Cell Banking Practices for Recombinant Biologics
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- INTRODUCTION
- GENERAL PROCESSES AND CONSIDERATIONS OF CELL BANKING
- PRODUCTION CELL LINE/STRAIN DEVELOPMENT AND RCB GENERATION
- MAMMALIAN CELL LINE CLONALITY
- CELL BANK CHARACTERIZATION
- CONCLUSION
- GLOSSARY
- REFERENCES
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
This general chapter is intended to provide users with practical guidelines regarding the derivation, production, storage, characterization, and qualification of bacterial and mammalian cell banks used in the manufacture of biological drug products. The chapter is primarily focused on the characterization of cell banks used to produce biological products that are subject to approval by the US FDA. Cell banking characterization and qualification should be based on sound scientific principles and current regulatory guidance. The relevant requirements and guidances for generating cell banks for biotherapeutics seeking FDA approval include the US Code of Federal Regulations (21 CFR Part 610.18) (1), FDA Points to Consider documents (2–4), FDA Guidance for Industry documents, and International Council for Harmonisation (ICH) guidelines (5–8). These guidances state that the nal biological product and production should be consistent, uniform, and free from cellular and adventitious contaminants. [Note—It is recognized that there are also relevant guidances from other international organizations such as the World Health Organization (WHO) and European Medicines Agency (EMA); however, this chapter focuses on US FDA, USP, and ICH regulations and guidances.]
Therefore, the intent of this chapter is to elaborate on principles noted in FDA and ICH guidances and in USP general chapters for the establishment and characterization of mammalian and bacterial cell banks used in the manufacture of biological products. This chapter represents current best practices for the production, characterization, and maintenance of controlled and consistent cell sources using risk based and product-phase-appropriate quality considerations.
The evaluation of the risk to quality (9) should be based on scientific knowledge and ultimately link to the protection of the patient; the level of effort, formality, and documentation of the quality risk management process should be commensurate with the level of risk. Regarding good cell line banking practice, risk analysis1 should be conducted on the basis of existing experience with the type of cell line and should be updated throughout the cell line’s product life cycle as further knowledge is gained to characterize the associated risks. The risk criteria for good cell line banking practices should include such quality risk factors as:
- Cell line history and origin
- Cell line growth and characteristics (relevant phenotypic and genotypic markers)
- Cell count
- Viability
- Authentication
- Absence of adventitious agents (e.g., microbial, fungal, viral, and mycoplasma contaminants)
- Genetic drift (e.g., transgene stability)
- Loss of cell line characteristics of interest (such as the host cell transformations, surface antigens, or monoclonal antibody expression)
- Process validation and capability
- In-process controls
- Change controls
- Cross contamination
- Testing
- Stability
- Storage
- Product and/or process trending that demonstrates the original cell line characteristics and resultant quality characteristics of the nal product that are retained throughout the cell line and product’s life cycle
Over time and experience—through feedback from the quality systems, trending, post-marketing surveillance, and pharmacovigilance systems—the risk assessment should continue to evolve to minimize the risks associated with the cell line’s performance during the product’s life cycle.1
1.1 Scope
Cell banks are the starting materials for manufacturing biological drug substances and therefore have a direct impact on the characteristics and safety of the resultant biological products. Cell banks preserve characteristics in a contamination-free process and ensure consistency of the quality attributes of the nal drug substance and drug product (DP). This chapter covers cell banking for biotechnology products for human use that are derived from cell lines of mammalian and microbial origin. In many instances, biological product development involves developing new recombinant prokaryote [e.g., Escherichia coli (E. coli)], eukaryote [e.g., Chinese hamster ovary (CHO)] or transgenic cell lines, and requirements may differ depending on the strain or cell line. Each new strain/cell line needs to be evaluated with respect to its own particular cellular characteristics and its associated master cell banks (MCBs) and working cell banks (WCBs) need to be manufactured under conditions consistent with good manufacturing practices (GMPs). Each GMP cell bank should be tested and characterized to satisfy quality standards for identity, purity, and genetic stability and ensured to be free from adventitious contaminants for use in drug substances for commercial applications. If possible, storage stability should also be performed. A risk-based approach should be applied for manufacturing. The bank can be conditionally released at risk before it is fully tested to start drug substance manufacturing; however, the bank has to be fully tested, especially for safety prior to a drug substance's release for clinical applications.
This chapter does not cover cell banking for antibiotics, viral seed banks, viruses that express vaccine antigens, bacterial seed banks, avian or insect cells, or cell banks for cell and gene therapies. For specific information on cell banking for vaccines, see Vaccines for Human Use—Bacterial Vaccines 〈1238〉 and Vaccines for Human Use—General Considerations 〈1235〉.
This chapter primarily focuses on cell banks derived from recombinant E. coli strains and CHO cell lines as they are the most common substrates for production of therapeutic proteins and centers on the cell line or strain history, GMP manufacturing and testing, and qualification of cell banks. Most, if not all, of the techniques and practices discussed are also applicable to other cell strains besides E. coli and CHO cell substrates. Based on the organism, suitable approaches can be selected to cover the cell line/strain history, GMP manufacturing and testing, as well as qualification of cell banks.
