals, viruses, and other analytes. The principle of agglutination is similar to that for precipitation except that the interaction takes place between an antibody and a particulate antigen and leads to a visible clump or agglutination. The most common example of this application is for blood typing (i.e., A, B, or O antigen).
4 CHOICE OF ITM
When choosing an ITM, analysts should consider sensitivity and specificity as well as the complexity of the sample. Table 1 provides an assay developer with a comparative view of the advantages and disadvantages of a variety of ITM formats. The intended application of the ITM should govern the choice of the most suitable format.
Table 1. ITMs Used in Biopharmaceutical Laboratories
| Method | Advantages | Disadvantages | Typical Industry Uses |
| ELISA |
|
|
|
| Western blot |
|
|
|
| Flow cytometry |
|
|
|
| SPR |
|
|
|
| Rate nephelometry |
|
|
|
| RIA |
|
|
|
| SRD |
|
|
|
| Precipitation |
|
|
|
| Agglutination |
|
|
|
Change to read:
5 KEY CONSIDERATIONS IN ITM DEVELOPMENT
The goal during method development is to produce an accurate assay that is practically feasible and possesses an acceptable degree of intra- and inter-assay precision. To minimize the overall imprecision, the sources of variability should be identified and minimized.
5.1 Reagent Selection
Immunoassays are subject to several sources of interference such as cross-reactivity, endogenous interfering substances, buffer matrices, sample components, exposed versus masked epitopes, conformation changes in the antigen of interest, and other factors. Hence, during method development, analysts must identify possible sources of interference both to develop a robust method and to aid future troubleshooting.
Cross-reactivity is a major obstacle during immunoassay development. This arises when the specificity of an antigen-antibody reaction is compromised by the cross-reactivity binding of structurally similar molecules with the reaction binder. Some common examples are protein isoforms, degraded analyte entities, molecules of the same class, precursor proteins, metabolites, etc. Cross-reactivity can be minimized by rigorous reagent characterization and selection.
Reagents used in ITM applications generally fall into one of two categories: critical reagents and noncritical reagents. Critical reagents are specific and unique to the particular ITM or reagents that are intolerant of very small changes in composition or stability. Examples of critical reagents generally include assay-specific antibodies and reference or method calibration standards. Equivalence in the assay format must be established before replacement with a new lot. Noncritical reagents are those that can vary to some degree in composition without adversely affecting ITM performance. Reagents are often assumed to be noncritical (e.g., buffers, water quality, blocking buffer, or substrate) but later may be identified as critical components if assay ruggedness fails and troubleshooting of ITM reagents begins. ITM-specific reagents, including vendor and catalog number, should be defined in test procedure documents.
Antibody selection is critical for development of a successful immunoassay because it defines the assay's specificity and sensitivity. Furthermore, during antibody generation, analysts should ensure that the immunization protocols support the end use of the antibodies. For some applications a more specific antibody can be generated by the selection of a small and specific immunogen and affinity purification of the antibody, resulting in highly defined epitope coverage. In other applications it may be critical to ensure broad coverage of the different available epitopes on the molecules of interest, and a polyclonal antibody (pAb) pool may be the best choice. Currently, monoclonal antibodies (mAb) are preferred for some applications for the detection of single analytes because of their high specificity, lot-to-lot consistency, and indefinite supply. Compared to polyclonal antibodies, mAb have a higher initial cost to produce, but for these applications, the advantages generally outweigh the initial cost. Other applications may require more comprehensive epitope selection to ensure that subtle changes in the molecule(s) do not prevent recognition of the entire antigen, and thus a pool of monoclonal antibodies, or a pAb pool, would be the preferred choice. The latter are widely used for detection in a complex mixture of analytes (e.g., host-cell proteins). However, the characteristics of a polyclonal antiserum are not solely determined by the immunogen. Different animals of the same species will not necessarily produce identical antisera to the same immunogen and they may even vary during the immunization protocol. This warrants performing qualification of each lot of reagent which should not be assumed to transfer between antisera produced in the same way. (USP 1-May-2021) Similarly, immunoassays may use two distinct epitopes on an antigen-one for capture and the other for detection-which greatly reduces cross-reactivity. Another approach to minimize cross-reactivity is to purify the antigen before immunoanalysis. Variations in incubation temperature and time can affect the reaction kinetics of antibody interactions with similar yet different antigens. Thus this property should be optimized to increase the specificity of antigen-antibody interactions.
