GUIDELINES ON THE ENDOTOXINS TEST
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- INTRODUCTION
- BACKGROUND: PYROGENS AND ENDOTOXINS
- ENDOTOXINS
- PREPARATORY REQUIREMENTS
- RSE: CSE Calibration
- CSE Calibration/Potency Determination Using the Gel-Clot Method
- CSE Calibration/Potency Determination Using Quantitative Kinetic and Endpoint Assays
- Activity Determination for a Liquid CSE
- Screening and Qualification of Consumables: Compendial Requirement
- Analyst Qualification
- Equipment and Instrumentation Calibration and Qualification
- Laboratory Environmental Conditions
- METHOD SUITABILITY
- Maximum Valid Dilution
- Method Suitability Testing
- Qualifying Test Preparation Methods Other than Dilution
- ROUTINE TESTING
- ALTERNATE TEST METHODS
- GLOSSARY
- REFERENCES
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
DOWNLOAD PDF HERE
1 INTRODUCTION
The first version of Bacterial Endotoxins Test 〈85〉 appeared in USP 20–NF 15 (1980). The chapter was subsequently harmonized with the Japanese and European Pharmacopeias, and the first harmonized chapter appeared in USP 25–NF 20 (2002). Since its first publication, the content has changed very little, but years of experience, increasing knowledge, and more complex parenteral formulations suggest that the basic methodologies described herein could benefit from additional supporting information. The purpose of this information chapter is to provide additional background information and guidance for the performance and proper application of the compendial bacterial endotoxins tests.
2 BACKGROUND: PYROGENS AND ENDOTOXINS
Pyrogens, or fever-causing agents, are possible contaminants in parenteral products. Many substances can cause fevers when injected, infused, implanted, or when they come in contact with the bloodstream or cerebrospinal fluid of mammals. Although some biologics, vaccines, and cell and gene therapies may, by their nature, elicit pyrogenic responses in patients, the predominant and most potent pyrogenic contaminants in the manufacturing of parenteral drugs and medical devices are bacterial endotoxins, which are components of the cell walls of Gram-negative bacteria (GNB).
Endotoxins are integral with the outer cell membrane of GNB. If GNB cannot grow, endotoxins cannot be generated. However, endotoxins may remain active in cell wall fragments after cells die, so a material may be sterile but may still contain quantifiable levels of endotoxins activity. Bacterial endotoxins, when present in parenteral products (including biological products) or medical devices, indicate that the growth of GNB occurred at some point during manufacture of the product. Endotoxins can be introduced into the process stream by pharmaceutical ingredients including water, raw materials (particularly from natural sources), the active pharmaceutical ingredients (API), drug product formulation excipients, primary (product contact) packaging components, improperly cleaned or stored manufacturing equipment, and/or ineffective microbial contamination control practices.
3 ENDOTOXINS
The terms "bacterial endotoxins" and "endotoxins" refer to a complex component of the outer cell membrane of GNB. Although still an active area of research, natural contaminants in sterile parenteral drug products or medical devices are thought to include outer membrane vesicles (OMV), which are mini-spheres of outer membrane material that are released from actively proliferating cells as part of the normal bacterial growth cycle (1–6) as well as outer membrane fragments that come from disrupted or dead GNB cells. The biologically active lipopolysaccharide (LPS) is embedded in or associated with other Gram-negative outer membrane components, including various proteins, phospholipids, and lipoproteins (1,3,6). The hydrophobic lipid A portion of the LPS molecule is embedded in the membrane and the hydrophilic polysaccharide portion is exposed to the external environment surrounding the cells. Some current research suggests that it is doubtful that pure LPS is released as part of the normal growth cycle of GNB (3).
The current USP Endotoxin Reference Standard (RSE) and commercially prepared control standard endotoxins (CSE) are extracted from the GNB cell membrane, generally by the Westphal hot phenol method, and are purified to remove any surrounding cell membrane components (7,8). Additionally, these purified preparations are often further formulated with stabilizing agents (8). As such, LPS prepared using Westphal, or other extraction methods, cannot contaminate pharmaceutical products because they do not exist in this form in nature. Endotoxin standards, attributed to differences in source and manner of preparation, could “react differently from native sources of endotoxins” (9). It is possible that standards extracted by the Westphal method or other denaturing methods and then formulated may not always be representative surrogates for modeling the behavior of natural endotoxins in some pharmaceutical, biopharma, or medical device extraction experiments (10,11).
Although general test methods in the compendial chapters numbered below 〈1000〉 are considered to be validated, all laboratories, including contract testing laboratories, must demonstrate that the chosen BET methodology is suitable for a specific product or material tested according to Bacterial Endotoxins Test 〈85〉, Gel-Clot Technique, Preparatory Testing, Test for Interfering Factors or Photometric Quantitative Techniques, Preparatory Testing, Test for Interfering Factors (also known as inhibition/enhancement testing). This suitability testing will require the following prerequisites:
- Calculation or assignment of an endotoxins specification or limit for the material under test, and calculation of the maximum valid dilution (MVD). See the following sections: Method Suitability, Calculating Endotoxin Limits for Drug and Biological Products, Calculating Endotoxin Limits for Medical Devices, Calculating Endotoxin Limits for Combination Products, and Calculating Endotoxin Limits for Active Substances and Excipients below.
- Demonstration that the positive product control (PPC) can be recovered at a dilution of material that does not exceed the MVD. See Bacterial Endotoxins Test 〈85〉, Gel-Clot Technique, Preparatory Testing, Test for Interfering Factors or Photometric Quantitative Techniques, Preparatory Testing, Test for Interfering Factors.
Once assay suitability has been demonstrated, the laboratory may perform routine testing according to the calculations and sample preparation conditions that were described in the suitability study. Changes to those conditions (e.g., the lysate and/or endotoxins source, product component(s), formulation, or changes in manufacturing) are subject to change control and may require that the laboratory repeat the suitability study.
4 PREPARATORY REQUIREMENTS
5 RSE: CSE Calibration
It is well known that purified LPS from different genera/species/strains of GNB, when experimentally administered to rabbits on a uniform weight or mass basis, can produce significantly different pyrogenic reactions (12,13). However, by relating these effects to the administered activity—meaning their ability to elicit a fever in the rabbit (Pyrogen Test 〈151〉) or initiate an LAL reaction (〈85〉)—rather than weight or mass, the structural variability of LPS molecules can be normalized to a defined unit of activity called the endotoxin unit (EU). Two notes apply here:
- One USP EU is equivalent to one International Unit (IU) as indicated in 〈85〉.
- Lysate reagents currently described in 〈85〉 are licensed in the United States by the FDA and have a sensitivity associated with them; however, the FDA does not license CSE preparations.
The RSE is the purified LPS primary standard that is formulated from a common bulk preparation of E. coli 0113:H10:K LPS that is shared among the World Health Organization (WHO), European Pharmacopoeia (Ph.Eur.), Japanese Pharmacopoeia (JP), and U.S. Pharmacopeia (USP). The RSE was originally developed by the FDA in the 1970s to calibrate lysate reagents from multiple manufacturers, and it remains the primary standard for lysate calibration, calibration of secondary standards (e.g., CSE), and the generation of assay parameters such as standard curves and PPC assay controls.
Although RSE is available from USP for routine use, most BET assays are performed using a CSE, which is a secondary calibration analyte that may be included in LAL test kits purchased from reagent manufacturers. Many CSEs are provided as lyophilized preparations of a purified LPS that was filled and packaged by weight, not by activity. The purpose of the standardization study is to determine the specific activity, or potency, of the CSE in EU/unit of weight of the material against the RSE primary standard.
All the reagents used in BET assays are biological in nature and, therefore, can exhibit some variability in sensitivity and potency, making calibration against the RSE an important task. Calibration against RSE is necessary for each unique combination of lysate lot and CSE lot. If a kit is purchased from a reagent vendor, the vendor will conduct the lot-specific standardization and provide a lot-specific certificate of analysis (CoA) in the kit, which should be retained for reference in the laboratory. However, there may be circumstances when a laboratory might choose to purchase CSE from a third party or to calibrate a liquid endotoxin preparation, which requires that the laboratory conduct its flown calibration study.
