Design, Evaluation, and Characterization of Viral Clearance Procedures
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- INTRODUCTION
- GOALS AND PRINCIPLES OF VIRAL CLEARANCE STUDIES
- CONSIDERATIONS FOR PERFORMANCE OF VIRAL CLEARANCE STUDIES
- Selection of Viruses
- Process Clearance Capability
- Downstream Processing Steps for Viral Clearance
- Qualification and Scale-Down of Purification Steps
- Selection of Sampling Points
- Selection of Assays for Viral Quantitation
- Sample Matrix Effects on Viral Quantitation Assays
- Effects of Storing and Freezing Viral Clearance Samples
- Qualification of Virus Stocks and Effects on Processing Steps
- Performance of Viral Spiking Studies
- GLOSSARY
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
This chapter is a companion document to Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (1050), which was adapted, essentially unchanged, from the International Conference on Harmonization Q5A. This chapter provides users with practical guidance regarding the design, evaluation, and characterization of viral clearance procedures. The chapter scope is the same as that described in (1050) and covers biotechnology products for human use that are derived from cell lines of human or animal origin. Viral clearance studies performed according to the principles outlined in this chapter will provide meaningful data about the ability of the overall production and purification processes to remove or inactivate a broad spectrum of viral types that may affect the safety of biotechnology-derived products. [NOTE-The Glossary contains definitions of terms used in this chapter that are not already defined in (1050).]
The regulations for licensing biotechnology products stipulate that cell banks, biologically derived raw materials, and bulk harvest must be controlled and tested for viral safety; however, many of the viral detection approaches that can be used have inherent limitations. In addition, certain detection methods may be so specific for a particular virus that they may fail to detect viral variants (e.g., noncytopathic strains, or non-laboratory-adapted wild-type strains, or mutated variants). Some newly emerging viruses may be missed. Finally, detection methods typically are limited by their sensitivities, and infectious viruses at a low titer may go undetected. Viral contamination of manufacturing processes can come from many sources, including cell substrates and raw materials of biological origin (such as cell culture supplements and other production raw materials), and contamination during production. In addition to the required viral safety testing of these materials, manufacturers must evaluate the effectiveness of downstream purification process steps, which together must remove or inactivate any viruses potentially present (see (1050)). USP encourages readers to maintain and keep current best practices in the field by reviewing current peer-reviewed publications, regulatory guidances, and key opinion leaders and organizations.
As suggested in the U.S. Food and Drug Administration "Points to Consider documents (Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use and Draft Points to Consider in the Characterization of Cell Lines Used to Produce Biologicals), as well as (1050), a multi-step approach is typically used to ensure viral safety. Clearance studies can generally be considered generic or modular. As described in the FDA's Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use, a generic clearance study is one in which virus removal and inactivation is demonstrated for several steps in the purification process of a model monoclonal antibody (mAb) in which the data may be extrapolated to other mAbs following the same purification and virus removal/inactivation scheme as the model mAb. A modular clearance study demonstrates virus removal or inactivation of individual steps during the purification process (column chromatography, filtration, pasteurization, solvent/detergent, low pH, and others) so that each module in the purification scheme may be studied independently of the other modules. If necessary, an alternative model mAb can be used to demonstrate viral clearance in different modules. Identical clearance modules can be extrapolated to the product mAb. This chapter complements these documents by providing users strategies to perform and assess viral clearance.
2 GOALS AND PRINCIPLES OF VIRAL CLEARANCE STUDIES
A viral clearance study should evaluate the ability of the overall purification process to remove or inactivate a broad spectrum of virus types, including viruses that are known to contaminate or have the potential to contaminate the raw materials, and those that can be introduced during manufacturing. Viral clearance studies typically involve substudies performed on specific and suitable individual steps of the manufacturing and purification process. The studies should be performed in a manner that generates quantitative data, allowing for identification of effective clearance steps and estimation of viral reduction factors (VRFs, also known as log reduction factors, LRF, or log reduction values, LRV). The VRFs of an individual clearance step represent the ratio of the viral load in the pretreatment material (used to challenge the clearance step) to the viral load in the post-treatment material. The VRFs derived from specific process steps are used to evaluate the overall capacity of the entire production process to remove or inactivate process-specific or nonspecific viruses.
One of the key goals of a manufacturing purification process is to achieve maximal viral clearance without compromising product quality. Critical attributes of strategic viral clearance steps in the manufacturing process must be characterized. In the context of viral clearance, robustness has two main components: 1) the ability of process steps to remove and/or inactivate viruses under worst-case conditions or over a wide operational range for parameters (e.g., temperature, protein concentration, pressure, flow rate, conductivity, and pH), and 2) the ability of process steps to consistently remove or inactivate nonspecific viruses that possess a broad spectrum of physical and chemical resistance characteristics (e.g., pH, heat, solvent/detergent treatment, or filtration). A validated process that provides robust viral clearance should establish the VRF achievable for a panel of "relevant" or "model" viruses (see next section for definitions). The validated process also should demonstrate robustness of dedicated inactivation or removal steps for which critical operational parameters that could affect viral clearance are well established. Therefore, a demonstration of viral clearance robustness provides confidence that the manufacturing process can remove or inactivate potential viral contaminants.
Viral clearance study protocols begin with an overall action plan and design of experiments. Chapter (1050). (see Section V and Table 4) describes five major action plans (Cases A-E) and can serve as a guide. Cell substrates categorized in Cases C, D, and E represent special cases, and manufacturers who use these substrates should discuss study design with the applicable regulatory authorities. Unique issues for each production process must be considered on a case-by-case basis. These issues may relate to the starting materials, production process, product, and intended use of the product. The overall action plan dictates the choice of viruses used in the viral clearance study, i.e., the study must include relevant, specific, and nonspecific model viruses. Further, the viral clearance study protocol may include the following elements: descriptions of the study facility and staff responsible for executing the study, the scaled-down purification models, justification of the appropriateness of the scaled-down model, and the study design. The study report may include these elements, as well as a stepwise analysis of calculated VRFs and the overall viral clearance capacity of the purification process. Viral clearance studies should be conducted in a well-documented, controlled manner that complies with current regulatory requirements.