2 GENERAL PROCESSES AND CONSIDERATIONS OF CELL BANKING
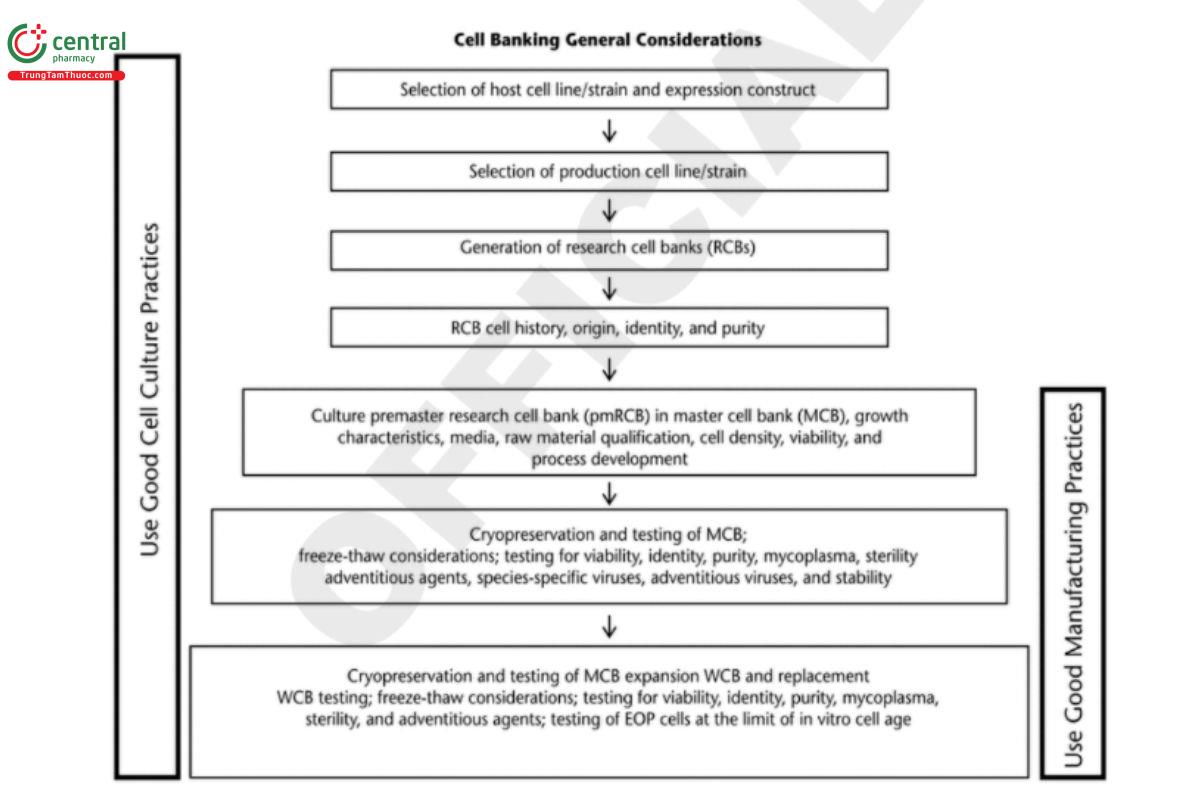
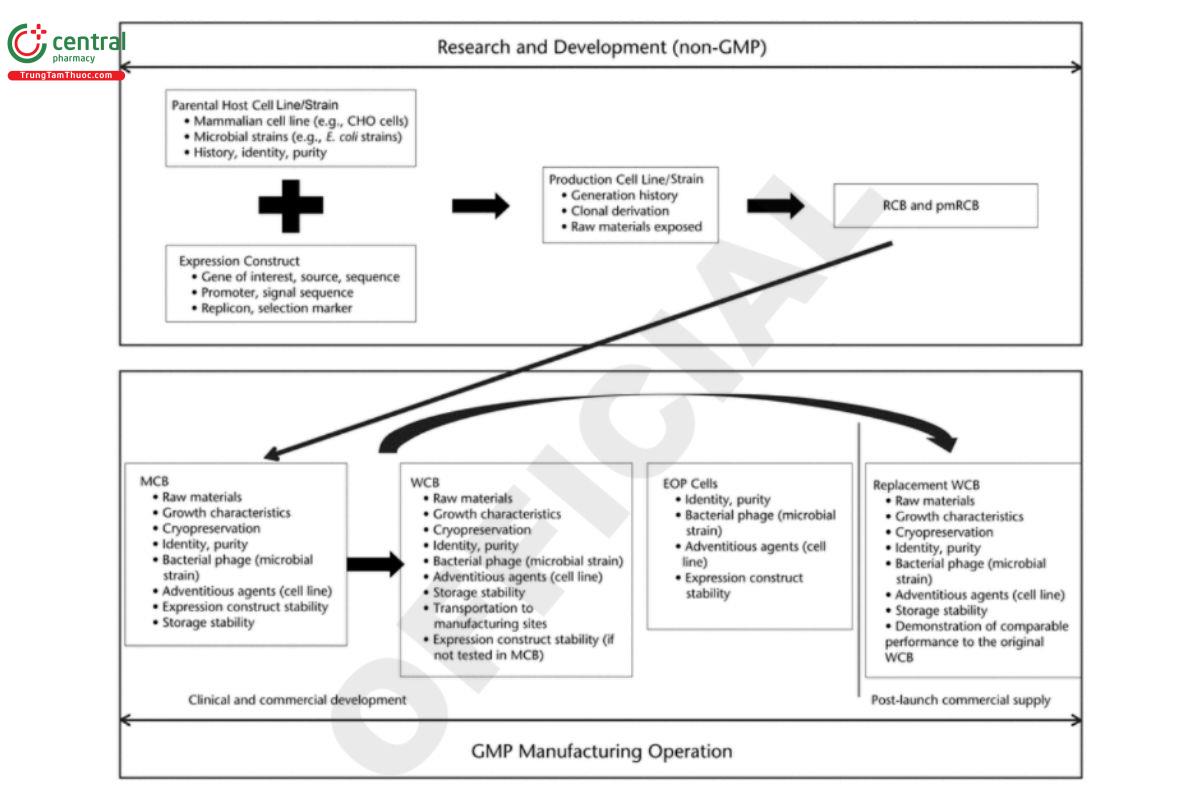
Cell banks used for manufacturing biological drug substances are usually prepared from research cell banks (RCBs) and use a two-tiered bank system, MCB and WCB or working seed lot. The MCB is generated as the source for the WCB, which is derived by expanding 1 or more vials of the MCB. Thus, the MCB is considered the starting raw material for the GMP manufacturing of drug substances. RCBs are usually generated under non-GMP conditions, whereas MCBs and WCBs are required to be produced in GMP facilities. Documentation of suitability of the RCB for GMP manufacture is needed. Figure 1 outlines the general practices and considerations of cell banking processes and how they evolve through development into GMP manufacturing. The critical steps shown in the gure are applicable to recombinant and non recombinant cell lines. More detailed best practices and recommendations are described in the sections that follow. For more detailed recommendations about preparation and preservation of cell banks, see ICH Q5D (7).
The generation of an RCB starts with choosing a suitable expression system that consists of host cells and expression constructs carrying the genes of interest (GOI).
Any facility including a research laboratory should be regarded as a potential source of contamination by viruses, bacteria, fungi, and mycoplasma, so the introduction of a new cell line should be handled under quarantine conditions until proven negative for such contaminants. The history, identity, and purity, as well as associated testing of the host cells, should be well documented (10).
An expression construct consists of the GOI and an expression vector carrying the genetic elements such as a promoter, signal sequences, selection markers, and other elements needed for the stable expression of the GOI in the host cells. Detailed descriptions of the GOI and those genetic elements carried by the vectors, as well as the steps in the assembly of the expression construct, should be documented.
The next step for RCB is the generation of the candidate production cell lines/strains. It is critical to document the details of the cell-line generation history, including:
- Description of the transfection/transformation of the GOI
- Screening and selection of the production cell lines/strains
- Demonstration of clonal derivation
- Information about the raw materials used for the generation of candidate production cell lines
The source, origin, generation, propagation, and selection of the cell line must be documented and described in detail. Multiple RCBs may be generated in relatively small quantities during the screening/selection processes. Only the nal, chosen production cell lines/strains and backup cell lines/strains are made in larger quantities. The RCBs chosen for the production cell lines/strains and backup cell lines/strains are sometimes referred to as premaster research cell banks (pmRCBs), which need to go through purity testing prior to the production of MCBs and subsequent WCBs.
