Evaluation of Plastic Packaging Systems for Pharmaceutical Use and Their Materials of Construction
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- 1. INTRODUCTION
- 2. SCOPE
- 3. GENERAL PRINCIPLES—THE OVERALL ASSESSMENT PROCESS
- 4. MATERIALS ASSESSMENT: CHARACTERIZATION, SCREENING, AND SELECTION, USP 〈661.1〉
- 5. PACKAGING SYSTEM ASSESSMENT AND QUALIFICATION, USP〈 661.2〉
- 6. APPLICABILITY AND APPLICATION OF 〈 661.1〉
- 7. APPLICABILITY AND APPLICATION OF 〈 661.2〉
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 1. INTRODUCTION
Drug products can chemically interact with their associated packaging systems and/or the system's plastic materials of construction while the drug product is being manufactured, shipped, stored, and administered. The magnitude of these interactions should not adversely affect the suitability of the packaged drug product, which includes both quality aspects and performance aspects such as efficacy, stability, purity, and compendial compliance.
Suitability for use, as determined by the impact of the interaction between a drug product and its packaging system, is assessed and established via the appropriate testing of the materials of construction, components, and packaging systems. Plastic Packaging Systems and Their Materials of Construction 〈661〉 establishes the tests and acceptance criteria that are necessary and appropriate for ensuring that such systems are suitable.
2 2. SCOPE
The purpose of this chapter is to communicate the key concepts behind 〈 661〉 and its related sub-chapters, Plastic Materials of Construction 〈661.1〉 and Plastic Packaging Systems for Pharmaceutical Use 〈661.2〉, and to provide additional information and guidance regarding the application and applicability of these chapters.
3 3. GENERAL PRINCIPLES—THE OVERALL ASSESSMENT PROCESS
The objective of USP plastic packaging standards is to establish the tests and acceptance criteria to ensure packaging systems do not materially impact the effectiveness of the drug product. Given the complex nature of packaging systems and their development and manufacturing processes, multiple testing procedures are needed to establish their suitability with a specific drug product. The logical development and manufacturing process for packaged drug products, starts with the packaging system's materials of construction, continues with the packaging system itself, and ends with the packaged drug product. This progression forms the basis of a three-stage approach to packaging systems qualification, as illustrated in Figure 1.
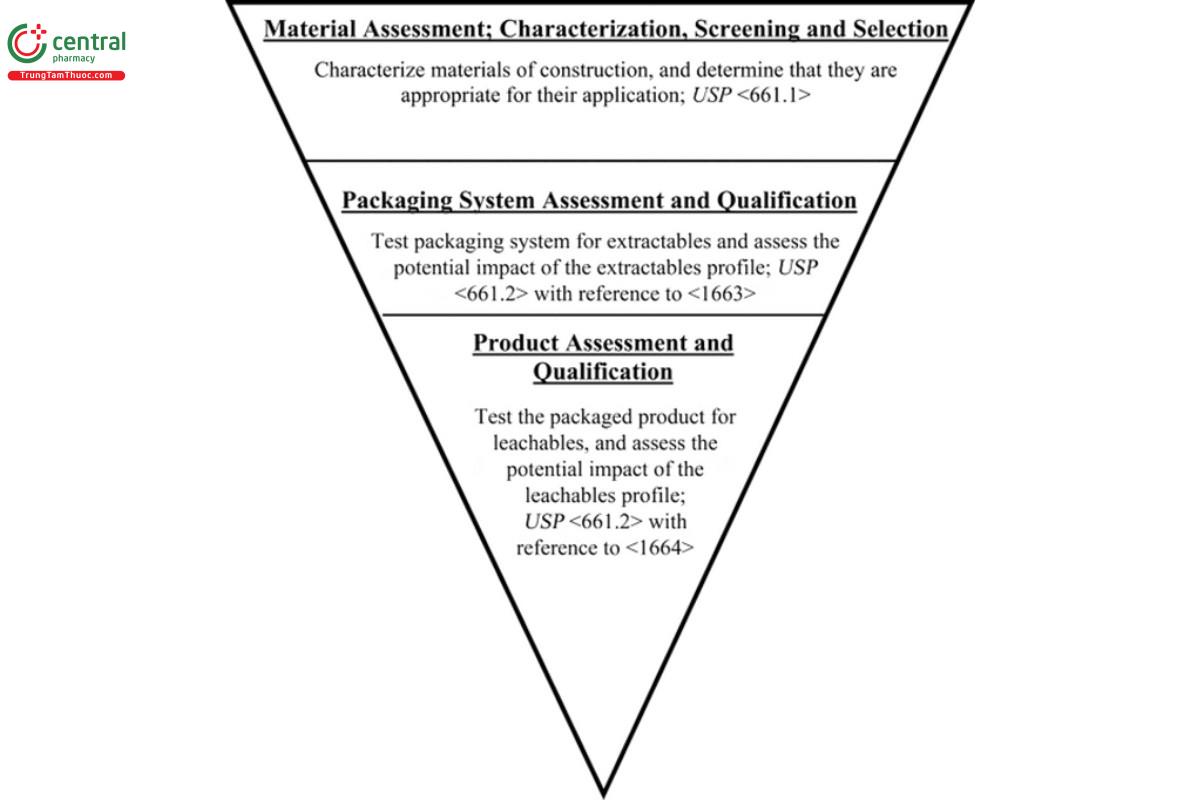