3 CONSIDERATIONS FOR PERFORMANCE OF VIRAL CLEARANCE STUDIES
3.1 Selection of Viruses
The choice and number of viruses that may be used in a viral clearance study are dictated by the nature and origin of the production cell line, as well as the nature and origin of the animal-derived materials used in production and purification. In general, at least two viruses, one enveloped (typically a retrovirus, e.g., MuLV) and one nonenveloped (preferably a parvovirus, e.g., MVM), are used in the early clinical phases of product development, but three or more viruses may be used to generate data for registration-enabling studies. At least two orthogonal virus removal/inactivation steps (steps with different mechanisms of clearance) should be evaluated per virus. The reproducibility of an effective step should be assessed by performing at least two independent experiments, or reproducibility should be supported by the process development history (or experiences). Model viruses for process evaluation and process characterization studies should be similar to the virus that may contaminate the product; however, other viruses with a wide range of physical and chemical characteristics should also be examined. The latter is important for showing that the purification process is capable of inactivating or removing a wide variety of viruses, including newly emerging viruses (e.g., vesivirus 2117, circoviruses, or newly discovered animal and human parvoviruses) or unexpected contaminants (e.g., BVDV, MVM, epizootic hemorrhagic disease virus, and Cache valley virus). The manufacturer should justify the choice of viruses in accordance with the aims of the process evaluation and process characterization studies and the guidance provided in this document, as well as relevant regulatory guidelines.
Viruses used in clearance studies fall into three categories: relevant viruses, specific model viruses, and nonspecific model viruses. "Relevant viruses" are viruses used in process evaluation studies, which are either the identified virus, or of the same species as the virus that is known, or likely to contaminate the cell substrate or any other reagents or materials used in the production process. When a relevant virus is not available or when it is not well adapted for this purpose (e.g., it cannot be grown in vitro to a sufficiently high titer or cannot be detected using cell-based viral titration endpoints required for assessing inactivation), a "specific model virus" can be used as a substitute. An appropriate specific model virus may be a virus that is closely related to the known or suspected virus (same genus or family), having similar physical and chemical properties to those of the observed or suspected virus. For example, cell lines derived from rodents usually contain endogenous retroviral particles or retroviral-like particles that can be infectious or noninfectious. The capacity of the manufacturing process to remove and/or inactivate rodent retroviruses present in products obtained from such cells should be determined. In the case of rodent-derived cells, one can use a murine leukemia virus as a model virus. Manufacturers using human-derived B-lymphocyte cells immortalized by infection with Epstein-Barr virus should demonstrate the ability of the manufacturing process to clear a herpesvirus. In this instance, pseudorabies virus or another herpesvirus can be used as a specific model virus.
Finally, when the goal is to characterize the capacity of the manufacturing process to remove and/or inactivate viruses in general (i.e., to characterize the robustness of the clearance process), viral clearance studies can be performed with a panel of "nonspecific model viruses" that have a wide range of physical and chemical characteristics. Data obtained from studies with relevant or specific model viruses also can contribute to this assessment. It is not necessary to test all types of viruses; preference should be given to viruses with diverse physical and chemical properties that would be representative of a wide range of viral families. The results obtained for such viruses provide useful information about the ability of the production process to remove or inactivate viruses in general. Table 1 gives examples of model viruses that represent a range of physicochemical properties for use in viral clearance studies, including those examples already described in (1050) (see Table A-1). Additional points to consider during viral selection are: 1) ability to grow the virus of interest to a sufficiently high titer, 2) ability to create stocks with minimal aggregates, 3) availability of a qualified/validated assay system for detection of the selected virus, 4) the selected viruses are not likely to pose a health hazard to personnel performing the study or the environment, and 5) potential to address new and emerging viruses.
Table 1. Examples of Viruses Used in Viral Clearance Studies for Biotechnology Products Derived from Cell Cultures
| Virus | Family | Genus | Natural Host | Genome | Enveloped | Size (nm) | Shape | Resistancea |
| Adenovirus 5 | Adeno | Mastadenovirus | Human | DNA | No | 70–90 | Spherical | High |
| BVDV | Flavi | Pestivirus | Bovine | RNA | Yes | 50–70 | Pleomorphic/ spherical | Low |
| Cache valley virus | Bunya | Bunyavirus | Bovine, ovine | RNA | Yes | 80–120 | Spherical | Low |
| Encephalom yocarditis virus (EMC) | Picorna | Cardiovirus | Mouse | RNA | No | 25–30 | Icosahedral | Medium |
| Feline calicivirus | Calici | Vesivirus | Feline | RNA | No | 35–40 | Icosahedral | High |
| Herpes simplex 1 | Herpes | Alphaherpesvirus | Human | DNA | Yes | 120–200 | Spherical | Medium |
| MuLV | Retro | Type C oncovirus | Mouse | RNA | Yes | 80–110 | Spherical | Low |
| Parainuenza virus | Paramyxo | Paramyxovirus | Various | RNA | Yes | 100–200 | Pleomorphic/ spherical | Low |
| Parvoviruses (MVM, PPV, CPV, BPV) | Parvo | Parvovirus | Murine, porcine, canine, bovine | DNA | No | 18–26 | Icosahedral | Very High |
| Poliovirus sabin type 1 | Picorna | Enterovirus | Human | RNA | No | 25–30 | Icosahedral | Medium |
| Porcine circovirus (PCV) | Circo | Circovirus | Porcine | DNA | No | 15–20 | Icosahedral | High |
| Pseudorabies virus | Herpes | — | Swine | DNA | Yes | 120–200 | Spherical | Medium |
| Reovirus 3 | Reo | Orthoreovirus | Various | RNA | No | 60–80 | Spherical | Medium |
| Simian virus 5 | Paramyxo | Rubulavirus | Simian | RNA | Yes | 150–300 | Pleomorphic/ spherical | Low |
| Sindbis virus | Toga | Alphavirus | Human | RNA | Yes | 60–70 | Spherical | Low |
| SV40 | Papova | Polyomavirus | Monkey | DNA | No | 40–50 | Icosahedral | Very high |
| Vesicular stomatitis virus | Rhabdo | Vesiculovirus | Equine, bovine | RNA | Yes | 70 x 150 | Bullet | Low |
| West Nile virus | Flavi | Flavivirus | Avian | RNA | Yes | 40–70 | Pleomorphic/ spherical | Medium |
a Resistance to physicochemical treatments based on studies of purification processes. Resistance is relative to the specific treatment, and it is used in the context of understanding the biology of the virus and the nature of the manufacturing process. Actual results will vary according to the treatment. These viruses are examples only, their use is not mandatory, and the table is not exhaustive.