6 CSE Calibration/Potency Determination Using the Gel-Clot Method
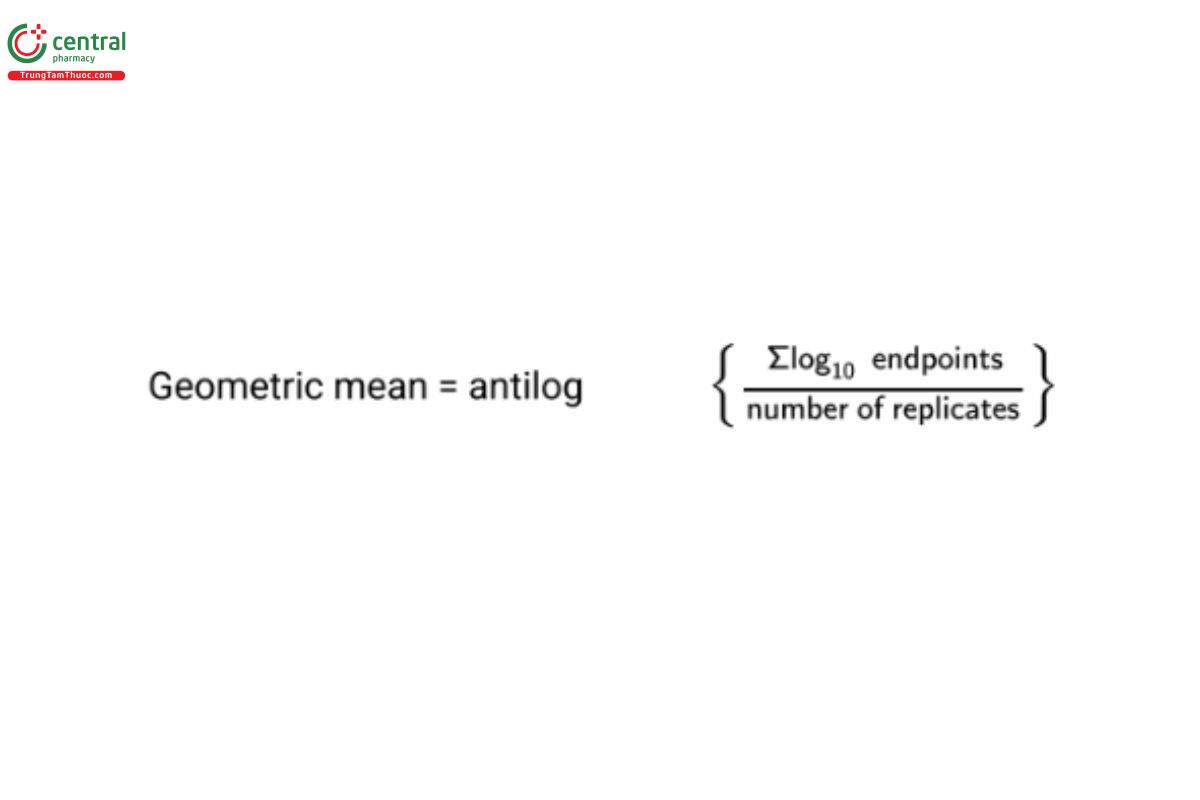
CSE potency determination in the gel-clot test is accomplished by comparing the geometric mean (GM) endpoints of separate dilution series of RSE and CSE made in Water for BET [see Bacterial Endotoxins Test 〈85〉, Reagents and Test Solutions, Water for Bacterial Endotoxins Test (BET)] and tested in quadruplicate. The endpoint is defined as the last tube in a two-fold series of RSE or CSE dilutions showing a positive reaction (gel). To have an endpoint, the last positive tube must be followed in the series by at least one negative dilution. The GM is calculated as follows:
The RSE is labeled in units of activity (EU/vial), and the units for the GM endpoint for the RSE are expressed as EU/mL. Because the CSE is filled by weight, the units for the GM endpoint for the CSE are expressed as ng/mL. The potency of the CSE in EU/ng is calculated as follows:

For example, to calibrate a new lot of CSE for use with a gel-clot lysate lot with a labeled sensitivity of 0.125 EU/mL, dilute the RSE in EU/mL to bracket the label claim of the lysate reagent. For this example, the GM of the RSE confirms the label claim of 0.125 EU/mL. Dilute the CSE in ng/mL so that an endpoint will be reached. For this example, the endpoint of the CSE series is 0.00625 ng/mL. The potency of the CSE for the particular lysate lot is calculated as follows:

For this lot of lysate, the potency of the new CSE lot is 20 EU/ng. If the lab wants to use this CSE lot with a different lysate lot, the calibration study must be repeated because potency determination references only a specific combination of lysate lot and CSE lot.
7 CSE Calibration/Potency Determination Using Quantitative Kinetic and Endpoint Assays
Determining the potency of CSE in a quantitative assay is performed much like a standard assay for an unknown. Onset/reaction times from dilutions of the CSE diluted in ng/mL are interpolated from an RSE standard curve to yield the activity of the CSE dilution in EU/mL.
Since the concentrations in ng/mL of these dilutions are known, calculation of EU/ng is a simple mathematical function. For any given concentration of CSE, calculate as follows:
EU/ng = (EU/mL) ÷ (ng/mL)
For example, to calibrate a new CSE lot for use with a chromogenic assay, the first step is to prepare a standard curve using RSE in Water for BET. The standard curve range is left to the discretion of the analyst, but appropriate choices would be either the same range as is routinely used in the laboratory or the maximum range suggested by the reagent manufacturer. Next, the analyst dilutes the CSE to a known concentration in ng/mL and tests the dilutions as unknowns (see Table 1). Points to consider:
1. Because of linearity requirements for a standard curve, data used in the calculation of potency may not be extrapolated beyond the range of the curve, but rather must fall within the range of the referenced standard curve
2. Standard curves should not exceed the maximum ranges that are suggested by the reagent manufacturer
3. For CSE calibration, it is recommended that replicate determinations for any given dilution be considered individually (not averaged) when calculating the correlation coefficient to assess variability when calibrating the CSE
Table 1. Example of Calibration of a CSE Analyte Against RSE in a Quantitative Assay
Example concentration (ng/mL) | Result (EU/mL) | Result (EU/ng) |
| 1 | 15 | 15 |
| 0.5 | 6 | 12 |
| 0.25 | 3 | 12 |
| 0.125 | 1.8 | 14 |
| - | - | Average: 13.25 |
In this case, the potency of the new CSE lot with kinetic chromogenic reagent is the mean of the four separate determinations. That calculation is 13.25 EU/ng, which is properly rounded down to 13 EU/ng. If the lab wants to use this CSE lot with a different lot of lysate—be it kinetic, endpoint, or gel clot—the calibration must be repeated because potency determination references a specific combination of lysate lot and CSE lot.
8 Activity Determination for a Liquid CSE
A liquid CSE such as a native endotoxin preparation does not have potency unless the actual weight of the source material is known (Endotoxin Indicators for Depyrogenation 〈1228.5〉). Typically, liquid CSE are described in terms of a concentration of activity expressed in EU/mL. The activity of a liquid CSE in EU/mL is determined by GM endpoint determination for replicate tubes in gel clot or by treating the solution as an unknown against an RSE standard curve for quantitative assays.
9 Screening and Qualification of Consumables: Compendial Requirement
The harmonized compendial chapter on the BET requires that the laboratory screen consumable plastics for test interferences. From 〈85〉, Apparatus:
If employing plastic apparatus, such as microplates and pipet tips for automatic pipettes, use apparatus that is shown to be free of detectable endotoxins and does not interfere in the test. [Note—In this chapter, the term “tube” includes any other receptacle such as a microtiter well.]
Plastics used in the performance of the test are molded, meaning that plastic pellets have been heated to a very high temperature to produce molten plastic in preparation for the molding process. This high temperature will destroy bacterial endotoxins, so molded plastics are free of detectable endotoxins when they are released from the molds. However, depending on the subsequent handling, recontamination is possible. Control measures should be in place to ensure appropriate storage conditions and avoid contact with potentially contaminating substances. However, unless depyrogenated plastics are stored under damp conditions or encounter substances during handling in which GNB could proliferate, the probability of recontamination is remote.
If a laboratory accepts a vendor CoA for the endotoxin content of a disposable component, there should be an understanding of the methods used to determine the reported test result. Two examples of CoAs are provided below:
If a shipment of dilution tubes is received from a vendor and is labeled “non-pyrogenic”, what does this really mean? Was the lot tested for pyrogens using a rabbit pyrogen test or a validated monocyte activation test? Was it tested using a standard 〈85〉, and if so, at what level of endotoxin does the vendor consider the material to be “non-pyrogenic”? It is important to ask the vendor how the material was prepared and tested.