The process for establishing a packaging system's suitability includes: characterization of its materials of construction (ingredients); testing and assessment of the system itself (extractables); and testing and assessment of the packaged drug product (leachables). The initial step of this process involves chemically characterizing candidate materials of construction so the selection of materials can be rationally made and scientifically justified. The intermediate step of system assessment is useful and necessary because it bridges the risk assessment gap between testing starting materials and testing the finished drug product, while providing a means for optimizing drug product testing. This intermediate test is necessary because materials of construction undergo considerable stress, such as exposure to high temperatures, as they are being converted into either components of the packaging system or the packaging system itself. Processing aids and additional additives may be introduced during the manufacturing process for a packaging system, so the extractables profile is likely to be different from, and potentially more complex than, the extractables profiles of its materials of construction. Therefore, the initial hazard identification performed during material selection is appropriately revisited by testing and qualification of the overall packaging system.
Ultimately, the effect the packaging system may have on the drug product end-user is mediated by packaging system-derived substances that are present in the drug product. The third stage of the process includes product assessment, specifically leachables testing of the packaged product and impact assessment, which considers the user's exposure to the leachables.
4 4. MATERIALS ASSESSMENT: CHARACTERIZATION, SCREENING, AND SELECTION, USP 〈661.1〉
Testing and characterizing materials of construction for attributes relevant to their suitability provides a rational basis for material selection in designing a packaging system and minimizes the risk that a system made from those materials will be unsuitable. Therefore, the characterization of materials of construction is the first step in the process of developing and qualifying suitable packaging materials.
Additionally, chemical characterization data may also provide the basis for effective and appropriate change control.
The intent of 〈661.1〉 is to establish whether potential material candidates could adversely affect the quality of packaged drug products. The basic tenet of materials assessment is that knowing the general composition and certain general characteristics of a material of construction allows one to:
Rationally assess the potential suitability of materials with a degree of certainty that is appropriate for early product development and/or manufacturing.
Forecast with some degree of accuracy the identity of extractables from that material of construction and from systems that use that material of construction.
Use the assessment and forecast to establish and justify the use (or rejection) of a particular material in a particular packaging system.
To this end, 〈 661.1〉 defines a well-characterized plastic material of construction as one whose: Identity has been definitively established.
Biological reactivity has been established.
General physicochemical properties have been established.
Extractable elements (when necessary). Additives have been quantified.