3.2 Process Clearance Capability
The viral clearance capability of a process is typically expressed as the logarithmic value of the virus reduction factor evaluated by the entire process. Although there are no specific requirements for the clearance capability that a process must achieve, the nature and origin of the cell type, raw materials used in production, as well as other factors may influence the need for greater or lower levels of viral clearance. For example, the target retroviral clearance capability for a CHO and NSO cell manufacturing process could be 4-6 LRF of total-process retroviral clearance above the level found in the maximum therapeutic dose, based on the retroviral-like particle titer in the production bioreactor material. Minimum clearance capability for other types of viruses might include >4 LRF of clearance for additional enveloped viruses and >4 LRF of clearance for nonenveloped viruses. Specific clearance targets must be established and justified on a case-by-case basis. In general, for a single virus clearance step to be considered effective, 4 LRF or more of clearance must be demonstrated. In contrast, a clearance step demonstrating 1-3 LRF is considered a supportive step. The titer of the virus input and output for any given process step evaluated should include 95% confidence (α = 0.05). A step can be supportive for some viruses and effective for others. When possible and as process knowledge allows, the analyst should perform viral clearance studies under expected worst-case conditions for the parameters defined as critical for that step (e.g., pH, temperature, and flow rate). Sufficient numbers of downstream purification process steps should be identified and evaluated so that the required overall clearance target can be reached.
3.3 Downstream Processing Steps for Viral Clearance
Downstream processing steps should employ different (orthogonal) mechanisms of viral clearance because consideration of multiple steps of the same or similar clearance mechanisms may lead to overestimation of the overall viral reduction capability. In general, a purification process should include at least one robust viral inactivation step and/or one robust viral removal step. For every purification step assessed, the mechanism of viral clearance should be described as inactivation, removal, or a combination of both; if both, the primary clearance mechanism should be identified. For inactivation steps, clearance studies should be planned in such a way that they can determine the kinetics of viral inactivation. Ideally, a manufacturing process includes steps that are dedicated to viral clearance; that is, they are present in the production or purification process specifically for the removal or inactivation of viruses. Examples of dedicated steps include solvent and detergent treatment, low-pH inactivation, and virus-removal filtration.
3.4 Qualification and Scale-Down of Purification Steps
Viral clearance studies using infectious virus spikes are not conducted at production scale in the manufacturing suite because this could contaminate the suite with viruses. Such studies should be performed in a segregated facility equipped for virological work and staffed with personnel who have virology expertise and familiarity with the operation of a scaled-down purification process. Each scaled-down step of the purification process must be qualified, i.e., found to be comparable to the full-scale production process by all relevant, measurable criteria. Comparability should be demonstrated using representative raw materials and intermediates from production, and equipment with process parameters through appropriate scale-down principles. The outputs should be measured with the appropriate analytical methods and statistical analyses.
In many instances, the monitored parameters are the same as those analyzed during performance of the actual manufacturing steps. For chromatography, the parameters should be representative of the respective clinical or commercial-scale manufacturing: [e.g., the column bed height, residence time, linear flow rate, chromatographic matrix, buffer composition (including pH, conductivity, and operating temperature), and product pool concentrations and composition]. Scaled-down and manufacturing-scale chromatography systems should produce similar elution profiles and step yields, and final product analytical profile (e.g., SEC-HPLC and/or SDS-PAGE analyses). Similar considerations apply for other types of procedures. Any unavoidable differences between scaled-down and manufacturing-scale procedures should be investigated to determine their potential influence on the viral clearance results.
3.5 Selection of Sampling Points
Sampling points for assessing viral inactivation should include the starting materials (virus-spiked process solutions and appropriate controls) as well as samples taken at several time points during the inactivation process. This approach allows the analyst to monitor the kinetics of viral inactivation. Sampling points for virus-removal steps, such as chromatography and filtration, should include the feed-stream process solution that will be applied to the step, as well as the resulting process pool that is then processed in the subsequent steps. Chromatographic fractions (e.g., flow-through, washes, pre-peak, peak, post-peak, strip) of the mainstream, as well as pre- and post-mainstream samples, typically are evaluated in studies that support registration-enabling studies. Analyzing such side fractions might also be helpful in cases where the mechanism of partitioning is not understood. Sampling points are discussed further in the section Performance of Viral Spiking Studies.
3.6 Selection of Assays for Viral Quantitation
Various types of viral quantitation assays can be used. Examples include infectivity assays such as tissue-culture infectious-dose assays and plaque quantitation assays. In addition, quantitative polymerase chain reaction (qPCR) assays detect and quantify the nucleic acids of both infectious and noninfectious viruses. These assays are described in Virology Test Methods (1237), Nucleic Acid-Based Techniques-Extraction, Detection, and Sequencing (1126), and Nucleic Acid Based Techniques-Amplification (1127). When using nucleic acid-based techniques (NAT), it is important to assure that the virus spike and the spiked samples to be analyzed do not contain significant amounts of free viral nucleic acids. The amount of free nucleic acids can be reduced by nuclease pretreatment of samples. It is important to characterize the impact of nuclease pretreatment on the virus spike since it could have a negative impact on the virus spike. Quantitative/quantal infectivity assays should have adequate sensitivity and reproducibility, and should be performed with a sufficient number of replicates to ensure adequate statistical validity of the results. All assays should include appropriate system suitability controls. Virus titration assays used in support of viral clearance studies must be shown to be suitable for this purpose by means of qualification or validation.
3.7 Sample Matrix Effects on Viral Quantitation Assays
When a virus is cleared by inactivation or removal (e.g., filtration or column chromatography) methods, the analysts must perform preliminary cytotoxicity and viral interference analyses to determine if their viral quantitation assays are sensitive to matrix components present in their samples. These studies should be performed before the spiking studies to confirm that the results of the viral clearance spiking studies are actually due to viral clearance and not to other intrinsic or extrinsic factors yielding a false-negative result. For example, some process intermediates may be cytotoxic to the detector (indicator) cells or may interfere with virus detection by a particular assay system. The process solutions used in these studies should be representative of those from a full-scale good manufacturing practice (GMP) manufacturing run. The dilution of the process sample solution that does not cause cytotoxicity or interference is determined and the process solution is diluted accordingly before storage or testing samples from that manufacturing step in the virus-spiking studies.
In a cytotoxicity evaluation, the cells used in the viral quantitation assay are exposed to a series of dilutions of the sample matrix solution for that process step. Then the cells are assessed for viability and changes in morphology that could interfere with evaluation of the indicator cells for viral cytopathic effects or plaques and hence could have an impact on assay performance. The dilution of the process sample solution that does not cause cytotoxicity is determined, and the process solution is diluted accordingly before storage or testing samples from that manufacturing step in the virus-spiking studies.