5.2 Development of Immunoassays
Development is an important stage in the establishment of a suitable ITM. During development of an ITM, analysts explore various settings of assay parameters and interactions between parameters to identify conditions under which the assay will consistently produce reliable results using minimal reagents, effort, and time. In Quality by Design terminology, the "possible operating space" is the collection of settings of assay parameters explored, and the "design space" refers to the conditions under which the assay performs well. The necessary performance properties of the ITM (precision, accuracy, specificity, etc.) required depend on the intended use(s). During ITM development, analysts should consider the following:
- Antigen-antibody ratio;
- In sandwich immunoassays, the ratio of capture antibody to detector antibody;
- Antigen-antibody reaction kinetics in the sample matrix (antigen-antibody binding generally is not linear);
- Selection of the standard (full-length antigen for the standard or just a small portion of the antigen containing the antibody-binding epitope, among other considerations); and
- Matrix effects.
The use of design of experiments (DOE) is strongly recommended, and different DOE methods may be appropriate in each stage of development. Early in development, screening designs are particularly useful (generally two-level geometric fractional factorial designs).
After screening (with a modest number of factors to study), full factorials or response surface designs are often appropriate. As development activities shift to qualification (ideally, if not typically, as the focus shifts to robustness), robust response surface designs often are a good choice. During qualification or validation, analysts may find it practical to simultaneously study robustness to assay operating conditions (using a small geometric fractional factorial) and validation parameters such as precision (via nested or crossed designs for random factors associated with repeatability, intermediate precision, and reproducibility).
Experiments that assess dilutional linearity and components of specificity, including matrix effects, usually involve construction of spiked samples. Although spiking often is performed in a dilution matrix, spiking a collection of actual samples or mixing actual samples is an important component of demonstrating robustness of dilutional linearity and components of specificity to the sample and matrix components.
Quantitative ITMs generate test sample data with an estimated concentration based on a calibration curve fit to reference (or standard) samples using an appropriate mathematical model. For data reporting, depending on the units of the reference standard, nature of the measured analyte, the degree of measurement standardization, the geographic region, and the history of the method, analysts may express concentration in terms of weight per volume, mole per volume, or weight of analyte per weight of product.
ITMs employ calibration curves prepared with reference standards of known (nominal) concentrations which helps control variation associated with repeatability, intermediate precision, and reproducibility and permits the estimation of results for unknown test samples.
Common simple statistical analyses assume that the (possibly transformed) data are normally distributed, have constant variance, are independent, and that an appropriate model has been used. Analysts should assess these assumptions using a substantial body of data (typically tens of assays) (USP 1-May-2021)
5.3 Reagent Considerations
A procedure for qualifying reagent sources and vendors (including audits), ordering, receiving, and disposing of commercial reagents and consumables should be outlined in a standard operating procedure (SOP). The preparation of internal reagents must be documented in a manner that allows reconstruction. Commercial and internally prepared reagents must be labeled with identity, concentration, lot number, expiration, and storage conditions. The stability and assignment of expiration dates for internally prepared reagents often are based on available literature and scientific experience, but analysts may need to confirm these empirically. An SOP for extending expiration dating of critical reagents is recommended. In addition, analysts should implement a mechanism for reagent tracking and linking lot numbers to analytical run numbers. Unacceptable reagent performance is detected by tracking quality control (QC) samples. Shifts in QC samples should prompt a review of analytical runs and changes in reagent lot numbers or review of possible deterioration of critical reagents. To avoid such shifts, analysts can cross-validate critical reagent lot changes.