Chapter 〈661.1〉 testing is not a guarantee that packaging systems constructed from plastic materials meeting 〈 661.1〉 requirements will be suitable for their intended use. Characterization of a material using 〈661.1〉 establishes the composition or characteristics of the material, which can be used to determine if the material is an appropriate candidate for use in a packaging system. Nevertheless, 〈661.1〉 testing leverages the logical connection between material additives, material extractables, and system extractables, and thus provides a useful indication of the probable suitability issues for materials and systems. The actual qualification of the material occurs when the entire system is qualified for use in a particular application via 〈 661.2〉 testing.
5 5. PACKAGING SYSTEM ASSESSMENT AND QUALIFICATION, USP〈 661.2〉
The impact of packaging systems on the chemical composition of packaged drug product can be established in two ways: 1) the packaging system itself can be characterized with respect to substances that can extract from it (extractables); or 2) the packaged drug product can be tested for packaging-derived substances that have leached into the drug product (leachables). In the case of extractables assessment, the impact is predicted based on a relationship that is established (or inferred) between extractables and leachables. In the case of leachables assessment, the impact is specifically measurable, assuming that all the relevant leachables can be discovered, identified, and quantified in the packaged drug product. In either case, 〈 661.2〉 establishes the tests and acceptance criteria for the packaging system, while referring users to relevant informational chapters (e.g., Assessment of Extractables Associated with Pharmaceutical Packaging/Delivery Systems 〈1663〉 for extractables and Assessment of Drug Product Leachables Associated with Pharmaceutical Packaging/Delivery Systems 〈1664〉 for leachables) for insights on how to design and execute relevant studies.
Considering the packaging system as the test article, the intent of 〈661.2〉 is to define and delineate the testing needed to produce the data required for establishing the packaging system's suitability. Chapter 〈 661.2〉 refers to this process of establishing the suitability of packaging systems as chemical suitability for use assessment and notes that a packaging system is suited for its intended use if:
The packaging system is constructed from well-characterized materials, as established by testing according to 〈661.1〉. The packaging system's general physicochemical properties have been established.
The packaging system's biological reactivity has been established.
The packaging system has been established to be suitable by means of the appropriate chemical suitability for use assessment.
The packaging system is chemically compatible with the packaged product, as established by appropriate compatibility assessments (e.g., stability studies).
5.1 5.1 Extractables and Leachables
For high risk dosage forms, in addition to being the basis for toxicological assessments, information about a packaging system's extractables can be used in several ways to optimize finished product testing for leachables. The potential quality or impact of extractables may facilitate identification of leachables that might adversely affect product quality. Such leachables of potential concern would necessarily be among the targeted analytes in testing of a final drug product within its packaging system. The targeting of specific leachables, as opposed to the screening of drug products for unspecified leachables, has significant analytical benefits, including the ability to develop, validate, and utilize test procedures that are appropriately sensitive, specific, and accurate. Extractables (and their accumulation levels in extracts) can be used to forecast the levels of leachables in the finished product, depending on how well the extraction conditions mimic the drug product's composition and actual conditions of clinical use. If the extraction conditions are such that they accelerate and modestly exaggerate the product's clinical use conditions, then the extractables and their levels in the extracts can be extrapolated to estimate the maximum levels of leachables in the finished product. If such extractables are assessed for their quality impact, the results of that assessment can also be extrapolated to, and considered to be relevant for, the drug product. Finally, if no adverse impact is found based on the extractables data, then no adverse impact can be inferred for the leachables in the packaged drug product.
Change to read:
6 6. APPLICABILITY AND APPLICATION OF 〈 661.1〉
6.1 6.1 Applicability
1. The holder of the drug product application and drug product manufacturer [in the case of many over-the-counter products (OTCs), where there is no application] bear primary responsibility and accountability for ensuring that the requirements of the chapters are met. The means by which the holder of the drug product application and drug product manufacturer obtain information to meet the requirement is at the discretion of the holder.