The study involves the following steps:
- Perform serial dilutions (e.g., 10-fold, 5-fold, or 2-fold) of the virus inactivation or filtration load using appropriate cell culture media as the diluent
- Inoculate the different dilutions of virus inactivation or filtration load alone onto the indicator cells that will be used for each virus titration
- Include appropriate buffer or media and cell controls
- Identify the dilution of the virus inactivation load that does not exhibit measurable detector cell toxicity attributable to the load matrix
Process solutions also can interfere with virus detection by the in vitro cell-based viral infectivity assay system, either by inactivating the virus itself or by altering the indicator cells in a way that delays or prevents occurrence of a productive viral infection. To assess a process solution for viral interference, analysts prepare dilutions of the process solution and spike them with a known quantity of virus. Each dilution is subsequently titrated in the quantitative/quantal viral infectivity assay. If the difference between the known titer and the titer determined for the spiked solution in the assay (the viral interference titer) differ by more than the predetermined assay variability (typically ±0.5 log), then viral interference is suspected.
To assess the inactivation step load for viral interference, analysts should follow these steps:
- Perform serial dilutions (e.g., 10-fold, 5-fold, or 2-fold) of the virus stock using appropriate cell culture media as the diluent. This will serve as the positive control for the virus titer
- Perform serial dilutions (e.g., 10-fold, 5-fold, or 2-fold) of the virus stock using the virus inactivation load as the diluent [NOTE-Information from the cytotoxicity study will determine the design of this step and the next one.]
- Inoculate the dilution series onto the indicator cells
- Include assay suitability controls (positive and negative) in the test
- Identify the dilution of the virus inactivation load that does not result in significant changes in virus stock titer compared to the positive control virus titer
If qPCR assays are used to quantitate the virus, each process solution should be tested for interference and recovery with the nucleic acid extraction method used, as well as for interference with the qPCR reaction itself. Each process solution must be diluted to a level that does not cause interference. Although diluting a process solution to achieve acceptable assay performance is a commonly used strategy, this reduces the maximum demonstrable VRF for the clearance step under evaluation.
3.8 Effects of Storing and Freezing Viral Clearance Samples
If process solutions are used within their established hold times, stability studies should be performed to better understand the effect on the clearance step of holding or storing the solutions, or on the virus (e.g., aggregation). For example, bacterial growth or precipitation of proteins in the solutions may occur, particularly after prolonged storage.
If the samples from the spiking study are stored frozen before virus quantitation, analysts should perform stability studies using the viral infectivity result as an endpoint. Ideally, this should be done before the viral spiking study by spiking dilutions of each process solution with a known amount of virus and then freezing each sample. After specified times in frozen storage, the samples are removed, and the titer of each sample dilution is determined. If the difference between the known titer and the titer determined from the spiked frozen solution is greater than the assay variability (typically ±0.5 log), this indicates that viral infectivity has been compromised by frozen storage.
3.9 Qualification of Virus Stocks and Effects on Processing Steps
The virus stocks used in viral clearance studies are critical reagents. Each virus stock should have a traceable, certified source with full documentation of the controlled production procedures. Virus stocks should meet predetermined criteria for identity, purity (e.g., sterility, mycoplasma, and adventitious virus testing), infectious titer, stability at freezing temperatures, and minimal viral aggregates. Analysts should minimize the number of passages from the master or working viral bank in order to reduce the chance of mutation. Purity of virus preparations should be taken into account in cases when virus preparation impurities may influence the performance of a certain unit operation (e.g., virus filtration).
During qualification, analysts should obtain an initial virus titer for each virus stock. Ideally, analysts should independently assay viral stocks for titer on separate days using different passage numbers of the indicator culture cells that were used in the viral quantification assay. The resulting titers should agree within the predetermined variability of the assay (typically the expectation is ±0.5 log), and if so, the analyst can average the titers to determine the certified titer of the stock.
It may be advisable to perform a mock spiking study before the true spiking study. This mock spiking study evaluates the effects of the spiking virus matrix on the process step that is under evaluation. Mock spiking studies are important for all steps that will be tested for viral clearance, but they are particularly important for those steps that will be evaluated for the clearance of virus from a stock that contains impurities or additives (e.g., stabilizing protein). Mock spiking studies are conducted by adding the viral suspension matrix (including all components except the virus) to the process solution at a virus-spike to feed-stream-solution (load) ratio of NMT 10% (v/v) and then executing the processing step. The mock-spiked step is monitored for expected performance, and if results are different than expected, then reducing the virus:load spiking ratio may be helpful. This adjusted virus-spike to feed-stream-solution (load) ratio would be used in performing the actual clearance study for the processing step under study.
3.10 Performance of Viral Spiking Studies
Viral clearance studies are usually conducted either at a site where both the spiking and viral quantitation assays are performed (e.g., a biosafety testing laboratory) or at two different sites: one where the spiking studies are done and a second where the samples are quantitated. Sponsors and analysts must gain knowledge and control factors that can influence each manipulation that is not part of the manufacturing process, for example, freezing and thawing (see Effects of Storing and Freezing Viral Clearance Samples) as well as shipping conditions.
Before initiating a clearance study, analysts should prepare a well-documented study design that clearly defines the following: the steps that will be tested, the sampling plan for each step, sample identification, sample handling and storage, sample shipment if required, critical operating parameters, process scale, rationale for worst-case conditions (if they have been established), and the appropriate controls for each step. At a minimum, the samples tested should include those used to determine: 1) the spiking virus titer, 2) the spiking virus titer after freeze-thaw (if applicable), 3) the virus titer in the production solution before processing, and 4) the virus titer after processing. The analyst may need to collect and assay additional samples, depending on the step under test and the phase of product development.