The impact of collection and storage containers on analytical performance often is overlooked. When defining the stability and expiration of in-house reagents, analysts should record information about the storage container vendor, catalog, and lot number. The importance of a suitable reference standard and its characterization cannot be overemphasized for ITMs for biological products. Because of their inherent complexity, reference and calibration standards of macromolecular biologics often are less well characterized than are conventional small-molecule drug reference standards. If the calibration standard represents a mixture of different antigens (e.g., host-cell proteins), it should be shown to be representative of the antigen profile in the samples being tested. Consistency in ITM results depends on the availability of a suitable representative reference standard material.
Change to read:
6 VALIDATION
Analytical validation involves the systematic execution of a defined protocol and prespecified analysis that includes prespecified acceptance criteria. A validation demonstrates that an analytical method is suitable for one or more intended uses [see Validation of Compendial Procedures (1225), Biological Assay Validation (1033), and International Council for Harmonisation (ICH) Q2(R1)]. Qualification may involve similar or identical experiments and procedures as validation, but qualification does not require prespecified protocols, analyses, or acceptance criteria. In certain situations (e.g., use of a commercial kit), assay development may not be required before qualification. Chapter (1225), discusses which assay performance characteristics must be examined during validation for four primary categories of intended uses. For example, analytical procedures that quantitate major bulk drug substances or active ingredients may not require validation of the detection and quantitation limits but do require validation of accuracy, precision, specificity, linearity, and range.
6.1 System Suitability or Assay Acceptance Criteria
The purpose of system suitability or assay acceptance criteria is to ensure that the complete system-including the instrumentation, software, reagents, and analyst-is qualified to perform the intended action for the intended purpose.
System suitability criteria should be selected based on method type and purpose. Identity methods should include positive and negative control samples. Methods intended for the measurement of sample activity should include appropriate criteria regarding performance of standards as well as response levels of blanks. If possible, a control of known activity can be included to help demonstrate suitable assay performance. Samples should be replicated and criteria for precision should be included in suitability criteria. Procedures for repeat sample analysis and identification and treatment of outliers, when applicable, should be included [see the FDA Guidance for Industry: Analytical Procedures and Methods Validation for Drugs and Biologics (2015)]. (USP 1-May-2021)
All processes should be controlled by well-defined SOPs that ensure consistency, reduce errors, and promote reproducibility of laboratory processes. Training files for all personnel should be contemporaneous and should include some demonstration that analysts are qualified to perform the method and the specific ITM.
Instrument and software qualification begins with a definition of the design qualifications, including a risk assessment and gap analysis that identify potential threats to the collection, integrity, and permanent capture of ITM data. Qualification also includes installation
qualifications (IQ) and operational qualifications (OQ) [see Analytical Instrument Qualification (1058), and Good Documentation Guidelines (1029)].
6.2 Data (USP 1-May-2021) Tracking
Regulatory agencies have strict requirements about maintaining the identity and integrity of both samples and data. A quality process driven by SOPs must be implemented to ensure the correct identity and integrity of test and reserve samples. Ideally, a bar code system should be used to track the collection, identity, location, chain of custody, number of sample freeze/thaw cycles, storage temperature, and length of time that a sample is stored. This information should be captured and should be auditable from the time of collection to disposal (or sample depletion). (USP 1-May-2021)
Quality systems must be in place to minimize data errors which might occur during the generation of data reports from the bioanalytical laboratories. (USP 1-May-2021) Errors may also be introduced by test sample misplacement or identification, incorrect data reduction, miscalculations, transcription errors, omissions, and other factors. Ideally, validated software and laboratory information management systems are used when possible to generate, transfer, and archive data[see the FDA Guidance for Industry: Data Integrity and Compliance with cGMP (2016)]. (USP 1-May-2021)