2. The testing required and acceptance criteria for materials of construction contained within 〈 661.1〉 are relevant to and applicable for all dosage forms, because it is the universal expectation that packaging materials be constructed from well-characterized materials, regardless of the potential interaction between a dosage form. It has been established that the concepts and principles of risk management has a strategic role in designing, implementing, and interpreting effective and efficient material suitability testing and assessments. It is well-established that risk-management tools and principles can be used to define the nature and magnitude of assessment (including testing), where low-risk situations require reduced or alternate assessment (testing) versus high-risk situations noted in Plastic Materials of Construction 〈661.1〉 , Table 1, which establishes biological reactivity and chemical tests that differ for low-risk dosage forms (such as oral and topical) versus high-risk dosage forms (such as inhalation and injections).
3. Application of 〈661.1〉 to materials of construction used for systems other than packaging systems for finished drug product is beyond the scope of this chapter at the present time, but the concepts and principles of the chapter may be applicable and relevant to other systems and their materials for construction (e.g., manufacturing systems for pharmaceutical products, and packaging/storage systems for drug substances).
4. The scope of 〈 661.1〉 is materials of construction for packaging systems for finished drug products and 〈 661.2〉 is packaging systems for finished drug products. A third type of test article is components and if component testing is considered to be necessary, the relevant testing and acceptance criteria for the component are contained within 〈661.2〉.
6.2 6.2 Application
1. There are two means of demonstrating that a plastic material of construction has met the requirements of 〈 661.1〉. The first is to perform the testing contained within 〈661.1〉 and meet the acceptance criteria. The second is the use of the material in a packaging system that meets the requirements of 〈661.2〉.
2. The outcome of 〈 661.1〉 testing is that the tested material of construction has been well-characterized. Characterization data generated during 〈661.1〉 testing can be used to support decisions on the proper use of the tested material. The characterization data do not specifically or universally qualify the material for use in packaging systems because the material's use can vary depending on the packaging applications.
3. It is the responsibility of the user of the tested material to decide if the material is appropriate for their intended application. It is the user's expert review of 〈 661.1〉 test results, coupled with additional information as necessary and appropriate, that establishes whether a well- characterized material is suitable for a specific application.
6.2.1 6.2.1 IDENTIFICATION TESTS
The identification tests required in 〈 661.1〉 serve the purpose of categorizing a material so that it is properly tested and evaluated against the appropriate acceptance criteria. The acceptance criteria for identification are based on a comparison of the test result obtained for the test material versus the relevant Reference Standard. This comparison is based on the concept of substantial equivalence as opposed to an exact quantitative acceptance criteria.
Establishing substantial equivalence requires that the test results and test material versus Reference Standard be considered to be equivalent. For example, although the infrared (IR) identification acceptance criteria may include wavenumber targets, these targets are not acceptance criteria but rather serve the purpose of establishing the expected general characteristics of the IR spectra. An identification test is considered successfully completed if the analytical results obtained for the test article and the appropriate Reference Standard are substantially equivalent, and where all differences between the test results for the article and the Standard are explained by the nature, processing, and/or composition of the test article.
6.2.2 6.2.2 CHEMICAL TESTING
The chemical testing prescribed in 〈 661.1〉 is orthogonal: physicochemical tests provide a general overview of extracted substances whereas plastic additives tests address potential organic extractables. It is also the case that chemical testing alone may not demonstrate all potential impacting attributes. Thus, chemical testing is augmented by the orthogonal approach of establishing biological reactivity.
6.2.3 6.2.3 EXTRACTABLE ELEMENTS
Knowledge of the potential extractable elements present in the materials of construction for plastic packaging components is important in establishing that the material is well-characterized. Knowledge of the elements that are likely to be present and the concentrations at which they may be observed provides information to determine potential drug product quality. Materials of construction can vary widely in terms of their intentionally and unintentionally added elements, the levels of the elements, and the chemical form of the elements. Because of this variety in composition, it is challenging to provide universally effective and efficient test methodologies for extracted elements (including extraction conditions), lists of elements to target, and reporting requirements. Therefore, it is the material user’s responsibility to evaluate the need for extractable elements testing and, if such testing is necessary, to establish and justify the means by which testing is accomplished, taking into account extraction conditions, target elements, extract analysis, and reporting requirements.