Cell substrate for manufacturing biological drug substances, MCBs, and WCBs must be produced in compliance with current GMPs (cGMPs). As illustrated in Figure 2, MCBs are often used for manufacturing drug substances for early clinical studies and as the source of the WCBs, which may not be available during the early phase of the development life cycle. Drug substances used for late-phase development and commercial applications are usually manufactured from WCBs. To ensure a consistent supply, it is generally recommended that additional replacement WCBs be produced as needed once a biologic is approved for marketing. Section 5. Cell Bank Characterization describes in detail cell bank characterization, testing and qualifications, as well as general considerations on raw materials, cell bank cryopreservation, storage and stability, and transportation.
3 PRODUCTION CELL LINE/STRAIN DEVELOPMENT AND RCB GENERATION
3.1 Common Cell Line Types and Associated Expression Constructs
Production cell line/strain development starts with choosing a suitable expression system including a host cell and an associated expression construct carrying the GOI. The choice of the host cell type for production of a recombinant protein is often a function of the molecular complexity of that protein. Prokaryotic production systems are often dedicated for simple, small proteins whereas eukaryotic expression systems are preferred for larger, more complex molecules because eukaryotic expression systems can perform necessary post translational modifications for a recombinant protein to be functional in vivo. In addition, eukaryotic expression systems can secrete the recombinant protein outside the cell, which makes purification easier than, for instance, isolation of recombinant proteins from inclusion bodies formed in prokaryotic systems. Mammalian cell lines such as CHO cells, non-secreting (NS0) mouse myeloma cells, and human embryonic kidney (HEK) cells and microbial strains such as E. coli and Bacillus are examples of cell types that have been used to generate production cell lines/strains. This chapter focuses on mammalian and bacterial cells because they are currently the predominant cell substrate types used for recombinant therapeutic production.
3.1.1 Chinese hamster ovary cells
General information: CHO cell lines are several related, but unique, cell lines originally derived from a clonal and spontaneously immortalized CHO cell line isolated in 1957. CHO cells were widely used in molecular and classical cell genetics studies until the 1980s. The knowledge derived from those studies facilitated the migration of CHO cells from research laboratories to industrial bioreactors. In 1986, CHO cells became the first mammalian cell-derived cell substrate used to produce complex proteins for human use and since then have remained the most common cell line for the production of therapeutic glycoproteins for the following reasons:
1. CHO cells pose a lower adventitious virus safety risk as only a few human pathogenic viruses are able to propagate in CHO cells (11).
2. CHO cells grow well in animal-component-free, chemically defined media, which further minimizes adventitious virus safety concerns.
3. As immortal cells, CHO cells can be propagated for a long time in vitro. This is an important characteristic for commercial production of recombinant proteins because it takes many cell passages to progress from cell line generation through cell banking to the nal, large-scale commercial production process. A non-immortalized cell line cannot be propagated long enough to generate a profitable and sustained commercial production process.
4. CHO cells are able to adapt and grow in suspension culture, which is ideal for large-scale manufacturing of biologics. 5. CHO cells have the capacity for efficient, post-translational modifications, such as glycosylation, which enable the CHO-derived therapeutic proteins to be both compatible with and bioactive in humans.
5. CHO cells are amenable to genetic manipulation, which allows for easy introduction of recombinant DNA resulting in expression/secretion of large quantities of the desired protein.
See Table 1 for a comparison of two common cell substrates for recombinant therapeutic products.
Table 1. Comparison of Two Common Cell Substrates for Recombinant Therapeutic Products
Cell Substrate | Mammalian Cell Lines (e.g., CHO Cell Lines) | Microbial Strains (e.g., E. coli Strains) |
Advantages |
|
|
Disadvantages
|
|
|
Because CHO cells are immortalized, they can pose a significant challenge, though, for the cell-line development process with respect to finding genetically stable cell lines needed for consistent commercial manufacturing processes. CHO cells have varied ploidy, and a culture of any CHO cell line, with or without any transgenes, consists of a population of cells that exhibit both genetic and phenotypic diversity and instability under continuous cultivation. Random integration of recombinant genes into the CHO genome adds new genetic variability to the CHO cell population. After transfection, implementation of a single-cell cloning step helps to minimize cell population heterogeneity by removing genetic variations that either existed prior to or were introduced during transfection.
All CHO cell substrates used for commercial recombinant protein production are expected to be single-cell derived. However, incorporation of the single-cell cloning step alone cannot prevent genetic and phenotypic heterogeneity within the population of cells that arises post-cloning, as these events are inherent to the use of immortalized mammalian cell lines (12). Thus, identifying an appropriate production cell line ultimately depends on the genotypic and phenotypic characterization of potential cell lines post-cloning at the limit of in vitro cell growth and the evaluation of the expressed product.
Common CHO cell lines: Some commonly used CHO cell lines include: CHO-DXB11, CHO-DG44, CHO-K1, and CHO-K1SV. These cell lines are transfected with expression plasmids carrying the GOI and corresponding selection markers specific to the CHO host cells. Their characteristics are briefly summarized below.
CHO-DXB11 and CHO-DG44: CHO-DXB11 (also known as CHO-DUK-XB11 or CHO-DUKX) and CHO-DG44 are dihydrofolate reductase (DHFR)- deficient mutants established in the early 1980s. CHO-DXB11 cells carry a deletion of one locus for DHFR and a missense mutation (T137R) of the second DHFR locus, rendering the cells incapable of reducing folate, a precursor for thymidine and hypoxanthine synthesis; CHO-DG44 cells carry the full deletion of both DHFR loci.
The DHFR-deficient cells are triple auxotrophs for glycine, hypoxanthine (a proline derivative), and thymidine. Introduction of the GOI into CHO cells can be accomplished by cotransfection with a functional DHFR gene, which obviates the need for these nutrients. Recombinant cell lines carrying the integrated GOI are selected in media devoid of those three nutrients. The GOI expression level can be further enhanced by gene amplification with Methotrexate (MTX), an antagonist of DHFR. Selection of recombinant cell lines can be performed using stepwise increases in the media concentration of MTX, which results in amplification of the transfected DHFR gene together with the GOI, possibly increasing the productivity of the GOI.
Sometimes MTX-induced gene amplification can result in large genomic reorganizations. New chromosomal structures, known as homogeneously staining regions (HSRs), can be found in the metaphase chromosome of MTX-selected CHO cells. These regions show multiple repetitions—up to thousands of smaller chromosomal regions (amplicons)—all containing DNA encoding at least in part sequences with DHFR activity as well as hundreds and even up to thousands of copies of the GOI. Gene amplification and high-level expression of a transgene product can challenge the cells with potential toxicity and metabolic burden. Additional DNA rearrangements within the GOI region may also occur during long-term subculturing in the absence of MTX, which may eventually lead to instability of the recombinant protein production. Therefore, genetic stability evaluation of the chosen CHO production cell line is important to ensure that the desired characteristics for the commercial production are acquired.