If a shipment of 10-mL tubes is received from a vendor with the label of “<0.5 EU/mL”, what does that mean? With what volume was the tube extracted (1, 5, or 10 mL)? A 1-mL extraction would mean <0.5 EU/tube, whereas a 5-mL extraction would mean <2.5 EU/tube and a 10-mL extraction would mean <5 EU/tube. The laboratory should ask the vendor about the method of endotoxin extraction from the tube as well as the BET test conditions that were used to generate the result.
A default method to confirm the CoA result is to treat the plastic disposable as if it were a medical device and proceed according to the methodology provided in Medical Devices—Bacterial Endotoxin and Pyrogen Tests 〈161〉. However, while 〈161〉 defines the endotoxin limit for medical devices as <20 EU/device, a laboratory may want to consider redefining the limits for disposables used in the test to be less than the value of the most sensitive test used in the laboratory. For example, if the laboratory is using sterile polystyrene tubes for sample dilution, and the most sensitive test is a kinetic chromogenic test with a λ = 0.05 EU/mL, then consider setting a limit low enough to prevent interference across all test methods.
Likewise, some materials could contain extractable or leachable substances that could inhibit the LAL-bacterial endotoxins assay. The PPC will indicate whether the normal, routine use of the plastic results in any leached inhibitory substance. If a laboratory is considering use of disposable plastic containers such as sterile polystyrene for long-term storage of materials that ultimately will be tested, it is suggested that they do testing to confirm that there are no inhibitory or enhancing factors that could affect the accuracy of the test.
10 Analyst Qualification
General laboratory training for analysts is good laboratory practice, as well as a current good manufacturing practice (cGMP) requirements (Microbiological Best Laboratory Practices 〈1117〉). Typically, training for performing any BET involves demonstration of acceptable proficiency for both sample preparation and assay method(s). It is suggested that training be divided into two parts:
1. Classroom training delivered by a subject matter expert (SME) develops an understanding of the principles and limitations of the test methods as well as the effects of the analyst’s technique on the test result. Differences between analysts in the accuracy and precision of executing basic laboratory tasks such as pipetting, weighing raw materials, and making dilutions may introduce bias. Due to the inherent variability of the reagents (lysate and CSE) routinely used in the laboratory, poor or inconsistent techniques may significantly affect the accuracy of test results. Analysts must be retrained if a new test method is introduced into the laboratory (e.g., a change in laboratory procedures from gel clot to a kinetic assay).
2. Training effectiveness should be confirmed by the demonstration of analyst competency in performing the test. Competency or proficiency training may be divided into two parts:
A. For compendial assays where confirmation of assay sensitivity is required, it is recommended one part of performance training follow the provision in Bacterial Endotoxins Test 〈85〉, Gel-Clot Technique, Preparatory Testing or Photometric Quantitative Techniques, Preparatory Testing. For the gel-clot method, this is the test for confirmation of labeled lysate sensitivity, and for quantitative tests, this requires the construction of a linear standard curve. For training on quantitative tests, it is recommended that replicate determinations for any given dilution be considered individually (not averaged) when calculating the correlation coeficient to assess the variability in the proficiency study. Both of these proficiency assessments require analysts to prepare and dilute RSE or CSE standards. If the laboratory uses a cartridge system with an on-board archived standard curve, it is recommended that the laboratory devise another method of ensuring that the analyst’s technique in sample preparation and dilution is sound. This proficiency training should be described, justified, and included in the firm’s standard operating procedures to ensure consistency across all analysts. Although there are many ways to determine proficiency, one possibility is to ask the analyst to prepare a sample with a known and confirmed level of endotoxin activity and ultimately compare the result to the known value. If the calculated values are incorrect, this may raise a concern about the analyst’s technique.
B. The second part of the proficiency training should be an "on the job" training where analysts are required to demonstrate their ability to calculate endotoxin limits, prepare samples according to the results of suitability testing, and appropriately execute positive and negative controls in accordance with product-specific instructions and 〈85〉.
The following should be stressed during training:
- Appropriate laboratory aseptic technique is important so the analyst does not contaminate samples, diluents, or accessories used to perform the test.
- Use of a vortex mixer or another validated method (e.g., sonication for sample preparation) is important to optimize the distribution of endotoxins in samples and the aggregation state of the purified standards in the standard series. Because the formulations of the CSEs provided in LAL test kits are proprietary, it is highly recommended that analysts follow the manufacturers’ instructions for vortexing time, both for reconstitution of the vial of LPS and in between dilutions. However, vortexing of lysate is not recommended as it may result in bubbles in the reagent.
- If reagents are saved, ensure that the “open” date and “expiration” date are clearly marked on the primary containers and that any holding of unused reagents follows manufacturers’ instructions.
- Do not store RSE or CSE dilutions without a validation study that includes vessel type and materials of manufacture, concentrations of RSE or CSE that are to be held, hold temperature, and volume of the dilutions to be held.
- When reading gel-clot results, pick tubes up one at a time and invert 180°. Picking up more than one tube could jostle the contents.
- Once a gel is broken, it will not re-form, and the result may be a false-negative.
- When inoculating a microplate, tube, or cartridge care should be taken to avoid the formation of bubbles, because they will impact the accuracy of the test result.
- When using heating equipment (e.g., bead baths, water baths, plate readers), be certain that the equipment is qualified.
- If using a water bath for gel-clot incubation, change the water frequently. The recommended frequency is at least once a week.
- Ensure that all mechanical pipettors are calibrated, and use them only within the calibrated range.
- When possible, use larger volumes (milliliters) for dilution rather than smaller volumes (microliters), as smaller volumes increase variability.
- Standard curves for photometric tests are constructed based on the log of the measured onset or reaction times as a function of log of the endotoxin concentration. Pay attention to the onset times of the standards to ensure that they are consistent from run to run, analyst to analyst, and day to day for any given combination of CSE lot and lysate lot. See Routine Testing, Standard Curve Control.
- If using a monocyte activation test, be sure to include at least one non-endotoxin control.
An analyst requires additional training if any of the following is noted by a supervisor:
- Failure to meet the requirements of the initial performance training.
- Frequent inability to meet system suitability parameters, yielding invalid test results (e.g., confirmation of label claim for gel clot, demonstration of linearity for quantitative assays, inability to ensure that negative controls are nonreactive). Note that the inability to recover the PPC within the required range may signal an issue with analyst technique or a change in the product’s manufacturing or formulation that changes the product’s interference profile.
- Erratic results for slope and y-intercept for quantitative assays. See Routine Testing, Standard Curve Control.
- Adverse trends for out-of-specification (OOS) or out-of-trend (OOT) test results.
11 Equipment and Instrumentation Calibration and Qualification
All instrumentation and equipment used in the performance of an LAL test, including (but not limited to) mechanical pipettors, water baths, heat blocks, and incubating plate readers, should be qualified using proper scientific standards and according to approved protocols and maintenance schedules. Where appropriate, user requirement specifications (URS), as well as IQ/OQ/PQ protocols and reports, should be written and ultimately approved by the quality unit. See Analytical Instrument Qualification 〈1058〉 for additional guidance. All equipment used in performing the BET should be properly calibrated and maintained at frequencies that are in accordance with the equipment manufacturer’s recommendations. Because BET incubation temperatures are critical, equipment such as incubating plate readers, heat blocks, and water baths should be evaluated for uniformity of heat distribution.
Incubating plate or tube readers should reference a URS, an installation qualification (IQ), an operational qualification (OQ), and a performance qualification (PQ). Very often the vendors of these instruments will provide the laboratory with IQ/OQ/PQ templates and hands-on assistance that are very useful for the execution of these studies.
If a laboratory or production dry heat oven is used to depyrogenate glassware or other heat-stable items used in the performance of any of the BET assays, it must be validated to ensure appropriate time/temperature exposure and load pattern (see Dry Heat Depyrogenation〈1228.1〉 for additional guidance).
Computer software must be compliant with all federal regulations and standards (21 CFR part 11 in the United States). It must allow for individual user passwords and audit trails. Quality control should understand how the vendors of the BET software have programmed their calculations. For example, if the instrument reports out an averaged result for the assay of three individual samples tested in duplicate, did the software program average the onset times to get the result or did it average the replicate results?