In general, however, the following points are relevant:
Extraction conditions should be such that elements that could be leached into pharmaceutical drug products are fully extracted from the test material. Thus, the combination of extraction solvent(s), extraction temperature, extraction duration, extraction process, and extracted surface area to extraction solution volume should be carefully considered. It is important to remember that the objective is to perform an extraction, not to digest or dissolve the test material. Furthermore, the extraction process should produce an extract that is analytically expedient, meaning that the extract is amenable to analysis by the chosen test methodology with the necessary levels of sensitivity, accuracy, and precision.
Target elements should minimally reflect those elements that have been intentionally added (e.g., catalyst and processing aids) to the material, those elements that could be unintentionally added to the material (e.g., processing contaminants, ingredient impurities), and those elements of potential toxicological concern, whether they are intentionally or unintentionally added.
Analytical test methods should be compatible with the extract and should have the specified sensitivity, accuracy, and precision.
Reporting requirements must reflect the analytical capabilities of the method and the levels at which an extracted element could have an adverse effect on drug product quality. The desired situation is that the test method is capable of producing reliable information at levels near those at which an element could have such an adverse effect. In this case, the reporting requirement is established considering the juxtaposition of analytical capability and potential effect.
6.2.4 6.2.4 PLASTIC ADDITIVES
The sole purpose of the tests for plastic additives is to establish which additives are present and to ensure that the levels of these additives are known. This information is relevant because additives are typically a source of extractables and leachables.
In 〈 661.1〉, the chapter requires that materials be tested for all relevant analytes. Clearly, an analyte will be present in a material if it is intentionally or knowingly added to the material during its production or if testing of the material has revealed the presence of the analyte. Although test methods included in 〈661.1〉 may be of sufficiently broad scope to detect all relevant additives, this is not always the case and one cannot rely on these methods to reveal all relevant analytes. It may be the case that the material's vendor has knowledge that may be unavailable to the material's user, which is germane to establishing relevant analytes. It is reasonable to expect that material vendors and users work together to produce a complete and robust list of relevant analytes.
It is reasonable to anticipate that there may be some information that the vendor is not in a position to share with a material’s user. Nevertheless, it is in the interest of both the vendor and the user that a material be well characterized and that the characterization include all relevant analytes. Thus, it is strongly recommended that the vendor and the user find a means of establishing all relevant analytes.
6.2.5 6.2.5 UNADDRESSED MATERIALS
For the materials that are not currently listed in 〈 661.1〉 (unaddressed plastics), it is the responsibility of the user to: 1) develop those test methods (e.g., ▲physicochemical▲ (ERR 1-Nov-2020) , biological reactivity, and plastic additives) and acceptance criteria; 2) justify those test methods and acceptance criteria, specifically considering their consistency with test methods and acceptance criteria that exist for materials that are listed in 〈661.1〉; and 3) possess the test results obtained when the material is tested.
6.2.6 6.2.6 CONCEPT OF NON-INTERACTION OF MATERIALS
Testing of materials of construction via 〈661.1〉 is predicated on the circumstance that the material will most likely interact with the packaged drug product when the material is used in a packaging system. It is not necessary for a material used in a packaging system to be well-characterized if there is little or no chance of the material and the packaged drug product interacting. Under these conditions the materials of construction would be considered non-interacting and would be exempt from 〈 661.1〉 and 〈661.2〉 testing.
It is relevant to differentiate between “no direct contact” and “non-interacting”, where the term “no direct contact” means that the material and the packaged drug product do not come into direct physical contact under the clinical conditions of use.