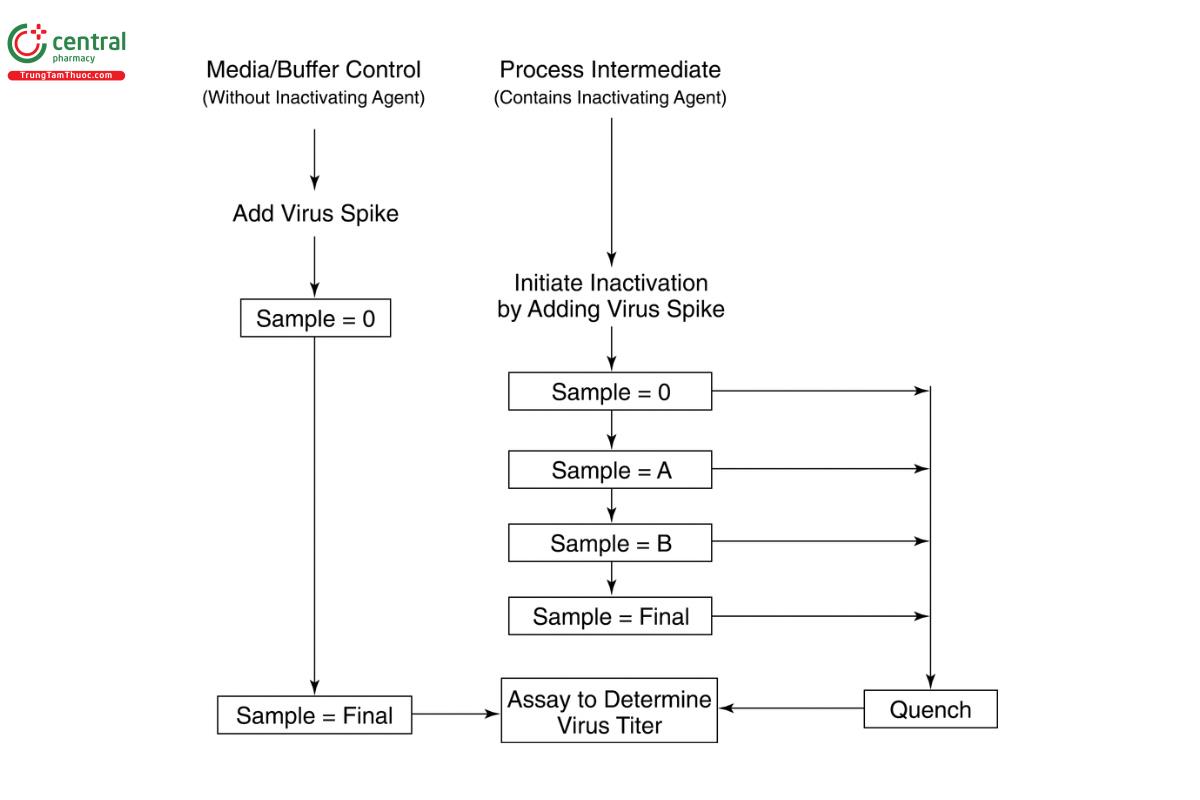
In a typical biomanufacturing process, viral clearance can be accomplished by virus inactivation (e.g., pH treatment, heat treatment, or solvent and detergent treatment) or by virus removal (e.g., filtration or column chromatography). Figures 1A and 18 show examples of experimental designs for virus inactivation, Figure 2 shows virus removal by filtration, and Figure 3 shows virus removal by column chromatography. These examples are intended for general reference and do not represent every specific condition. In developing their study designs, sponsors should consider their own process conditions in order to plan their own specific steps.
The overall concept of viral clearance procedures can be expressed as follows:
Viral Clearance = Viral Inactivation + Viral Removal, where
Viral Removal = Removal by Filtration + Removal by Column Chromatography
3.10.1 VIRAL CLEARANCE BY VIRUS INACTIVATION
General considerations: Analysts should recognize that virus inactivation is not always a simple, first-order reaction. Typically, virus inactivation is complex and involves a fast initial phase followed by a slower phase. Thus, analysts should plan the inactivation process in such a way that samples are taken at different times in order to construct an inactivation time curve. Samples collected for inactivation studies should include the planned process time, and at a minimum, time zero; a time point or a suitable number of greater than zero but less than the minimum inactivating-agent exposure time; a time point equal to the minimum inactivating-agent exposure time; and in addition a time point beyond the minimum exposure time might be helpful in cases where there is a slow inactivation curve. Additional time points may be particularly important when there is no prior experience with virus inactivation kinetics.
When viral clearance by inactivation occurs, two scenarios generally are possible: 1) the inactivating agent is not present in the load material and inactivation is initiated by addition of the inactivating agent after virus spiking (Figure 1A); and 2) the load material already contains the inactivating agent before the virus spike, and inactivation is initiated by addition of the virus spike (Figure 18). An example of the latter is a Protein A column eluate at low pH. Whenever possible, analysts should determine the initial virus load from the virus-spiked load material before they add an inactivating agent. However, this is not possible when the inactivating agent is already present in the load material at the start of the experiment. In these situations, the initial virus load can be calculated using the certified titer value of the spiking virus preparation and the virus:load spiking ratio. When viral inactivation occurs too rapidly to allow for plotting an inactivation time curve under normal process conditions, analysts should include appropriate controls to show that activity is indeed lost due to the inactivating agent present.