12 Laboratory Environmental Conditions
The BET can be performed in most modern laboratories under controlled conditions. Appropriate aseptic technique is important when preparing and diluting standards and handling samples. Gowning practice outside of normal laboratory personal protective equipment (PPE) requirements is not a concern unless the product under test demands specific analyst safety considerations due to toxicity or infectiousness. Gloves should be talc-free, as the talc may contain significant levels of endotoxins. Plate readers, water baths, and dry heat blocks used for sample incubation should be on a laboratory bench away from heating, ventilation, and air conditioning (HVAC) ducts, significant vibration, and laboratory traffic that could affect the test results. Sample hold times and conditions should be determined and subsequently documented, if necessary, to ensure that accurate test results can be generated in the qualified time. For example, if the laboratory receives a Water for Injection (WFI) or in-process sample, must it be refrigerated or can it remain at room temperature, and for how long? Prior to testing, it is recommended that the primary sample container(s) be adequately mixed before removing the test aliquot(s) for either direct testing or subsequent dilution.
Change to read:
13 METHOD SUITABILITY
13.1 Calculating Endotoxin Limits for Drug and Biological Products
An endotoxin limit specification for a compendial article is the allowable amount of endotoxin activity that can be safely contained in a parenteral product, according to current understanding and experimental evidence (5). The endotoxin limit calculation for any drug product is dependent on three variables: 1) the route of administration, which largely defines K, the numerator in the endotoxin limit formula, 2) the dose of the product per kilogram of body weight, and 3) the duration (time) of administration. This information can be found in the package insert for an approved drug product or can be obtained from the product development group for a product that is either still in development or in early clinical trials.
An endotoxin limit specification is calculated for each drug product formulation and set of administration conditions as follows:
K/M
K = threshold pyrogenic dose of 5 EU/kg for most routes of administration or 0.2 EU/kg for intrathecally administered drugs (see Table 2)
M = maximum recommended dose of product per kilogram of body weight of the patient. This dose relates to the concentration of active ingredient (potency of the active ingredient) in the finished product formulation. If a product is infused or injected into a patient at frequent intervals over an extended time, then M is based on the maximum total dose administered in a 1-hour period. If the pediatric dose per kilogram per hour is higher than the adult dose, the pediatric dose must be used for the calculation.
When calculating the endotoxin limit specification, body weight is defined according to the intended patient population, which can differ in terms of geographical regions or patient populations. For example, the average adult in the United States is assumed to weigh 70 kg, whereas the average adult in Japan is assumed to weigh 60 kg (14). Pediatric patients could be 30 kg or below. The average weights for children can be found on the Centers for Disease Control and Prevention clinical growth charts page (see www.cdc.gov/growthcharts/clinical_charts.htm). The body weight factor selected for pediatric and other special category patients should consider the worst case, i.e., lowest body weight in targeted patient populations that can receive the greatest recommended dose. There is also a special consideration with respect to body weights for veterinary products. Veterinary drug products may be administered to a variety of different species or subspecies. Generally, the smallest animal will have the greatest dose per kilogram. Reference to the product package insert is highly recommended when establishing veterinary endotoxin limit specifications.
Different routes of administration or types of product, e.g., radiopharmaceuticals or oncology products administered per square meter of body surface, have defined values for K in the endotoxin limit calculation as described in 〈85〉 and above. Where the product package insert describes multiple patient populations, indications, and routes of administration, it is suggested that the laboratory calculates the limit specification for each administration and chooses the most conservative one as the endotoxin limit specification for the product. A summary is shown in Table 2.
Table 2. Defined Values for K in Terms of Route of Administration
| Route of Administration | K | M |
| Intravenous (IV) for parenteral products | 5 EU/kg of body weight | Maximum dose per kilogram administered in 1 h |
| IV for radiopharmaceuticals | 175 EU | Volume of the maximum recommended dose |
| Intrathecal (IT) for parenteral products | 0.2 EU/kg of body weight | Maximum dose per kilogram administered in 1 h |
| IT for radiopharmaceuticals | 14 EU | Volume of the maximum recommended dose |
Parenterals administered per square meter of body surface | 100 EU/m2 | Maximum dose per square meter per hour |
Injections other than IV (intramuscular, subcutaneous, etc.) | 5 EU/kg of body weight | Maximum dose per kilogram administered in 1 h |
| Intraocular fluids | 0.2 EU/mL (15) | - |
| Anterior segment solid devices | 0.2 EU/device (15) | - |
| Ophthalmic irrigation products | 0.5 EU/mL (〈771〉) | - |
Injected or implanted ophthalmic drug product | 2 EU/dose (〈771〉) | - |
Some USP product monographs have endotoxin specifications defined at a targeted concentration for administered product. However, endotoxin limit specifications should be calculated for all indications in the product’s package insert because indications and administrations for the product may be different from the data used to calculate the original USP monograph limit. If a firm’s most stringent limit is lower than the USP limit, the firm should use its lower calculated endotoxin limit.
It is important that the endotoxin limit specification, as a critical quality attribute for a new product, be calculated early in development and monitored throughout development and early-stage clinical trials. If a dose has not been established, the limit should be calculated based on the worst case (highest) dose that is anticipated for the product with respect to the target patient population and the route of administration.
Early phase endotoxin limits may change based on dosing and/or formulation changes prior to commercialization.
13.2 Relevance of Limits for Compounded Sterile Preparations
When sterile compounding pharmacies prepare therapies for injection or infusion, care must be taken to avoid the addition of endotoxins to the preparations. The compounding pharmacy should use only product contact materials that they have depyrogenated in house or that they have received as sterile and free of detectable endotoxins (see Screening and Qualification of Consumables: Compendial Requirement earlier in the chapter). When diluents or intravenous (IV) solutions are used for preparing a product intended for IV, IM, intraocular, or intrathecal (IT) administration, the diluents should be commercially obtained and should meet the compendial limits, which most commonly are 0.5 EU/mL. If the required diluent is not a USP monograph article, it should be manufactured to meet the compendial limit of 0.5 EU/mL. If the required diluent is to be used for IT administration, it is essential that the laboratory ensure that the diluent plus drug product does not exceed the more stringent IT endotoxin limit.
13.3 Calculating Endotoxin Limits for Active Substances and Excipients
The control of the levels of endotoxins in excipients and active substances can minimize the risk of finished drug product contamination.
Suppliers and drug manufacturers should perform risk assessments of these substances based on raw material origins, production methods, representative sampling and testing, and storage conditions. For example, materials of natural origin such as sugars, heparins, and enzymes may contain significant levels of endotoxins and/or glucans. Suppliers of these materials should be audited or closely evaluated to ensure control of bioburden and endotoxins in their manufacturing operations. If these suppliers provide a CoA regarding the endotoxins or glucan content for individual lots of material, the evaluation should also include an assessment of the validity of test methods and the accuracy of test results and any testing history.
Consideration should be given to using a glucan blocker for products or materials that may contain glucans in addition to endotoxins. Glucan blockers are used to ensure that all tests are specific to endotoxins, preventing a false-positive reaction of the test to glucans. Because products do not have glucan specifications, glucan-blocking reagents are specific to assay methods and lysate formulations and are generally offered by lysate reagent manufacturers. Because glucan-blocking reagents are used in different ways, it is recommended that laboratories follow the reagent manufacturers' instructions for the use of these reagents. If noncompendial materials or articles are tested and released in-house, endotoxin limits should be assigned after a thorough understanding of their potential contribution to the formulation. Working backwards from the calculated drug product specification for endotoxins, limits can be assigned to each individual component in the formulation with the assurance that the drug product limit would not be exceeded if each component were at its limit. If the same component is used in multiple formulations, the lowest limit should be used for the testing and release of that component.
13.4 Calculating Endotoxin Limits for Combination Products
A combination product is defined as a product comprised of two or more regulated components (i.e. drug/device, biologic/device, drug/biologic, or drug/device/biologic) that are physically, chemically, or otherwise combined or mixed and produced as a single entity. It also is defined as two or more separate products packaged together in a single package or as a unit and comprised of drug and device products, device and biological products, or biological and drug products (16,17). Typically, these products can be two or more regulated components presented as a single-entity product (e.g., prefilled syringe), or a kit where two or more regulated products are packaged for use together as a single-entity product (e.g., a lyophilized drug packaged with a diluent and a syringe). Although a manufacturer can propose and justify unique limits for review in regulatory submissions, there are some general points to consider:
- If the combination product is a drug/device combination such as a prefilled syringe, the prefilled syringe may be tested as a filled unit and the endotoxin limit for the drug product prevails. Any endotoxins contributed by the container (device) are assumed to be eluted with the drug product during sample preparation and, therefore, are accounted for in the product's assayable endotoxins.