CHO-K1 and CHO-K1SV: CHO-K1 was derived as a sub-clone of a parental CHO cell line lacking the proline synthesis gene. CHO-K1 was adapted to grow in suspension culture in protein-free medium, resulting in the cell line CHO-K1SV. Introduction of a GOI into the CHO-K1SV cells can be accomplished by cotransfection of the GOI with a functional glutamine synthetase (GS) gene followed by selection in the absence of glutamine. In addition, application of a GS inhibitor [methionine sulfoximine (MSX)] allows an increase in the stringency of selection. Although the copy number of the GOI and GS is significantly lower in the GS-based selection system compared to that seen in the DHFR-based system, instability of the GS-based CHO cell lines is still often observed during long-term cultivation. Therefore, it is important to evaluate genetic stability of the chosen CHO production cell line over the proposed in vitro cell age needed for commercial manufacturing.
Expression vectors for CHO cell substrate: Expression vectors used for CHO-based cell substrates consist of multiple components that contribute to the successful expression of the therapeutic proteins. These components include:
- The GOI and associated transcriptional and translational genetic elements (e.g., promoters, enhancers, Kozak translation initiation sequences, leader sequences, and polyadenylation signals)
- Selection markers such as the commonly used DHFR or GS for recombinant cell line selection
- Selection markers such as the β-lactamase gene (bla) and replicons such as ColE1 for propagation of the plasmid in E. coli
Expression plasmids are often linearized prior to transfection and are generally integrated into the CHO host cell genome by random integration. Resulting transfectants are a mixture of CHO host cells carrying variable copy numbers of expression plasmids randomly inserted in single or multiple loci in the host genome. A single-cell cloning process enables the clonal derivation of future production cell lines. Significant effort is spent on screening the clonally derived transfectants to identify candidates for nal production cell lines that possess high productivity. Furthermore, the identified cell lines must be capable of maintaining the integrity and stability of the expression construct integrated into the host genome in order to be used for large-scale manufacturing processes.
To reduce the heterogeneity associated with traditional random integration approaches, new expression systems have been developed to facilitate targeted integration of the GOI into a specific desirable locus within the CHO genome. Targeted integration systems described in the literature include site-specific recombinases, integrases (ØC31), transposases (piggyBac), zinc finger nucleases, transcription activator like effector nucleases (TALENs), and CRISPR-Cas RNA-guided nucleases. Detailed descriptions of the genetic components carried by the expression plasmids, the specific integration method, and identification of the preferred production cell line should be included in the history of the nal production cell line.
CHO host cell engineering: In addition to expressing the GOI, recombinant CHO cell lines can be engineered to further enhance productivity and quality of the recombinant product as well as the robustness of the cell culture performance. Host engineering includes overexpression or knock-out of specific genes that may impact the desired recombinant product characteristics and thereby the performance of the production cell line. Examples of overexpressed genes are those involved in:
- Glycosylation [e.g., α-2,6-sialyltransferase (ST6Gal)]
- Cellular metabolism [e.g., vitreoscilla hemoglobin (VHb)]
- Anti-apoptotic pathways [e.g., B-cell lymphoma 2 (BCL2)]
- Pro-proliferative pathways [e.g., valosin-containing protein (VCP)], as well as genes that may enhance cellular protein biosynthesis machinery and secretion [e.g., transcription factor ATF4, heat shock 70 kDa protein 5, binding immunoglobulin protein (BiP), etc.]
Alternatively, there are some host cell proteins that can decrease the quality or yield of recombinant proteins [e.g., α-1,6- fucosyltransferase (FUT8) and peptidylglycine α-amidating monooxygenase]. In these cases, genomic knockouts of the host cell genes can yield a more homogeneous product.
Although the overexpressed or knocked out genes do not encode sequences for the nal therapeutic protein, they play critical roles in the productivity and quality of the expressed therapeutic proteins. Therefore, evaluation of the integrity and stability of the overexpressed genes for the intended purposes should be included as a part of the cell substrate genetic stability characterization. In addition, knocked out genes should be characterized.
3.1.2 Escherichia coli
General information: E. coli is one of the best-understood organisms in terms of genetics, physiology, and biochemistry. The key advantages of the E. coli system include:
- The GOI can be easily inserted into an expression plasmid using recombinant DNA technology. The resulting recombinant plasmid, which is usually small and simple, can be inserted into the E. coli host by transformation and maintained as an extrachromosomal segment of the host DNA or a piece of the host genome after integration. Numerous E. coli expression systems are commercially available, making this organism easy to test for production of a new GOI.
- E. coli is well-characterized genetically with versatile comprehensive engineering tools.
- E. coli strains are capable of rapid growth in inexpensive media in small shake flasks as well as in large-scale fermenters used to produce therapeutics with high yield and quality. E. coli expression systems continue to evolve, and the advances in biotechnology and bioengineering as well as high-density fermentation technology have further enabled E. coli systems to produce a variety of complex proteins, including antibody fragments and Fc-fusion proteins.
However, E. coli strains also have limitations as cell substrates for therapeutics production, including their inability to perform certain post-translational modifications. In addition, endotoxins need to be thoroughly removed during purification of the recombinant protein produced by an E. coli host (also see Table 1).
Sometimes the recombinant protein itself can be an obstacle for production in E. coli systems because it can be toxic to the host. In such cases, a well-designed but more sophisticated expression system may be required, for instance, an inducible promoter to produce the recombinant protein at the end of cell expansions. In the following sections, the characteristics of some E. coli-based cell substrates consisting of E. coli host strains and vectors carrying the genes encoded for the therapeutic proteins are briefly summarized. E. coli host strains: The most commonly utilized strains are the K12 strain and its derivatives including E. coli RV308 and W3110, which are all genetically modified so they cannot colonize a human host or produce toxins. These strains may qualify for an exempt status according to the requirements of the National Institutes of Health (NIH) guidelines for research involving recombinant DNA molecules.
The other commonly used E. coli hosts are the B strain and its derivatives, such as BL21, C41, and C43. Although these strains do not qualify for exempt status and require full Institutional Biosafety Committee (IBC) review, they are still desirable hosts for therapeutic protein production in the industry because they are able to express various types of proteins. Advantages of the B strain and its derivatives are the low levels of acetate accumulation under high Glucose concentrations, the specific protease deficiencies, and the high outer membrane permeability.
Expression vectors for E. coli strains: The expression vectors often used are the result of multiple combinations of replicons, promoters, selection markers, and other genetic elements that are essential or beneficial for the desired efficiency of transcription, translation, and protein secretion, as well as the stability of the plasmid.