In certain applications a “no direct contact” material of construction is also a “non-interacting” material of construction, this does not ensure no interaction, especially when the conditions of contact include long durations and/or substantially elevated temperatures. To explain the concepts of “no direct contact” and “non-interacting”, consider the following example. An aqueous drug product is packaged in a flexible plastic container, which is placed in a foil overpouch and the combination is terminally sterilized. An adhesive label is then applied to the outside of the foil overpouch.
Here, both the foil overpouch and the label are examples of “no direct contact”, because there is at least one physical barrier (the primary container) between the packaged drug product and these two items. However, if the flexible plastic primary container is permeable, the foil overpouch can be considered a “potentially interacting” component, as substances from the overpouch could migrate through the primary packaging, especially under the high-temperature conditions of terminal sterilization. The label is a “non-interacting” component because 1) the foil overpouch is impermeable and 2) the label is applied after the thermal stress associated with terminal sterilization.
The difference between a “potentially interacting” and “non-interacting” “no direct contact” component is the permeability of the barrier that separates the “no direct contact” components from the drug product. If the barrier is incomplete, then the component (and its materials of construction) is “potentially interacting” and the materials should be tested per 〈661.1〉. If the barrier is complete, then the component (and its materials of construction) is “non-interacting” and the materials need not be tested per 〈661.1〉.
6.3 6.3 Description of Plastics Contained in 〈661.1〉
6.3.1 6.3.1 CYCLIC OLEFINS
Cyclic olefin copolymers are manufactured by the copolymerization of cyclic monomers, such as tetracyclododecene or norbornene, with an olefin such as ethylene or propylene; or by ring-opening metathesis polymerization (ROMP) of cyclic monomers, followed by hydrogenation facilitated by Ziegler-Natta catalysts to produce cyclic olefin polymers. Cyclic olefins are commonly supplied in pellet form and are suited for standard polymer processing techniques such as extrusion, injection molding, injection blow molding, compression molding, thermoforming, and others. As they are amorphous, and given their high purity, moisture barrier, clarity, and sterilization compatibility, cyclic olefins are an excellent alternative to glass in a wide range of medical products, including packaging. Cyclic olefins exhibit good chemical resistance and are generally considered to be of high purity with low levels of extractables. Nevertheless, cyclic olefins may contain residual processing aids, colorants, and antioxidants.
6.3.2 6.3.2 POLYAMIDE 6
Polyamide 6 is synthesized by ring-opening polymerization of caprolactam. Hydrolytic or catalytic ring-opening polymerization of caprolactam produces epsilon-aminocaproic acid, which readily condenses to polyamide 6 at high temperatures and under a vacuum. The high strength, flexibility, and chemical resistance of crystalline polyamide 6 make it well suited for pharmaceutical applications ranging from soft and flexible tubing to catheters and containers to stiff components for surgical and dental instruments. Entities present in commercial polyamide 6 include residual monomers, residual reaction intermediates, residual catalysts (copper and chromium oxides) and activators (acetyl lactams, oxazolines, ethylenebisamides, and isocyanates) and certain additives including stabilizers (mixtures of metal and alkali metal halides), processing aids (nucleating agents and lubricants), and modifiers (chain extenders, plasticizers, and impact modifiers).
6.3.3 6.3.3 POLYCARBONATES
Polycarbonates are a group of thermoplastic polymers containing carbonate groups in their chemical structures. In interfacial polymerization, the polycarbonate material is produced by the reaction of bisphenol A (BPA) and phosgene. This process is being replaced by an alternative, termed “melt polymerization”, which entails transesterification from BPA and diphenyl carbonate, thus avoiding the use of phosgene. Entities present in commercial polycarbonates include residual monomers, solvents or catalysts (triethylamine or lithium halides, and hydroxides or aluminum hydrides), processing aids (e.g., mold release agents), UV stabilizers, impact modifiers, flame retardants, colorants, and sterilization stabilizers (free radical scavengers including propylene glycol, aromatic bromate, or disulfide compounds).