- If the combination product is two drugs to be administered simultaneously [IV or intramuscular (IM)], then the endotoxin content of the combined dose may not exceed the endotoxin limit for drugs of 5 EU/kg/h for IV or IM administrations or 0.2 EU/kg/h for IT administration.
- If the combination product is a kit containing multiple components that are administered as a single entity (e.g., a lyophilized product/diluent/syringe) the endotoxin content of the combined dose may not exceed the endotoxin limit for drugs of 5 EU/kg/h for IV or IM administrations or 0.2 EU/kg/h for IT administration.
13.5 Calculating Endotoxin Limits for Medical Devices
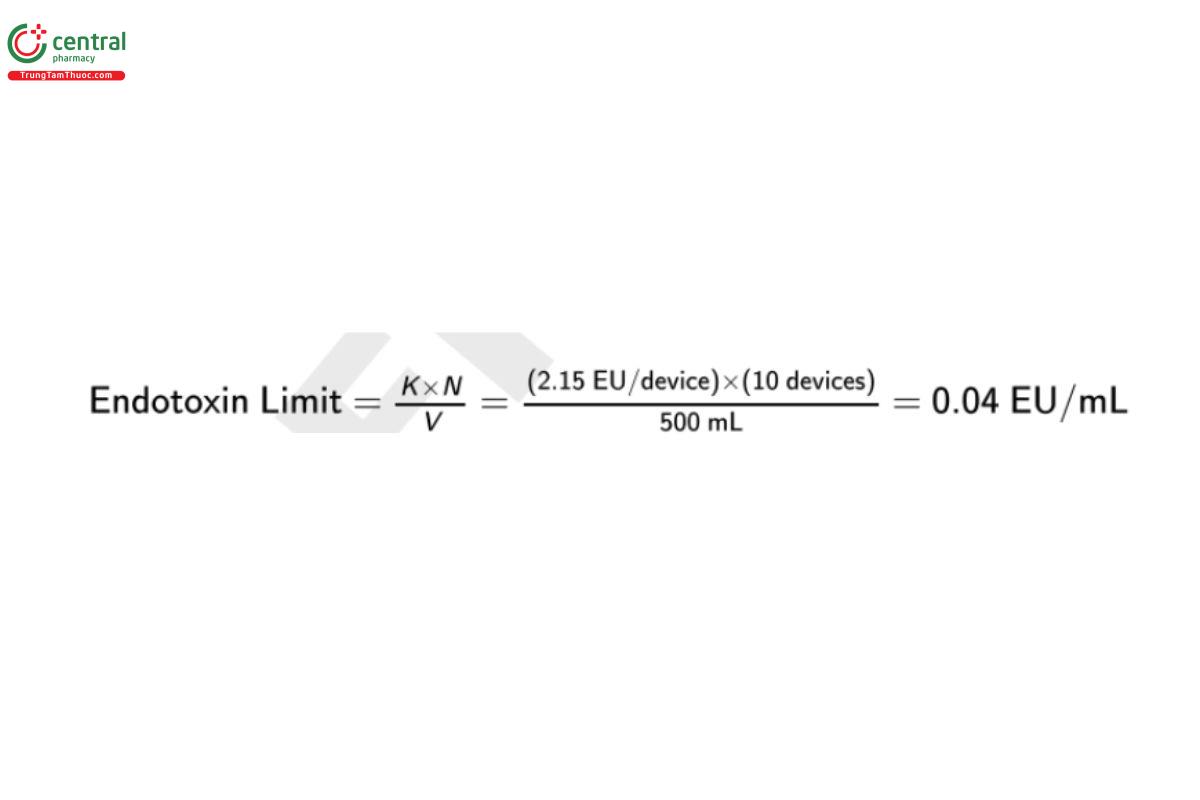
Chapter 〈161〉 assigns the endotoxin limit for medical devices as 20 EU/device except for those medical devices that come in contact with the cerebrospinal fluid, which has an assigned limit of 2.15 EU/device. Devices that contact the anterior segment of the eye should not exceed a limit of 0.2 EU/mL or 0.2 EU/device, as appropriate. Endotoxins in or on solid matrix medical devices are not measured directly, but rather the device is rinsed, soaked, or extracted in an appropriate volume of solvent (generally WFI) and the extracts are tested; in some cases extracts from several devices are pooled for testing. As a result, the endotoxin limit for a device extract is expressed in EU/mL, which can later be converted mathematically to EU/device. The endotoxin limit for an extract is inversely proportional to the volume of solvent used for the extraction. The relationship is:
Endotoxin Limit = (K x N)/ V
K = where endotoxin limit per device (20 EU unless otherwise defined and justified; 2.15 EU/device for IT devices)
N = number of devices represented in the pool
V = total volume of solvent used to extract the devices
For example, if the laboratory tests 10 IT devices, each with an extraction volume of 50 mL, the endotoxin limit specification for the pooled extract is:
[Note—Although primary packaging components such as vials or stoppers may be tested using the techniques described in 〈161〉, they are not considered to be medical devices. The laboratory should assign endotoxin limits to these components that are much lower than the limits for standard medical device, but appropriate for the drug formulation and presentation. Also some medical devices are liquid (e.g., dialysis fluid) or a solid (e.g., an enzyme). For these products, the endotoxin limit is calculated or assigned and tested as if the device were a drug.]
14 Maximum Valid Dilution
As product-associated interferences are diluted, so will any endotoxins in the sample be diluted. Therefore a calculation called the MVD is included in the compendial chapter to define the upper bound of allowable product dilution. The MVD is dependent on the endotoxin limit for the product, the starting concentration of the product (generally the concentration of the active ingredient), and the sensitivity of the test method. The MVD is defined as:
MVD = (Endotoxin Limit)×(Product Concentration)/ λ
where
- The endotoxin limit is the calculated limit for the product or device.
- The product concentration equals the concentration of the active ingredient in units per milliliter. For those products administered on a milliliter per kilogram basis or for medical device extracts, the product concentration equals 1.
- λ = the confirmed label claim sensitivity for gel-clot method or the lowest point on the referenced standard curve for the quantitative tests
Additional information on the MVD includes the following:
- The endotoxin limit is constant for any given formulation/dose/administration.
- The MVD is directly related to the starting concentration of active ingredient, and the higher the starting concentration, the higher the MVD.
- The MVD is inversely related to the numerical value given to the test method sensitivity. The more sensitive the test (λ as the denominator in the MVD formula gets lower), the higher the MVD. Changing the test sensitivity to a lower number (more sensitive test) will assist in providing additional dilution room for interfering products.
- The units in the formula cancel out. The resulting calculated value is a dilution factor with no units. For example, an MVD of 100 means that maximum dilution for the product concentration can be diluted no further than 1:100 to have a valid test.
- The MVD does not limit the necessary product dilution when determining the true amount of product contamination in a sample that fails to meet the endotoxin limit. When a product fails at the MVD, it does not meet the endotoxin limit requirement. In this case, it is recommended that the laboratory determine the total bacterial endotoxin content in that product by dilution to extinction (a negative test for gel clot and a valid reading on the quantitative standard curve). The actual level of endotoxin contamination in the product may prove very helpful for trending purposes and to determine the root cause of the contamination during the OOS investigation.
15 Method Suitability Testing
15.1 Interference screening for drug products, including biologic drugs
The principles and practice for performing inhibition/enhancement testing are provided in 〈85〉, Gel-Clot Technique, Preparatory Testing, Test for Interfering Factors or 〈85〉, Photometric Quantitative Techniques, Preparatory Testing, Test for Interfering Factors. Although historically suitability tests utilizing three consecutive lots of drug product were considered sufficient to assess suitability, it is recommended that an appropriate number of lots of product be determined prospectively for suitability testing to enable a valid assessment for the potential of lot-to-lot variability in endotoxins content. This is especially important for biological products or products where product development and process validation has indicated significant lot-to-lot variability. Products with greater variability in their starting material, API, and manufacturing process will typically require more than three lots for suitability testing, whereas for products with little or no process or product variability, three lots may suffice. The number of lots tested for suitability should be supported by a risk assessment including information on the life cycle stage of the product (clinical/commercial), known sources of variability, and material testing history that could support an increased or decreased number of lots chosen for suitability testing. All materials that are being tested using methods described in 〈85〉 including excipients and raw materials, should have a suitability study to assure that any interferences and variability are identified, mitigated (if necessary) and taken into account in the testing procedures.