A replicon in a vector consists of one origin of replication together with its associated cis-acting control elements. It controls the copy number of a plasmid replicated in an E. coli host. Commonly used replicons include:
- pMB1 and its derivative origins used in pUC-based vectors (500–700 copies/genome)
- pBR322-based vectors (10–20 copies/genome)
- p15A origin used in pACYC and pSC101, which have low copy numbers (about 5 copies/genome)
For large-scale therapeutic protein production, an optimal copy number should be identified because too few gene copies may result in low levels of mRNA leading to low protein productivity while too many copies may impose metabolic burdens on cells. In addition, high-copy number plasmids have been shown to possess higher segregation instability, especially in the absence of selective markers. This can lead to the overgrowth of plasmid-free cells, resulting in a significant loss of protein productivity. Therefore, it is usually not desirable to use high copy-number plasmids, and it is critical to monitor plasmid retention during large-scale production. Testing to qualify the microbial cell bank includes testing to demonstrate purity and freedom from phage contamination and to confirm strain identity. Additional testing includes viability, plasmid copy number, plasmid retention, and restriction enzyme mapping.
Promoters control and regulate the efficiency of transcription. Commonly used promoters include:
- lac and its derivatives such as lac UV5, tat and trc, which are induced by isopropyl-β-D-thiogalactopyranoside (IPTG) Bacteriophage λ p and/or p promoters, which are induced by temperature shift via the regulation of the temperature-sensitive cI857 L R repressor
- T7 promoter, specifically recognized by T7 bacteriophage RNA polymerase from T7 bacteriophage
- phoA, trp, and ara PBAD, which are nutritionally inducible promoters
Selection markers, usually antibiotic-resistant genes such as TetR, are included in the plasmid to determine the growth of plasmid-free cells. Therefore, antibiotics are usually included in the growth media. Some common examples include Neomycin, Polymyxin B, Streptomycin, and Gentamicin. However, the use of antibiotics in clinical manufacturing can lead to adverse events in some patients and raises concerns over the development of drug-resistant pathogens, so it is desirable to develop antibiotic-free plasmid systems, if possible.
In addition to the coding sequence encoding the protein, genetic elements in expression vectors that impact protein expression include the sequence and structure of the translation initiation region (TIR) at the 5′ end of each mRNA and the stability (half-life) of mRNA. Another genetic element used in some expression vectors is the signal sequence fused to the N-terminus of the protein, which leads the protein to the periplasmic space of the host or the cell culture medium. Without signal sequences, recombinant proteins mainly accumulate in the bacterial cytoplasm. Due to the lack of glycosylation and disulfide-bond formation, many human therapeutic proteins expressed in E. coli tend to aggregate and form inclusion bodies due to misfolding. Correct folding of eukaryotic proteins containing multiple disulfide bonds is more likely to occur in the oxidative environment of the periplasm. The secretory process allows removal of the N-terminus signal peptide and yield of the authentic N-terminus of the expressed protein. Co-expression of periplasmic chaperones, fusion with secretory partners, and modification of the outer membrane structure by host engineering may further promote release of periplasmic proteins.
Overall, E. coli expression systems have been preferred choices as cell substrates for the expression of non-glycosylated therapeutic proteins due to their well-characterized genetics, rapid growth, and high-yield production capability. Both the host strains and the genetic elements carried by the expression vectors should be well described in the strain development histories and documented for the cGMP cell bank generation. Together with the therapeutic protein-coding sequence, the key genetic elements that may potentially impact the drug substance quality and the consistency of drug substance production should be evaluated during cell bank characterization and qualification.
Clonal selection of E. coli host strains: As with CHO-derived cell lines, it is expected that E. coli strains used for therapeutics production be clonally derived. Microbial clonality is easily achieved and accepted by picking transformants from well-isolated single colonies grown on solid media. Additional rounds (usually two) of single-colony isolation and phenotypic confirmation may be performed to ensure the clonality.
Table 1 compares two commonly used mammalian cell and microbial strain substrates for generating cell banks for biological products.
bacillus
Other useful bacterial systems are Gram-positive bacilli, including Bacillus megaterium, B. subtilis, B. licheniformis, and B. brevis. Bacillus is very often considered an attractive alternative to E. coli for the following three reasons:
1. Bacillus strains usually secrete proteins
2. Bacillus strains are suitable for high-density cultures in fermenters
3. Most Bacillus strains lack membrane proteases
In addition, the genomes of B. subtilis and B. licheniformis have been sequenced, and there is no production of harmful exotoxins or endotoxins. With these bacterial systems, secretion of the desired proteins into the fermentation medium results in easy downstream processing, thereby eliminating the need for cell disruption or chemical processing techniques. Thus, the recovery is relatively efficient and cost effective. Host strains used for successful expression of recombinant proteins are often deleted for the genes amyE, aprE, nprE, spoIIAC, and srfC and are easily transformed. These bacteria are often preferred for homologous expression of enzymes such as proteases and amylases.
3.2 Production Cell Lines/Strains and Recommended Documentation
A production strain/cell line refers to the transformed strain or transfected cell line with the expression plasmid. The history of the production strain/cell line generation is an essential part of cell banking documentation and should include the following: Raw materials used
- Expression plasmid
- Host strain/cell line
- Generation of the production strains/cell lines
- Generation of RCB/pmRCB
This information should enable an overall evaluation of the risks associated with the use of the cell substrate derived from the strain/cell line and a detailed analysis of potential entry routes for adventitious agents into the MCBs.
description of expression construct
The description of the expression plasmid used to generate the production cell line should include details on the origin of the coding genes used for the recombinant protein as well as all other genetic components carried by the plasmid. Additionally, the steps in the assembly of the expression construct should be provided. The recommended details are described below.
The name and the origin of the nucleotide sequence coding for the protein should be provided with a brief summary of the rationale for the development efforts. It is important to include descriptions of the engineering methods used to optimize the protein functions and biochemical/biophysical properties along with figures displaying the engineered residuals compared to the original sequences. For example, if the gene is for a therapeutic antibody (including bi-specic antibodies and Fc-fusion proteins), literature references to the constant regions/Fc regions such as the publicly known gene identification should be provided. In addition, if any particular mutations or residual substitutions in the constant regions have been performed, then these should also be provided with a rationale for such modifications. It is recommended that the well-annotated DNA sequence as well as the deduced Amino acid sequence of the GOI be provided.
For example, for a therapeutic antibody, the annotations include the indications of the complementarity-determining regions (CDRs), variable and constant regions, linkers, signal sequences, and the sequence modifications applied to the molecule.
The steps in the GOI assembly from the original sources to the nal expression plasmid should be described in detail and are often summarized using a flow diagram. In addition, the source and function of all genetic components carried by the expression plasmid should be provided. The components include:
- The GOI and associated transcriptional and translational genetic elements (promoters, enhancers, Kozak translation initiation sequences, leader sequences, and polyadenylation signals)
- Selection markers such as DHFR or GS for recombinant cell line selection, or antibiotic resistance genes for recombinant strain selection
- Selection markers such as the bla gene and the replicon such as ColE1 for propagation of the plasmid in E. coli
- Any other expressed proteins encoded by the plasmid
Furthermore, a well-annotated plasmid map clearly indicating the size of the plasmid, all genetic components described above, and the restriction enzymes used for major cloning steps and for genetic stability characterization should be provided. The DNA sequence of the whole construct/plasmid, or at least the sequences encoding the recombinant protein that have been confirmed by DNA sequencing, should be included.