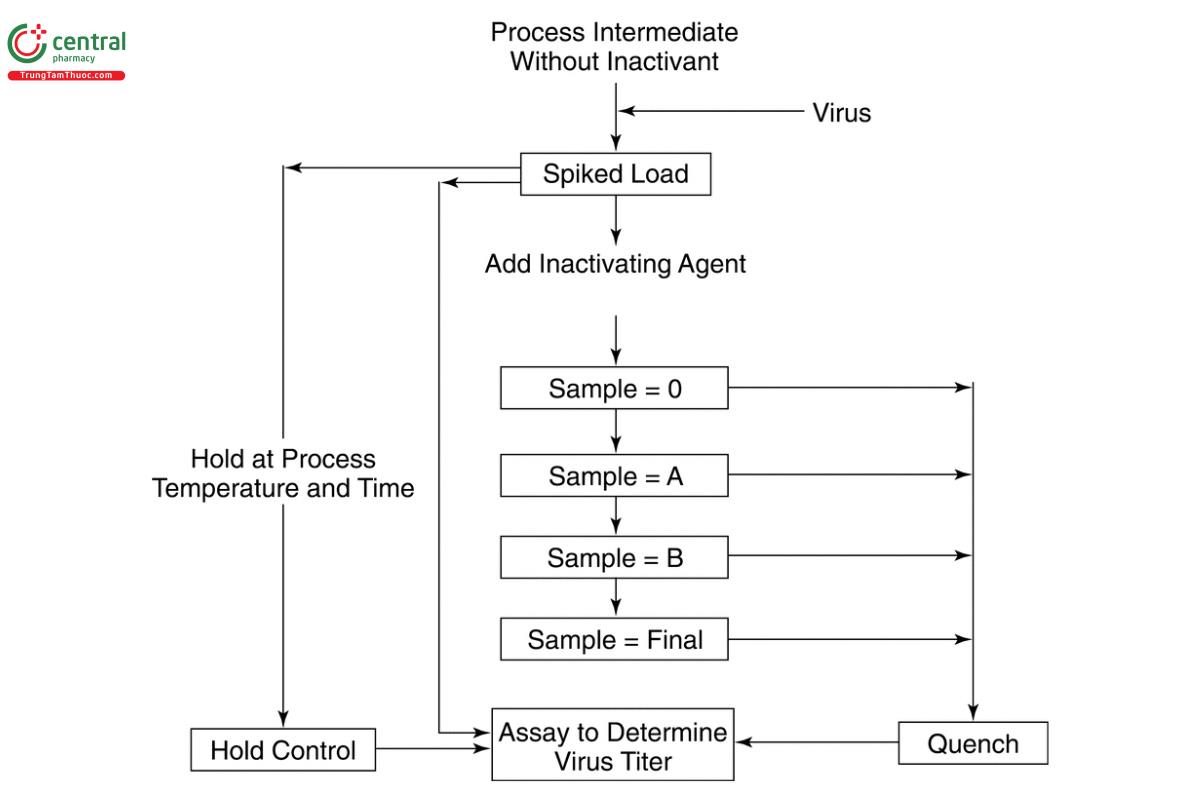
Study design: The general approach to the study design for assessing viral clearance by inactivation is outlined in Figures 1A and 1B. In Figure 1A, the virus spike is added to the load material (process intermediate) before initiation of inactivation. The virus and load material are mixed well, and the spiked load and hold control samples are pulled. The spiked load should be tested immediately to determine virus titer. The actual titer of the spiked load (obtained experimentally) should be compared to the theoretical titer of the spiked load (obtained from the certified titer of the lot of virus used and the spiking ratio) to verify performance of virus detection assay and that the appropriate amount of virus was added to the load material. The hold control should be treated the same as the spiked load (i.e., prior to neutralization of the hold control, the sample should be pH adjusted in the same manner as the spiked load and then neutralized and spiked with virus). In addition, the hold control should remain at process temperature until the final sample in the experiment is pulled. The viral titer of the hold control then is assessed and compared to the actual titer of the spiked load to determine if any virus was inactivated in the presence of the load material over time and at the temperature of the inactivation study. This may be considered when determining the final VRF.
Inactivation should be initiated in a way that mimics the manufacturing process (e.g., with constant mixing) so that the inactivating agent (e.g., detergent, or acid utilized to lower pH) is mixed homogeneously into the solution as soon as possible. This minimizes the presence of localized high concentrations of the inactivating agent. If heat is used for inactivation, the heat should be applied in a way that mimics manufacturing (e.g., the ramp to target temperature should be identical) and controls should be kept unheated. However, the exact reproduction of addition (mixing) of inactivating agents or reproduction of a heating ramp can be very difficult at down-scale. In such cases it is advised to spike the virus directly into the inactivation agent-containing (or heated) material and to follow inactivation kinetics as described in Figure 1B. For control of the added virus, virus is spiked into process material without the inactivating agent or into unheated material.
Worst-case process conditions (e.g., pH, time, and temperature) should be applied if they have been established. Time points (samples) are pulled and the reaction is immediately quenched (e.g., by neutralization, dilution, or immediate cooling of the sample) so that further viral inactivation cannot occur. This allows the analyst to determine the kinetics of virus inactivation and then construct a virus inactivation versus time curve. Large-volume sampling can be applied to maximize clearance values.
In Figure 1B, the load material contains the inactivating agent and thus the addition of the virus spike starts the inactivation. In this scenario, it may not be possible to neutralize the process intermediate solution before the virus-spiking study because large fluctuations in pH may cause the protein product to fall out of solution. Also, the spiked load and hold control samples cannot be pulled from the virus-spiked load material, thus the control samples are pulled from a media or buffer control that does not contain the inactivating agent.
3.10.2 VIRAL CLEARANCE BY FILTRATION
Analysts must perform preliminary cytotoxicity and viral interference analyses when preparing to perform viral clearance by filtration (see Sample Matrix Effects on Viral Quantitation Assays). The viral filter load and pool dilution that will be used in the clearance study is that dilution shown not to cause cytotoxicity or interference.
Study design: The general approach to the study design for assessing viral clearance by filtration is outlined in Figure 2. Once the process intermediate is spiked with virus, the spiked load sample should be assayed immediately for virus titer. Virus spiking can be associated with aggregation effects. The aggregation status of a contaminating virus in the respective product intermediate is difficult to predict and therefore, as a worst-case approach, a mono-disperse virus preparation is advised for spiking. Filtration of virus before spiking (e.g., 0.45 µm or 0.22 µm) reduces larger virus aggregates from frozen virus stocks. A pure high-titer virus preparation can be helpful to reduce virus spike-induced effects influencing performance of virus filtration. The hold control sample should be maintained at processing conditions (temperature and time) and then assayed to evaluate the impact of these conditions, as well as the process intermediate components, on virus titer. Worst-case process conditions should be applied, if they have been established. The virus titers obtained from the spiked load and hold control should be within the experimental variability of the virus titration assay. If there is a negative impact on virus titer because of the processing conditions (temperature, time, and process intermediate composition), then an investigation should begin to understand the process. If warranted, the final VRF may need to be adjusted to account for the impact of hold conditions on viral preparations. However, this approach might underestimate the removal capacity from virus filtration. Alternatively, use of another detection method (e.g., NAT) or another, more stable, model virus can be discussed.