15.2 Common test interferences
Most pharmaceutical products have been found to interfere to some extent with BET performance (18). Because of the high assay sensitivity, these product-specific interferences can usually be overcome by dilution in Water for BET [see Bacterial Endotoxins Test 〈85〉,
Reagents and Test Solutions, Water for Bacterial Endotoxins Test (BET)], not to exceed the product-specific MVD. Interferences may affect either the enzyme cascade of the LAL reaction itself or the analyte used as the PPC (e.g., purified LPS), standard, or both. Table 3 is a listing of common interferences and mitigations that may be considered and implemented beyond mere dilution (19). Where buffers or solvents other than Water for BET are used for dilution they should be free of detectable endotoxins.
Table 3. Common Interferences and Mitigations
| Interference | Interferes With | Mitigation |
| pH | LAL cascade | Dilution in Water for BET Adjust pH of the product with hydrochloric acid (ERR 1-Dec-2020) , sodium hydroxide, or dilution in tris buffer so that lysate + sample is within the optimal range specified by the lysate manufacturer. |
| Osmolarity | LAL cascade | Dilution in Water for BET is usually sufficient. |
| Chelating agents | LAL cascade LPS aggregation | Dilution in Water for BET Add a magnesium ion-containing buffer. |
| Glucan | LAL cascade | Use a glucan blocker supplied by the reagent manufacturer. |
| Nonspecific protein interference (e.g., serine proteases) | LAL cascade | Dilution in Water for BET Dilution in Water for BET combined with heat |
| Heavy metals | LAL cascade LPS aggregation | Dilution in Water for BET or diluents containing 1 mM chelating agent ethylenediaminetetraacetic acid (EDTA) |
| Proteins | LAL cascade LPS aggregation | Dilution in Water for BET or normal saline Dilution in Water for BET combined with heat |
| High detergent | LAL cascade LPS aggregation | Dilution in Water for BET is usually sufficient. |
| Calcium cation | LAL cascade | Dilution in Water for BET or diluents containing 1 mM chelating agent EDTA |
A majority of test interferences are overcome by simple dilution. Dilution can be in Water for BET, buffers (e.g., tris or HEPES), buffers containing glucan-blocking agents, and buffers containing divalent cations or chelating agents as required. Dispersing agents may be added to diluents to further mitigate interference, and their value as an additive is typically considered during method development. It is noteworthy that glucan interference is a common interference in some biological products. Where glucan interference is likely, the use of a glucan blocker should be considered during assay suitability test method development. Diluents other than Water for BET and common buffers described above should be checked to assure that they do not interfere with the BET assay. For example, some neutralizing agents (e.g., proteinase K) may not be manufactured with appropriate controls against their contamination with GNB. If these reagents are used, they should be checked to assure that they do not contain endotoxins at levels that may contribute to the levels ultimately reported for the product. All BET assays must be conducted at neutral pH. Neutral pH for BET assays is generally defined as 6.0–8.0, but because all lysates are different in their formulations, the laboratory should confirm the proper range as described in the lysate manufacturer’s package insert.
However, the reagent itself may provide buffering capacity so the mixture of lysate and product dilution should be tested for pH during the suitability study. pH may be adjusted to neutrality by the use of buffers as diluents or endotoxin-free hydrochloric acid (ERR 1-Dec-2020) (HCl) or sodium hydroxide (NaOH). If adjustment of pH is required using acid or base, it is suggested that the pH be measured during the suitability study to assess lot-to-lot variation in the product.
Lysates from different manufacturers have different proprietary formulations, and all are calibrated for sensitivity against the RSE to ensure that they all detect LPS activity equally well in water. However, LPS activity may not be recovered equally in different products, or by using different compendial test methods. If there is significant product-specific interference that cannot be overcome by dilution to within the MVD or by standard mitigation methodologies (Table 3), the laboratory should consider a different lysate formulation or test method before concluding that the LAL-bacterial endotoxins test is either invalid or unacceptable (20).
16 Qualifying Test Preparation Methods Other than Dilution
For sample preparation that requires manipulation other than simple dilution, e.g., heating or ultra filtration, 〈85〉 instructs the analyst to proceed as follows:
Interference may be overcome by suitable treatment such as filtration, neutralization, dialysis, or heating. To establish that the chosen treatment effectively eliminates interference without loss of endotoxins, perform the assay described above using the preparation to be examined to which Standard Endotoxin has been added and which has then been submitted to the chosen treatment.
17 ROUTINE TESTING
17.1 Sampling
All materials used to sample materials for endotoxin content (scoops, pipettes, bottles) must be inert with respect to the material(s) being sampled, and they must be sterile and free of detectable endotoxins. Even though samples are often taken in the field without the aid of a filaminar flow hood, samplers must take precautions that a) they do not contaminate the sample itself, and b) in taking the sample, they do not contaminate the rest of the material in its original container.
Historically, the sampling scheme for finished drug products is to take at least 3 units representing the beginning, middle, and end of the batch. However, for a standard run of a small-volume parenteral, biological therapy, or large-volume parenteral, 3 units may not be a representative sample and may identify only those lots that are uniformly and highly contaminated. Sampling schemes should be justified and should be based on the known variability of the process, the unit operations of the process, historical knowledge of the process, and materials used in manufacture.
Bacteria and endotoxins are generally not homogeneously distributed in any product or material, liquid or powder. Care must be taken to flush ports in a circulating water loop before sampling and use the same equipment for sampling that is used for manufacture to assure a representative sample. For example, if a hose is attached to a port for the purpose of transferring water from a circulating loop to another formulation vessel, the water sample must be taken from the same hose with the same flush times as used by manufacturing. So, while the water within a properly constructed and controlled loop may theoretically be homogeneous, differences in the ports along the distribution loop may add endotoxins to samples.
For highly viscous materials, a suitable inert rod free of detectable endotoxins may be used to mix the material prior to sampling. Sterile, pyrogen-free spatulas and scoops are used for powdered and granular solids. Because of the lack of assurance of homogeneity, the number of samples from viscous materials and powders should be determined relative to the number of units initially received using appropriate statistical procedures.
Particularly for samples of viscous and powdered materials, samplers should have training on how to spot signs of non-uniformity. Such indicators may include differences in shape, size, or color or evidence of moisture in powdered materials. For viscous materials, samplers should be trained to look for stratification of the material, differences in viscosity or colors, and particulate contamination. Samples of raw materials that could exhibit variability in endotoxin content should not be pooled (see Pooling).
In the case of a new vendor of a raw material, the methodology and calculations used in the determination of the endotoxin content of the material should be reviewed. If the material is designated as critical for manufacturing, a site audit of the vendor may be in order. In addition, it is highly suggested that an in-house confirmation of the accuracy of the endotoxin level stated on the CoA be performed. For critical materials, the quality agreement should require that manufacturers of these materials inform the pharmaceutical manufacturer of changes in processes, controls, or raw materials so that the changes can be discussed at change control and the need for re-confirmation of materials can be determined. Once the CoAs have been successfully confirmed for a predetermined number of shipments justified in the sampling plan by history, and depending on the variability of the manufacturing process, the laboratory can consider accepting the CoA with periodic testing of the incoming materials, as long as the manufacture of the materials has not changed.
17.2 Pooling
The term "pooling" refers to creating a composite sample preparation that includes the total contents of several individual units or equal aliquots from the units taken from the same lot or batch. Pooling is often done for batch-release testing. Pooling is an acceptable option for laboratories performing any of the BET assays on drug products (9). However, pooling has a number of drawbacks; the most obvious is that it will dilute endotoxins that may be in any one of the units, potentially concealing the variability in endotoxin content and distribution among the units that were pooled. Sampling plans, including instructions to pool samples, should be scientifically justified.