3.2.1 Parental E. coli strain/cho cell line
A parental strain/cell line is a cell line from which the MCB will be derived following introduction of the expression construct. The origin of the host cells should be stated, together with a detailed record of their history, with relevant references from the scientific literature. This includes classifications such as genus, species, and strain designation as well as specific phenotypic and/or genotypic traits. Three key aspects of information should be provided:
1. Any biohazardous components that are produced by the parental cells
2. Any exposure of the parental cells to materials of human or animal origin such as serum, enzymes, hydrolysates, or other living cells 3. Genetic and phenotypic characteristics of the parental cells that may enhance the evaluation of the safety and characteristics of the subsequently derived cell banks
The lineage of the parental cells traced back to the original source or related literature references should be provided. If the parental cells undergo additional genetic modifications (e.g., targeted gene knockout or overexpression) that are aimed at impacting the quality of the future recombinant proteins, then the methodology and characterization results should be described in detail.
Characterization of the initial cell line should take risk into consideration. Use of low-risk cells (e.g., cells shown to be sterile and phage or mycoplasma-free) is recommended. Cell line strain ID can be assessed with polymerase chain reaction (PCR) or using short tandem repeat (STR) profiling or DNA fingerprinting to verify the identity of the host cells. When higher-risk cells (e.g., a mixture of cells with varying degrees of genetic instability) are used, a more thorough screening is necessary, unless the supplier of the cells can provide documentation that minimizes the associated risk.
3.2.2 Generation of the production cell lines/strains
Generation of the production cell lines/strains usually consists of three steps:
1. Transferring the expression plasmid to the parental cells and selecting recombinant strains/cell lines
2. Single-cell cloning to produce clonally derived recombinant strains/cell lines
3. Choosing the production strains/cell lines through screening/characterization
Recommendations for documentation to include in regulatory submissions are as follows:
- Details on transformation/transfection conditions
- Manipulations of the plasmid DNA prior to the transfer (e.g., linearization by restriction enzymes often applied prior to transfection)
- Procedures/methods used for the selection of recombinant strains/cell lines
- Detailed description demonstrating the single-cell derivation of the production strain/cell line
For an E. coli strain, 2 or more rounds of single-colony streaking are commonly applied to ensure the clonal derivation of the production strains
For CHO cell lines, numerous approaches can be used to achieve clonal derivation (see 4. Mammalian Cell Line Clonality)
- Description of how the production strains/cell lines were screened and identified including the following three parts:
1. Detailed cell culture conditions
2. Selection criteria
3. All raw materials and reagents used throughout the generation process
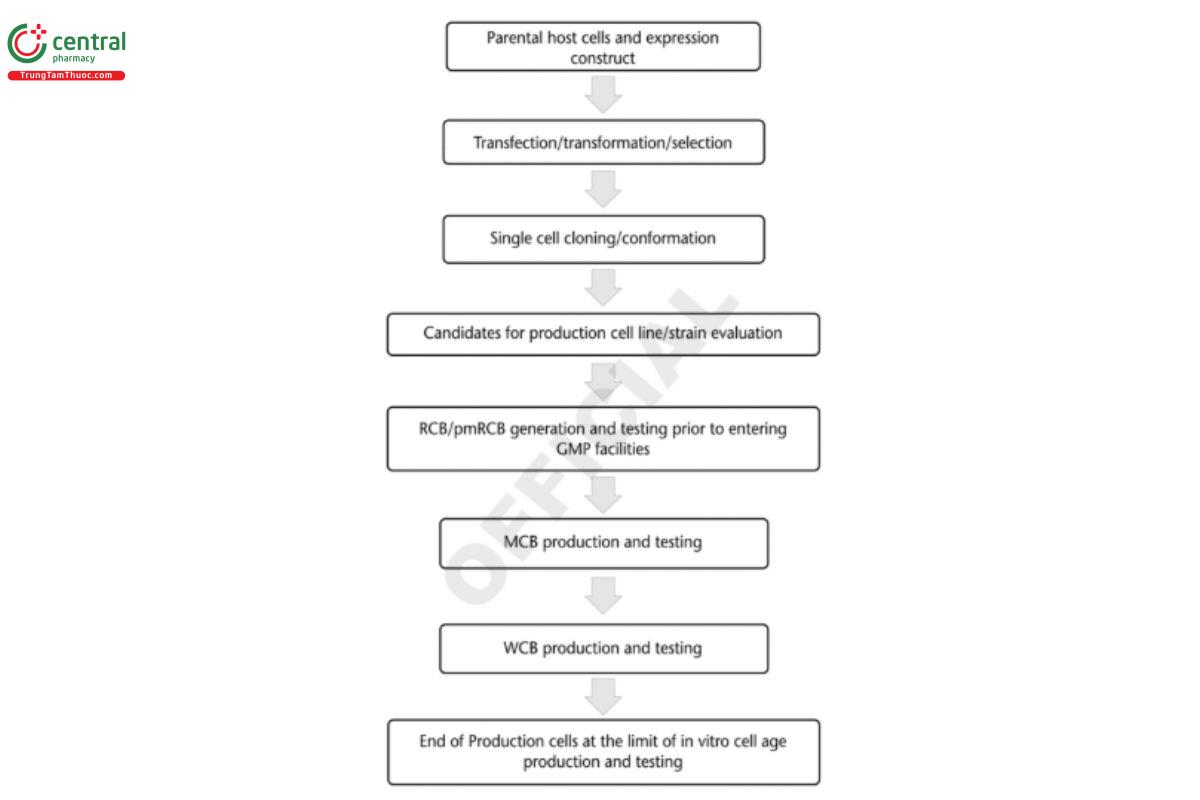
It is helpful to provide a flow diagram (see Figure 3) of the production strain/cell line generation process, which includes the above three parts and ends with the MCB and WCB production.
3.2.3 Generation and considerations of rcb/pmrcb
A common practice is to bank multiple potential production strains/cell lines in small quantities to generate RCBs or MCB as appropriate. Once the final production strains/cell lines are identified, the corresponding RCBs are thawed and expanded to generate the pmRCB. Descriptions of the cell culture medium/cryopreservation processes used to generate the pmRCB should be provided.