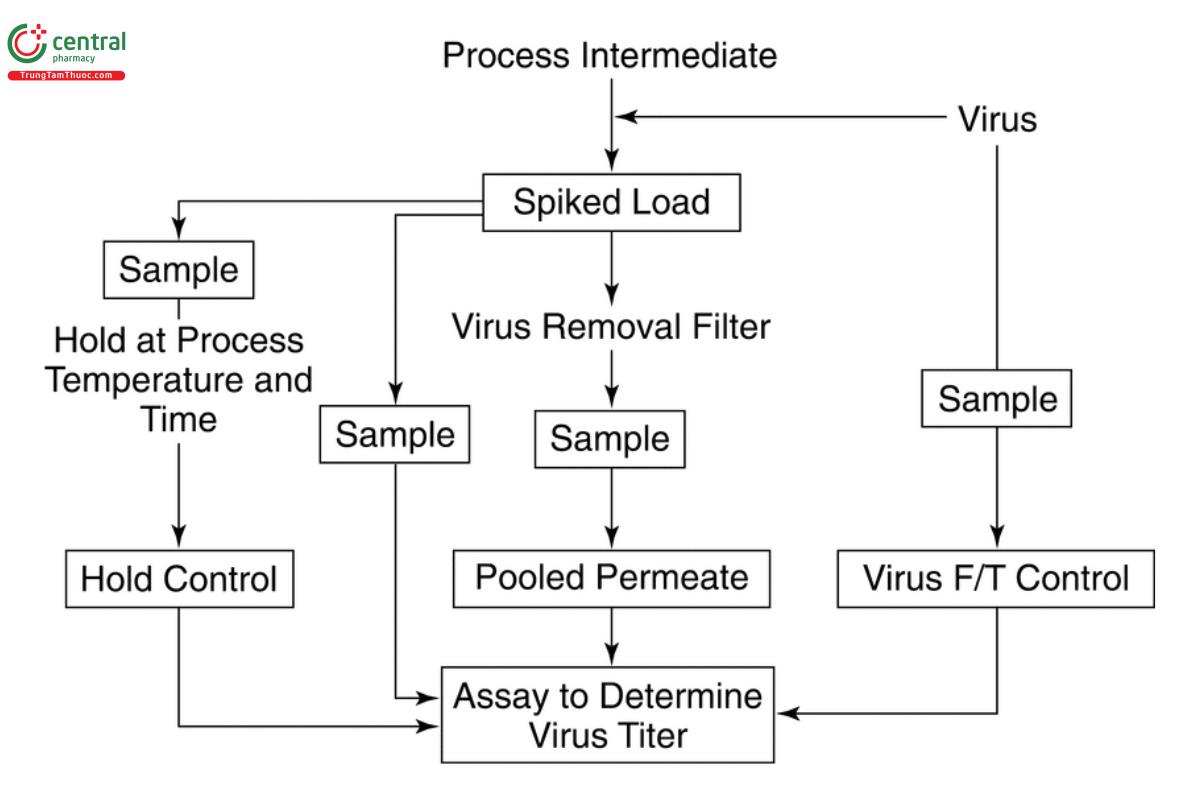
Appropriate pre- and post-filtration controls should be used to assess removal of the virus spike by the prefiltration step. Samples are evaluated using virus titration assays. Test samples may require neutralization to appropriate pH levels for cell-culture-based titration assays. If a 0.22-µm or 0.45-µm prefilter is used during production in line with the virus-reduction filter, spiking could be performed on the intermediate before prefiltration. However, a sample should be taken from the stage after prefiltration in order to determine the VRF. Alternatively, the virus spike can be added to the intermediate after this prefiltration (using a similarly filtered virus spike) in order to determine virus reduction caused by the virus filter. To gain additional understanding of the virus clearance process, analysts can take samples at various time points throughout the run. In the event that samples are stored frozen prior to virus quantitation, a virus freeze/thaw (F/T) control is suggested to determine if there is any impact of the F/T cycle on virus titer (see also Effects of Storing and Freezing Viral Clearance Samples).
3.10.3 VIRAL CLEARANCE BY COLUMN CHROMATOGRAPHY
General considerations: For registration-enabling studies, analysts should investigate the distribution of the virus load among different chromatography fractions. Generally, analysts add virus to the starting intermediate (feed stream or load) for each step tested and determine the virus titer of the spiked load material and product pool spiked load material before and after the step. Fractions, in addition to the main product pool, may require testing at the later stages of product development (phase 3 trials and beyond). As is usual for viral clearance, these studies should be performed in duplicate. With column chromatography, the ability of the columns as well as other devices to clear virus may increase or decrease after repeated use. Evidence of consistent viral clearance after multiple uses may provide support for repeated use of such columns. Typically, this evidence is obtained when analysts compare clearance on new resin versus resin that has been cycled to or slightly beyond the targeted column lifetime, i.e., the number of times the column will be used in commercial manufacturing. For registration-enabling studies, data should also be provided to show that any virus potentially retained by the resin (carry-over) will be adequately inactivated or removed by cleaning procedures before reuse of the column.
For each manufacturing step assessed for viral clearance, the probable mechanism of reduction in viral infectivity should be known and described as virus inactivation or virus removal, or both. In some situations, it may be necessary to distinguish between removal and inactivation. For example, a column chromatography step that physically separates virus from product also may use a buffer capable of inactivating virus. In such situations, it may be possible to combine the use of viral infectivity assays with NAT assays to measure the individual contributions of the inactivation and removal mechanisms. Dissection of each step to determine the relative contribution of each mechanism of clearance allows for a thorough understanding of how viral clearance is achieved. This understanding may help to identify critical variables in each clearance step that should be controlled to support reproducible clearance. The testing of each critical variable (when it is well understood) at worst-case conditions helps to evaluate the process robustness of the step.
To measure virus clearance by a column chromatography system, analysts must perform preliminary cytotoxicity and viral interference analyses (see Sample Matrix Effects on Viral Quantitation Assays for details). The column load dilution that will be used in the clearance study is that dilution shown not to cause cytotoxicity or interference. [NOTE-Interference and cytotoxicity studies also can be performed on collected fractions, if warranted.]