If units are pooled, the MVD must be adjusted to account for the possibility of endotoxins in just one of the samples (9). The adjusted MVD is calculated by dividing the originally calculated MVD by the number of units contributing to the pool. For example, if the MVD for a small volume parenteral is 240 and a manufacturer chooses to pool three vials for testing, the adjusted MVD is 240/3, which is 80. This reduction in MVD effectively reduces the endotoxin limit for the product by a factor of three to compensate for pooling. While pooling may result in an incremental savings in reagent usage and therefore save some money, there are a number of points to consider when pooling:
- Pooling may obscure any non-uniformity in endotoxin content between the individual sample units. Information on variability may be valuable in troubleshooting or investigations. For example, random contamination in one of multiple filling needles may cause some vials to contain endotoxins and others not.
- Taking aliquots of samples for pooling should always be performed using aseptic technique and with individual units vigorously mixed prior to removing the aliquots. The original containers with remaining product should be retained for investigation in the event of an OOS test result. Removing the aliquot through the disinfected rubber stopper using a pyrogen-free syringe is advisable to maintain the integrity of the unit container during subsequent storage for investigative testing.
- The concept of adjusted MVD does not apply to medical devices as they are, by convention, commonly pooled for testing.
- Any sampling scheme for drug products must represent the beginning, middle, and end of the batch. Additional samples may be taken if interventions in manufacturing raise concerns about possible endotoxin contamination.
- If testing at the adjusted MVD causes an unacceptable increase in product-specific interference, samples should be tested individually.
- Products with low calculated MVDs, or suspensions where there is no assurance of homogeneity in the removed aliquots, may not be good candidates for pooling.
- Pooling is not appropriate for in-process samples, particularly those representing different stages of manufacturing.
17.3 Calculation of Endotoxin Content
All BET results are provided in EU/mL. If the product being tested has a limit expressed in EU per unit of weight or activity (e.g., EU/mg), a calculation must be made to convert EU/mL to EU/mg. For example, the endotoxin content for a product with a starting concentration of 10 mg/mL, after adjustment for the dilution factor, is determined by gel clot to be 5 EU/mL. The endotoxin content expressed in EU/mg is:
(5 EU/mL) ÷ (10 mg/mL) = 0.5 EU/mg
17.4 Out-of-Specification Results and Retesting Considerations
An endotoxins test result that does not meet product specifications necessitates an investigation (21). The suspect result is considered valid unless a comprehensive investigation clearly demonstrates otherwise.
Invalid tests, however, are not OOS results. Invalid tests are those where system suitability parameters such as negative controls, PPCs, confirmation of label claim in gel clot, or generation of a linear standard curve do not function as expected and therefore may affect the accuracy of the test results. Although invalid tests should be tracked and trended to look for patterns and trends that might require a corrective action, a true OOS test failure only exists when a valid assay has been performed and generates results that exceed the specification.
A finding of a nonconforming test result requires a laboratory assessment to assure the accuracy of the data. The FDA OOS guidance for industry terms this a Phase I investigation (21). Despite the fact that a failed BET assay is rare, the investigation should be done in a timely manner to ascertain if there was anything atypical about this sample, its preparation, calculations, or performance of the test that may affect the accuracy of the test result. Phase I should include a critical and in-depth review of sampling methods and techniques, sample hold conditions, sample preparation, test method parameters, and any documentation, including real-time notations of testing errors by analysts and a review to find any errors in calculation. An organizing tool, such as a fault tree analysis (FTA) or Ishikawa (shbone) diagram, may help the team members to organize their thoughts and complete the investigation in a timely and compliant manner.
The use of a checklist approach will promote completeness and consistency across all assessments.
The purpose of the Phase I laboratory investigation is to confirm the accuracy of the original test result. A Phase I checklist should include the following, at a minimum:
- The testing history of the material under test: Has a specific assay problem or repeated failures for the same product been previously observed?
- Sampling procedures, sample container integrity, and sample preparation: Were samples taken, transported, and stored correctly, following approved methods?
- Reagent preparation, storage, and use.
- Calibration and maintenance status of dispensing, incubation, and reading instrumentation.
- Appropriateness of sample contact equipment and disposables.
- The test procedure: Is it consistent with the suitability data, and was it precisely followed?
- Were all controls and other system suitability indicators within normal limits? If not, then the test is not OOS and is invalid.
- Were all calculations (product endotoxin limits, MVD, and endotoxin contamination levels) performed correctly? Were all transcriptions performed accurately?
- Was the analyst adequately trained? What is the analyst’s OOS history?
An approved BET standard operating procedure (SOP) should include two important directions to analysts:
1. If an analyst realizes and documents that a mistake has been made during the testing, including an error in calculation, sample preparation, dilution, or performance of the assay that could affect the accuracy of the test results, the test should be stopped and the reason for the termination of the test documented at the time that the error was identified.
2. Analysts should not discard the original sample, any of the sample preparation dilution tubes, or the reagents used until the results of the assay are known and evaluated against the product specification. The tubes and reagents from an OOS assay may be an important part of the laboratory investigation.
If there is no analytical failure or documentation error found, the result is considered valid and the product is OOS. On the other hand, if the analysis was conducted incorrectly and documented error(s) are the root cause of the failure, the initial assay is invalid and the product may be tested again with appropriate controls.
Investigation of production practices, otherwise known as Phase II investigations, require assessment of all manufacturing activities that could have impacted the endotoxin content of the failed product sample. Phase II may be more easily conducted by a cross-functional team including operations, facilities, and engineering, with assistance from a microbiologist to identify potential sources of contamination relating to contamination control requirements during manufacturing.
The Phase II investigation is actually a reassessment of validated process control, particularly if the failure was not the first failure for the product. A product manufactured under a state of control should not fail endotoxins testing, so the Phase II investigation should focus on anything that may have been different about the lot in question.
At any point in the investigation, testing can be performed to challenge hypotheses regarding the reason for the failure of the material to meet its limit. However, it is important to note that these investigational tests are not considered to be re-tests and should not be used to release material. Rather, they are used to identify conditions that might have contributed to an inaccuracy in the testing of the product. A laboratory SOP or policy should also be written that precisely describes the procedure(s) that must be followed for the purposes of investigational testing. Such a plan eliminates situational decisions regarding the appropriateness of investigational testing, retesting, or resampling; the plan should be approved by the quality unit.
Investigational testing should be well-defined and justified, pre-approved, and well-documented. If an investigational second test of the original sample preparation finds that the product meets the specifications for the test, it is possible that an error was made in some aspect of the execution of the original assay or just as easily in the execution of the second test. If this is the case, any hypotheses should be proposed and testing executed to confirm that an error was made. The intended purpose of the investigational tests on the sample is not to reverse the original OOS result but rather to gain information regarding a potential failure so that a more effective effort can be made to pinpoint the problem and implement any necessary corrective and preventive actions.
The statement in 〈85〉 that reads “In the event of doubt or dispute, the final decision is made based upon the gel-clot limit test unless otherwise indicated in the monograph for the product being tested” has been widely misinterpreted and is relevant only to the specific case of a conficting result between a regulatory agency using the gel-clot method for lot release testing and a testing laboratory using a different LAL-bacterial endotoxins test platform. It is not intended to allow a laboratory to use a gel-clot result that provides passing results when the laboratory's customary assay method for the product shows an OOS test result.
In the event of a product failure, the total level of endotoxins activity in the failed sample(s) should be determined. For gel-clot assays, this may involve testing of dilutions beyond the MVD until an endpoint is reached for gel clot or a valid test result is obtained for quantitative tests. These values may provide a clue as to the source of potential or actual product contamination, or a case of mishandling of a positive control, or a tube mix-up.
Standard Curve Control
Standard curves are created for endpoint chromogenic assay by evaluating the direct relationship between color intensity and standard concentration. The resulting curve has a positive slope, and 〈85〉 indicates that the correlation coefficient of the curve must be ≥0.980.
The standard curve is created by plotting the log of the onset or reaction time required for each standard to reach a predetermined optical density for kinetic turbidimetric assays or color intensity for kinetic chromogenic assays, as a function of the log of the standard concentration. This transformation of the data results in a standard curve with a negative slope, and the correlation coefficient must be ≤−0.980. Therefore, it can be stated that the linearity requirement for all compendial quantitative BET assays is |r| ≥ 0.980. The requirement is expressed to three significant gures because the last “0” is important; it is inappropriate to have a standard curve with |r| = 0.979 and round up. However, in a well-controlled laboratory, correlation coefficients should routinely be greater than 0.980.