Because the RCB or pmRCB is usually generated in a non-GMP facility, it is recommended that RCBs/pmRCBs from mammalian cell lines should be tested and found sterile for contaminants and negative for mycoplasma before transfer to a GMP facility for the generation of the MCB. Appropriate qualification testing of RCB/pmRCB is recommended to be performed prior to moving into GMP. For an E. coli-derived RCB/pmRCB, testing for purity, identity, and bacteriophages is recommended to be performed before entry to a GMP facility. In addition, post-banking evaluation of the RCB/pmRCB for growth, viability, and viable cell density should be provided to aid MCB production and characterization. The MCB could be prepared by expansion of the production cell line over a suitable number of passages, dependent on the cell line. When the cell culture has reached an adequate cell density and total number of viable cells, the culture is prepared for freezing.
Numerous considerations for cell cryopreservation and management can be found in Cryopreservation of Cells 〈1044〉. Mammalian cells can be propagated in fermenters, roller bottles, shaker flasks, or wave bags and are frozen in a controlled process to optimize viability upon thawing. Typically, the cells are carefully mixed with a low concentration of Dimethyl sulfoxide (DMSO) in cell culture media and/or other ingredients appropriate for that specific cell strain. The mixture is frozen by lowering the temperature over an appropriate time period to avoid cell osmotic shock, which can lead to disruption of the cell membrane. The vials must be labeled in advance with the cell bank lot number, tube number, and production date. After the carefully controlled freezing process, the new cell bank is moved to appropriate storage conditions. To avoid any unexpected loss of the cell bank, the containers are typically kept in at least two different geographical locations.
Post-banking controls after thawing include measuring cell number and viability. Because these parameters vary between individual cell banks, appropriate post-banking quality control for viability can be established and justified for each bank.
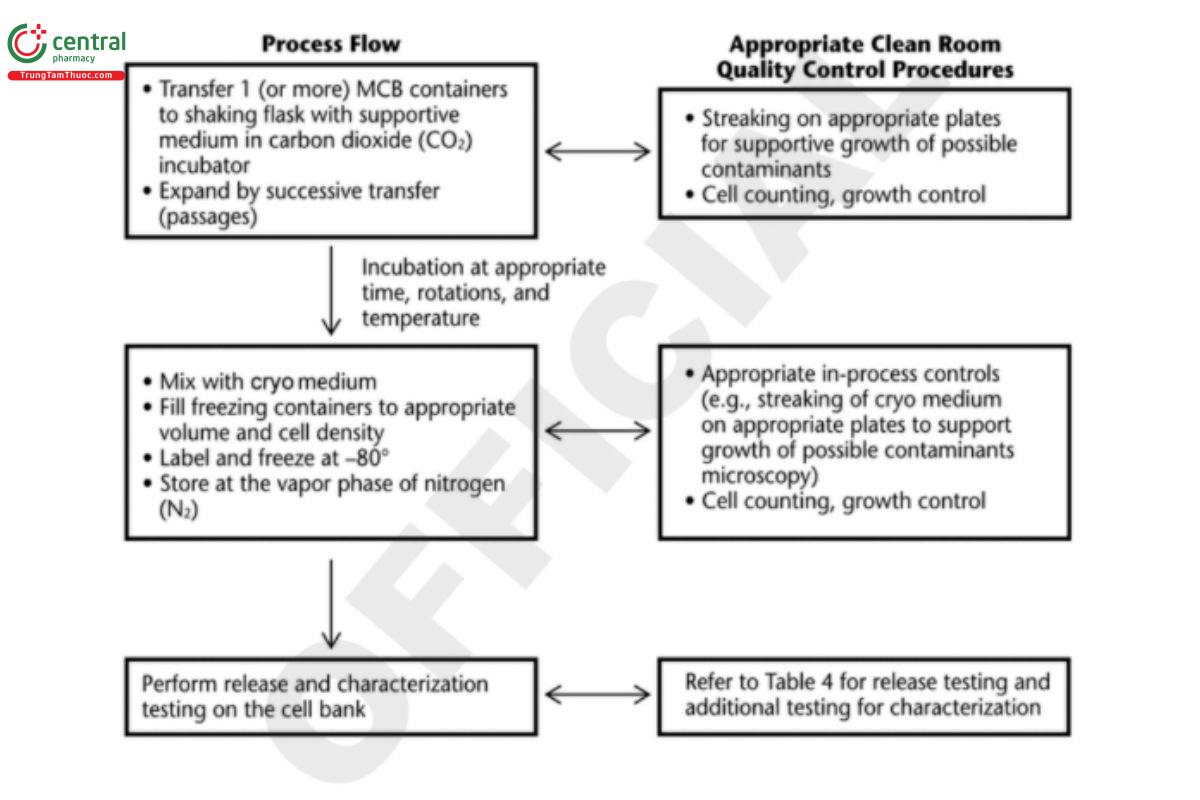
After the production of an MCB or WCB, each bank must be tested against specific acceptance criteria before that specific cell bank can be released for use. Mammalian cell banks should be stored in multiple geographical locations. Figure 4 shows an example of a cell process for a mammalian (CHO) cell bank.
Similarly, for bacterial cell lines used for therapeutic protein manufacturing, the chosen clone is subjected to growth propagation experiments to ensure that the best growth conditions are chosen for this specific clone. The growth conditions for an E. coli clone can be evaluated using shake-ask culturing. The optimal cell density is established by following growth curves in order to harvest the cells at an exponential growth. The cells are harvested in vials containing sterile 20%–30% Glycerol under sterile conditions. E. coli cells harvested at a living cell density of 106 to 1010 are adequate for production; however, this depends on the specific strains and selection pressure chosen.
All viability tests should be done with care, keeping in mind the actual selection system as well as how the harvest of any produced protein will be performed (e.g., from inclusion bodies). This is not a concern for the frozen E. coli cells after thoroughly mixing with glycerol; however, without adequate knowledge of the optimal growth point for harvesting and freezing as well as the actual needed cell density, the viability and expected growth rate can create problems at the production site.
The optimization process shall be described and followed in detail each time a new growth of the strain is necessary. From these optimized conditions, the first cell bank will be produced, which can be used for research purposes, followed by the production of an MCB. For the general process flow or production of a microbial WCB, see Figure 5.