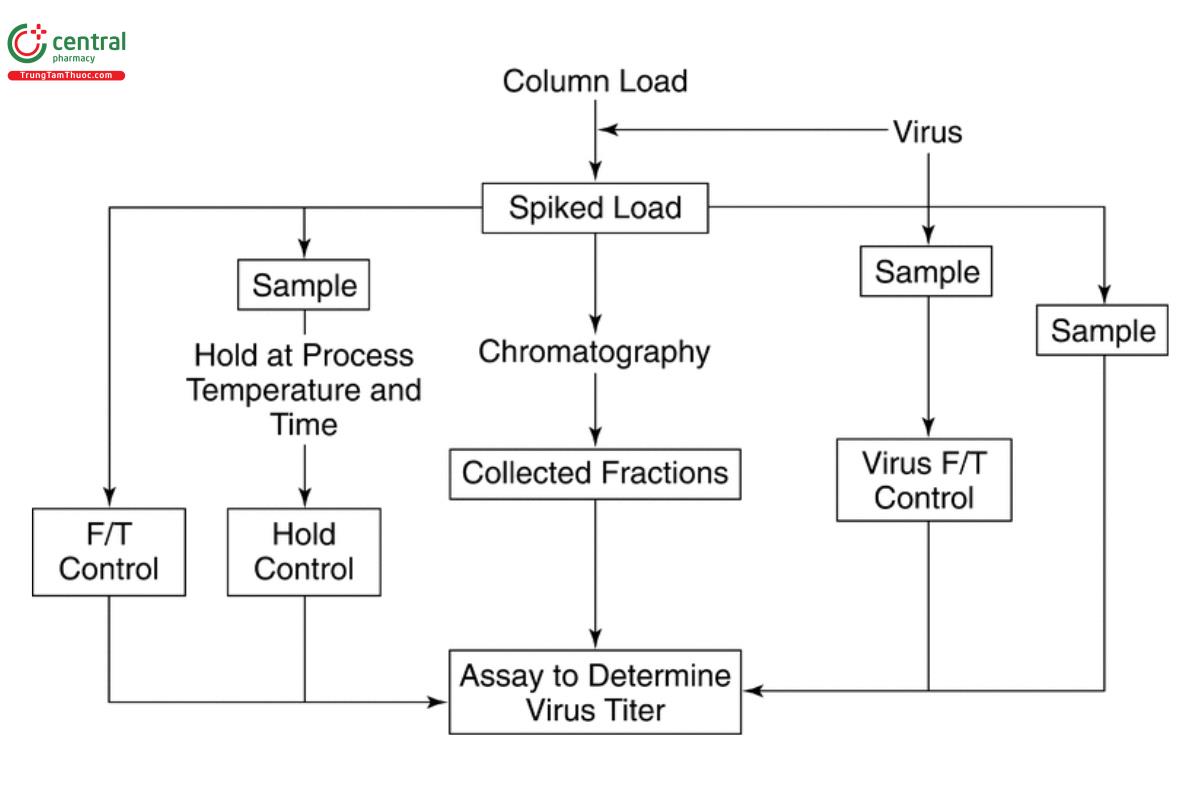
Study design: Figure 3 shows the general approach to study design for assessing viral clearance by chromatography. The column load is the material applied to the chromatography column. The load material in small-scale viral clearance studies should be comparable to material from clinical or commercial manufacturing in terms of composition and physicochemical attributes. The virus stock is a freshly thawed virus material of predetermined infectivity titer that is used to perform virus spiking of the column load material and also is used in the interference study. The virus stock can be sampled to determine the actual virus titer immediately after thaw. The spiked load control and additional samples should be tested immediately or immediately frozen. The titer of the frozen sample and other samples will be determined at the end of the chromatography step run. In certain cases, filtration of the spiked load with a 0.22-µm or 0.45-µm prefilter may be necessary to remove any viral aggregates formed when the process intermediate was spiked with the model virus.
The chromatography step experiment for viral clearance should closely mimic the actual manufacturing process step parameters. It should be performed under worst-case conditions (e.g., flow, protein concentration, and elution conditions) if they have been established. The product pool or multiple fractions can be collected, and the virus titer can be determined in each collected fraction (such as flow-through, washes, pre-peak, peak, post-peak, strip, and others). The titers of virus in all the fractions that comprise the product pool are used to calculate the clearance factor of the step. Interference and cytotoxicity studies also can be performed on collected fractions, if warranted. For the duration of the step, the hold control (the load plus virus) should be kept at the temperature at which the chromatography is performed. At the end of the chromatography run, the hold control should be titrated along with other collected samples/fractions (these can be titrated immediately, or if not feasible, can be frozen and tested at a later time). Samples are evaluated using virus titration assays. Test samples may require neutralization to appropriate pH levels for cell-culture-based titration assays.
4 GLOSSARY
(See also Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin (1050), Glossary.)1
Process Robustness of Viral Clearance:
Ability of the downstream process or a single-unit operation to exhibit the characteristic performance and effectiveness in clearing viruses after minor or moderate changes of standard operating parameters or conditions.
Process Solutions:
Production intermediates that are obtained for viral clearance studies. They should be obtained from a full-scale production facility or from a scaled-down process that has been shown to be representative of full-scale production.
Production Process:
The manufacturing processes for biotechnology products are often varied and complex, and they are typically divided into upstream and downstream processing. Upstream activities produce the protein of interest, usually by cell culture or fermentation, and considerations include integrity and quality of the process, cell banks, expression systems, cultivation, media, process/product purity, impurities, and contaminants. Downstream processing involves separation and purification of the bulk bioproduct to make it suitable for its end use, e.g., purification, sterilization, and final formulation. Downstream activities include filtration, centrifugation, precipitation, numerous chromatographic separations, sterilization by terminal filtration, or lyophilization.
Purification Process:
Separating and isolating the product of interest, in its desired form, from the fermentation supernatant or cell homogenate.
Viral Clearance Effectiveness:
The efficacy of the entire purification process or an individual process step within a purification process to clear viruses, as determined by the viral reduction factor. In general, for a virus clearance step to be considered effective 4 logs or more of clearance must be demonstrated with 95% confidence (a = 0.05).
Viral Load:
The amount of virus added to the load material of a purification step and then subjected to an inactivation/removal treatment. The experimental determination of viral load before and after treatment enables calculation of a reduction factor that is specific to the virus and the treatment used.
Viral Reduction Factor (VRF):
The VRF of an individual clearance step represents the log10 of the ratio of the virus load in the pretreatment material (used to challenge the clearance step) to the virus load in the post-treatment material. The overall VRF is calculated by adding all the individual steps with a log10 reduction factor >1. The log10 viral reduction factor is determined by the following equation:
Log10initial virus load - log10final virus load = Log10Viral Reduction Factor
The log10 viral reduction value expresses levels of decreased viral contamination by factors of 10 that could be easily converted to percent reduction. For instance, a 1-log reduction is equivalent to a 90% reduction, a 2-log reduction is a 99% reduction, a 3-log reduction is a 99.9% reduction, and a 4-log reduction is a 99.99% reduction.
Virus Removal/Inactivation Validation:
Virus removal/inactivation validation studies evaluate the capacity of the process to eliminate and/or to inactivate the viruses. Typically, this involves spiking the product with a known virus and then subjecting the product to inactivation/removal processes.
1 Cf. glossary in 〈1050〉.