6.3.4 6.3.4 POLYETHYLENES
High- and low-density polyethylenes are long-chain ethylene-based polymers synthesized under controlled conditions of heat and pressure with the aid of catalysts from NLT 85.0% ethylene and NLT 95.0% total olefins. Other olefin ingredients that are most frequently used are butene, hexene, and propylene. Low-density polyethylene (LDPE) contains many long-chain branches along the polymer backbone, preventing the alignment and packing of the chains, thus forming a low-density material. Linear low-density polyethylene (LLDPE) contains several short chains along the polymer backbone that prevent the alignment and packing of the polymer chains, thus creating a poor crystalline material. High-density polyethylene (HDPE) contains relatively few side chains, allowing the polymer backbone to align and pack together, thus forming a crystalline, high-density plastic. High-, low-, and linear low-density polyethylene all have an IR absorption spectrum that is distinctive for polyethylene, and each possesses characteristic thermal properties. High-density polyethylene has a density between 0.941 and 0.965 g/cm3. Low-density polyethylene has a density between 0.850 and 0.940 g/cm3. Additives are added to the polymer in order to optimize its chemical, physical, and mechanical properties, thereby rendering it suitable for its intended use. These additives may include nucleating agents, clarifying agents, antioxidants, colorants, lubricants, antiblocking agents, and others. These additives typically are present individually in the polyethylene at levels of 0.01–0.3 weight %, and the total levels of the antioxidants typically are < 0.3%. Other additives, specifically amides and stearates, typically are present in polyethylenes individually at levels of NMT 0.5 weight %. Polyethylene materials that provide light protection can contain as much as 4% by weight titanium oxide.
6.3.5 6.3.5 POLYETHYLENE TEREPHTHALATE AND POLYETHYLENE TEREPHTHALATE G
Polyethylene terephthalate (PET) polymers are long-chain crystalline [e.g., polyethylene terephthalate glycol-modified (PETG)] polymers prepared by the condensation of ethylene glycol with dimethyl terephthalate or terephthalic acid. PET copolymer resins are prepared in a similar way except that they may also contain a small amount of either isophthalic acid (NMT 3 mole %) or 1,4-cyclohexanedimethanol (NMT 5 mole %). Polymerization is conducted with the aid of catalysts and stabilizers. PET polymers may contain silica or silicates (NMT 0.5% by weight) and may contain colorants.
6.3.6 6.3.6 POLY(ETHYLENE-VINYL ACETATE)
Poly(ethylene-vinyl acetate) polymers are typically obtained by copolymerization of mixtures of ethylene and vinyl acetate. Poly(ethylene- vinyl acetate) used in containers has a defined quantity of vinyl acetate of NMT 25%. Poly(ethylene-vinyl acetate) used in tubing has a defined quantity of vinyl acetate of NMT 30%. A certain number of additives are present in the polymer to optimize its chemical, physical, and mechanical properties, thereby rendering it suitable for its intended use. These additives may include antioxidants, amides, stearic acid salts, a source of base (calcium carbonate or potassium hydroxide), and inorganic fillers. Poly(ethylene-vinyl acetate) can contain NMT 3 antioxidants with individual levels of NMT 0.2 weight %. This material may also contain: 1) oleamide and/or erucamide at individual levels of 0.5 weight %; 2) calcium stearate, zinc stearate, or both at levels NMT 0.5 weight %; 3) sources of base at levels NMT 0.5 weight %; and 4) colloidal silica at 0.2 weight %.
6.3.7 6.3.7 POLYPROPYLENE
Propylene polymers are long-chain polymers synthesized from propylene or other olefins, for example, ethylene or butene, under controlled conditions of heat and pressure with the aid of catalysts. A certain number of additives are added to the polymer in order to optimize its chemical, physical, and mechanical properties, thereby rendering it suitable for its intended use. These additives may include nucleating agents, clarifying agents, antioxidants, colorants, lubricants, antiblocking agents, and others. These additives typically are present individually in the polypropylene at levels of 0.01–0.3 weight %, and the total levels of the antioxidants typically are less than 0.3%. Polypropylene that provides light protection can contain as much as 4% by weight titanium dioxide.