All standard curves have a corresponding linear equation y = mx + b where m is the slope of the standard curve regression line and b is the fly-intercept. Because the y-intercept is the point where x = 0, and because in a kinetic assay the log of 1 is 0, the y-intercept is really at the 1 EU standard. Because of the transformation of data to log , standard curves for quantitative assays can be very sensitive to small changes in onset times.
Accuracy of test results depends on the accuracy of the standard curve. Therefore, the onset times for standards representing unique combinations of lysate lot and CSE lot from run to run, instrument to instrument and analyst to analyst should all be monitored. For example, an analyst can make a twofold or tenfold dilution error in the dilution of the standards, yet still produce a linear standard curve that meets the requirement |r| ≥ 0.980. The dilution error would not be noticed by looking at the correlation coefficient alone, but would be evident in the onset times of the standards. An overdiluted set of standards would run more slowly (longer onset/reaction times) than a properly diluted standard series, and an underdiluted set of standards would run more quickly (shorter onset/reaction times) than a properly diluted standard series. The dilution error would also be refiected in the values generated for the y-intercept. Changes in slope can also affect the accuracy of the test data. It is suggested that part of an analyst’s training in quantitative assay performance is to understand the impact of variability on the accuracy of the test result (22,23).
18 ALTERNATE TEST METHODS
The methods listed in 〈85〉 for the detection of bacterial endotoxins (gel-clot limits test, gel-clot assay, kinetic chromogenic, endpoint chromogenic, kinetic turbidimetric) are considered to be validated. However, a laboratory may choose to use an assay methodology that is not listed in 〈85〉. If such a choice is made, the alternate test for the detection of bacterial endotoxins must be fully validated to ensure that decisions made using the alternate methodology are equivalent to or better than decisions made using the validated USP methods and ultimately approved by the appropriate regulatory authority. Although endotoxin testing is not specifically cited, guidance on how to think about the validation of alternate methods can be found in Validation of Alternative Microbiological Methods 〈1223〉 and Validation of Compendial Procedures 〈1225〉.
19 GLOSSARY
Bacterial endotoxins: A GNB outer membrane macromolecular complex of polysaccharide, lipid, and protein. Extracted, purified, and highly concentrated (protein free) endotoxin is generally referred to as lipopolysaccharide or LPS to distinguish it from the more natural complexed cell membrane associated form (24).
Bacterial endotoxins test (BET): Compendial bacterial endotoxins test.
Control standard endotoxin (CSE): A preparation with a stable endotoxin concentration calibrated against RSE. CSEs are generally prepared by vendors or by laboratories, and although called "standards," are not certified for either purity or activity by a third party. CSEs are best described as calibration analytes. CSEs may be purified or they may be a preparation of native endotoxins. If a CSE is a preparation not already adequately characterized, its evaluation should include characterizing parameters such as activity, uniformity, and stability, which would demonstrate the suitability of the material to serve in the calibration of a BET. Detailed procedures for its preparation and use to ensure consistency in performance should also be included.
Intrathecal: A parenteral injection that results in the product coming in contact with the cerebrospinal fluid.
Lysate or limulus amebocyte lysate (LAL): The reagent used in the performance of the BET
Parenteral: Drugs or medical devices that are injected, infused, or implanted or that may otherwise come in contact with the bloodstream or cerebrospinal fluid.
USP Endotoxin Reference Standard (RSE): Primary endotoxin standard. RSE is an extracted, purified, and formulated preparation of E. coli 0113:H10:K LPS.
Suitability: Demonstration that the chosen assay is appropriate for its defined purpose in testing the material.
20 REFERENCES
1. Brogden KA, Phillips M. The ultrastructural morphology of endotoxins and lipopolysaccharides. Electron Microsc Rev. 1988;1:261–277.
2. Beveridge TJ. Structures of Gram-negative cell walls and their derived membrane vesicles. J Bacteriol. 1999;181:4725–4733.
3. Ellis TN, Leiman SA, Kuehn MJ. Naturally produced outer membrane vesicles from Peudomonas aeruginosa elicit a potent innate immune 1212 response via combined sensing of both lipopolysaccharide and protein components. Infect Immun. 2010;78(9):3822–3831.
4. Kulp A, Kuehn MJ. Biological functions and biogenesis of secreted bacterial outer membrane vesicles. Annu Rev Microbiol. 2010;64:163–184.
5. Schwechheimer C, Kuehn MJ. Outer-membrane vesicles from Gram-negative bacteria: biogenesis and functions. Nat Rev Microbiol. 2015;13(10):605–619.
6. Bonnington KE, Kuehn MJ. Outer membrane vesicle production facilitates LPS remodeling and outer membrane maintenance in Salmonella during environmental transitions. mBio. 2016;(5):e01532–16.
7. Rudbach JA, Akiya FI, Elin FJ, Hochstein HD, Luoma MK, Milner EC, et al. Preparation and properties of a national reference endotoxin. J Clin Microbiol. 1976;3(1):21–25.
8. Poole S, Dawson P, Gaines D. Second international standard for endotoxins: calibration in an international collaborative study. J Endotoxin Res. 1997;4(3):221–231.
9. Food and Drug Administration. Guidance for industry. Pyrogens and endotoxins testing: questions and answers; 2012.
10. Bolden J, Knight M, Stockman S, Omokoko B. Results of a harmonized endotoxin recovery study protocol evaluation by 14 BioPhorum Operations Group (BPOG) member companies. Biologicals. 2017;48:74–81.
11. Bolden J, Platco C, Dubczak J, Cooper JF, McCullough KZ. The use of endotoxin as an analyte in biopharmaceutical product hold-time studies. Pharm Forum. 2015;41(5).
12. Griesman SE, Hornick RB. Comparative pyrogenic reactivity of rabbit and man to bacterial endotoxin. Exp Biol Med. 1969;131(4):1154– 1158.
13. Weary ME, Donahue G, Pearson III FC, Story K. Relative potencies of four reference endotoxin standards as measured by the limulus amebocyte lysate and USP rabbit pyrogen tests. Appl Environ Microbiol. 1980;40(6):1148–1151.
14. G4 microorganisms: decision of limit for bacterial endotoxins. In: Japanese Pharmacopeia 17. Tokyo: Pharmaceuticals and Medical Devices Agency; 2016:2481.
15. Food and Drug Administration. Endotoxin testing recommendations for single-use intraocular ophthalmic devices: guidance for industry and food and drug administration staff; 2015.
www.fda.gov/downloads/medicaldevices/deviceregulationandguidance/guidancedocuments/ucm393376.pdf.
16. 21 CFR3 Part 3 (1) and (2)
17. Food and Drug Administration. Guidance for Industry and FDA Staff. Current good manufacturing practice requirements for combination products; 2017. www.fda.gov/downloads/RegulatoryInformation/Guidances/UCM429304.pdf.
18. Twohy CW, Duran AP, Munson TE. Endotoxins contamination of parenteral drugs and radiopharmaceuticals as determined by the limulus amebocyte lysate method. J Parenter Sci Technol. 1984;38(5):190–201.
19. Dubczak J. Resolving test interferences. In: McCullough KZ, ed. The Bacterial Endotoxins Test: A Practical Guide. DHI Publishing; 2011.
20. McCullough KZ, Wiedner-Loven C. Variability in the LAL test: comparison of three kinetic methods for the testing of pharmaceutical products. J Parenter Sci Technol. 1992;46(3):69–72.
21. Food and Drug Administration. Guidance for industry. Investigating out-of-specification (OOS) test results for pharmaceutical production; 2006.
22. McCullough KZ. Constructing and interpreting standard curves for quantitative BET assays. In: McCullough KZ, ed. The Bacterial Endotoxins Test: A Practical Guide. DHI Publishing; 2011:31–62.
23. Bolden JS, Clarebout ME, Matthey KM, Murphy MA, Smith KR, Warburton RE. Evidence against a bacterial endotoxins masking effect in biologic drug products by limulus amebocyte lysate detection. J Parenter Sci Technol. 2014;68(5):472–477.
24. Hitchcock PJ, Lieve L, Makela PH, Rietschel ET, Stritmatter W, Morrison DC. Lipopolysaccharide nomenclature—Past, Present and Future. J Bacteriol. 1986;166(3):699–703.