6.3.8 6.3.8 POLYVINYL CHLORIDE
Polyvinyl chloride (PVC) is produced by polymerization of the vinyl chloride monomer in a process that uses initiators that break down to start the radical chain reaction. The polymerization reactions used to produce PVC are designed to produce levels of residual vinyl chloride monomer of <1 ppm on a weight basis. Polyvinyl chloride may contain NMT 15% by weight of: 1) copolymers based on Acrylic and/or methacrylic acids and/or their esters, 2) styrene, and/or 3) butadiene.
To obtain the required mechanical and stability characteristics, materials based on PVC may contain: 1) epoxidized oils at levels up to 8 weight %; 2) calcium and/or zinc salts of long chain fatty acids at levels NMT 1.5 weight %; 3) liquid paraffin, waxes, and/or hydrogenated oils at individual levels NMT 2 weight %; 4) nonylphenyl phosphite-type compounds at levels NMT 1 weight %; 5) Sorbitol and macrogel esters, NMT 1.5 weight % each; 6) stabilizers at levels of either 0.25 weight % (for tin-based stabilizers) or 1 weight % (for other stabilizers); and 7) colorants, pigments, or opacifiers. The additives used and their allowed levels are dictated by the application of the polyvinyl chloride.
6.3.9 6.3.9 POLYVINYL CHLORIDE, PLASTICIZED
PVC polymers are long-chain vinyl chloride polymers synthesized from vinyl chloride monomers via free radical polymerization. Various additives are compounded into PVC to provide the materials with properties that render it suitable for its intended use. These additives may include heat stabilizers, primary and secondary plasticizers, stabilizers, impact modifiers, lubricants, pigments, and others. These additives typically are present individually in the PVC at levels ranging from 0.1 to 45 weight %. The additives used and their allowed levels are dictated by the application of the plasticized PVC.
7 7. APPLICABILITY AND APPLICATION OF 〈 661.2〉
7.1 7.1 Applicability
1. The holder of the drug product application and drug product manufacturer [in the case of many OTCs, where there is no application] bear primary responsibility and accountability for ensuring the requirements of the chapter are met. The means by which the holder of the drug product application and drug product manufacturer obtain information to meet the requirement are at the discretion of the holder.
2. The scope of 〈 661.2〉 is packaging systems for finished drug products. If component testing is considered to be necessary, the relevant testing and acceptance criteria for components are contained within 〈 661.2〉.
3. Application of 〈661.2〉 to systems other than packaging systems for finished drug products is beyond the scope of these chapters at
the present time, but the concepts and principles of these chapters may be applicable and relevant to other systems.
7.2 7.2 Application
1. Chapter 〈661.2〉 requires that all packaging systems be demonstrated to be suitable by performing a chemical suitability for use assessment but does not specify the details of the chemical suitability for use assessment process acceptance criteria. Rather, for the chemical suitability for use assessment, 〈661.2〉 discusses the use of a risk-based approach in determining the level of testing that is deemed necessary for a packaging system. It is currently a regulatory expectation that extractables and leachables testing be performed for high risk dosage forms. Thus, 〈661.2〉 references chapters 〈1663〉 for extractables and 〈1664〉 for leachables, thereby providing users with a means for designing and implementing an effective, efficient, risk-based, and more or less customized extractables or leachables assessments that comply with regulatory requirements.
2. Chapter 〈661.2〉 provides holders of packaging system or drug product applications and/or packaged drug products manufacturers with the flexibility to operate within the context of their own specific situation and their own specific risk-management philosophy. The trade-off for having such flexibility is that it is the responsibility of the holders and manufacturers to justify their test methods and acceptance criteria. It is proper and appropriate that the justification exists and that it be judged (and approved) on the basis of its individual scientific and risk-management merits.