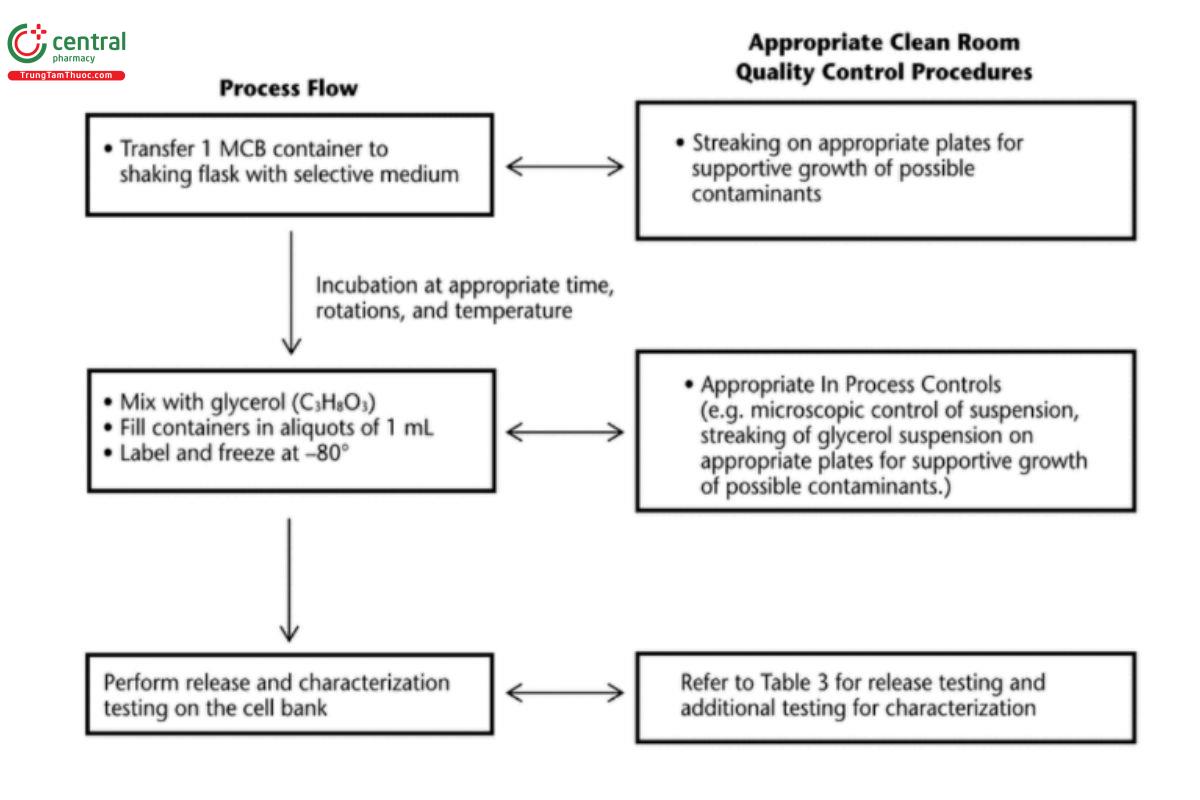
As illustrated in Figure 5, in-process control testing is performed by checking the culture broth under a microscope before the filling of containers. After filling, the suspension of culture broth and glycerol (or DMSO) is checked for contamination by monitoring growth on appropriate plates. The containers are labeled individually with batch number, cell bank identification, and container number and then filled. The WCB containers are kept frozen under well-defined and controlled conditions.
3.2.4 Additional considerations
During development, the production strains/cell lines used for manufacturing clinical materials may continue to evolve to meet the commercial manufacturing needs. For example, new expression plasmids or new parental cells (or both) may be used to generate higher yielding production strains/cell lines for commercial applications. Additional testing considerations may be needed when retroviruses (e.g., lentiviruses) are used to insert the gene of interest. The new cell lines may be from a new species (e.g., a change from NS0 to CHO) or from the same species with additional genetic modifications to improve the cell culture performance and/or the quality/productivity of the therapeutic proteins. In such cases, it may be helpful to provide a summary of the linkage among the expression plasmids/parental cells used throughout product development. In addition, comparability and other product impacts should be evaluated. To avoid any unexpected loss of the cell bank, the containers are typically kept in at least two different geographical locations. During transportation, the containers are shipped and are continuously monitored for temperature to confirm that they remain acceptably frozen.
4 MAMMALIAN CELL LINE CLONALITY
4.1 Overview
This section discusses clonality of production cell lines derived from mammalian cells. Obtaining a stable cell line that maintains high levels of recombinant therapeutic protein expression and consistent product quality is critical for commercial development. Typically, production cell lines are derived from immortalized cells, such as CHO cells, which exhibit a relatively high degree of genetic plasticity (6). Therefore, the addition of a cloning step is important to the overall cell line generation process. Incorporation of a cloning step serves two core purposes:
1. Minimizing cell population heterogeneity
2. Isolating a highly productive, stable cell population
It is recognized that incorporation of a cloning step does not prevent genetic and phenotypic heterogeneity within the population of cells that arises post-cloning due to genetic features of immortalized mammalian cell lines. However, non-clonally derived cell lines have been used for toxicology studies. It is important to demonstrate that critical quality attributes (CQAs) of the preclinical and clinical proteins are similar enough to assure safety based upon the toxicology studies.
This section describes relevant regulatory guidances regarding clonality as well as practices to achieve clonally derived production cell lines. These practices include limiting dilution, fluorescence-activated cell sorting (FACS), semisolid plating, and single-cell printing (see Table 2). In addition, this section describes principles and types of data used to support clonal derivation of a cell line.
The concept of clonality is recognized in multiple regulatory guidance documents. For instance, ICH Q5D (7) states: “For recombinant protein products, the cell substrate is the transfected cell containing the desired sequences, which has been cloned from a single cell progenitor.” ICH Q5B (6) defines the MCB as: “An aliquot of a single pool of cells which generally has been prepared from the selected cell clone.” The guidance goes on to indicate that “In addition, methods used to amplify the expression construct and criteria used to select the cell clone for production should be described in detail.” Likewise, the FDA's Points to Consider in the Manufacture and Testing of Monoclonal Antibody Products for Human Use (4) indicates that a “description of the cell cloning procedures” should be captured in documentation.
The aforementioned guidance documents indicate a general expectation that a clonally derived cell line be used to produce recombinant therapeutic products. Using a cell line derived from a single progenitor minimizes the variability of the starting cryopreserved cell population, which would be anticipated to have the effect of minimizing product heterogeneity. However, sustained culturing of immortalized cells can result in genetic alterations, which may be further exacerbated by amplification procedures. Therefore, it is not feasible to term a population as truly genetically homogeneous following sustained culture. Despite this, the reason for generating a clonally derived cell line relates to the ability of a controlled process to produce a consistent product with minimal heterogeneity. Thus, for these reasons, any adaptations (e.g., switching to serum-free conditions) should be considered prior to cloning. In contrast, use of an entirely non-clonal cell population as a starting point may give rise to outgrowth of a subpopulation of cells that generate products with different CQAs. For instance, this could affect glycosylation, which could then impact the mechanism of action if the product is an antibody that functions by antibody-dependent cell-mediated cytotoxicity (ADCC) or complement-dependent cytotoxicity (CDC). Likewise, a different population with a different integration site might have altered expression levels, growth metrics, and stability, which could have the potential to lead to drug shortages if a cell bank is no longer performing as expected. Such adverse end points could be exacerbated in conditions where cell culture parameters or raw materials have been altered in a way that places selective pressure on the system.
4.2 Methodology
Several methodologies used to generate clonally derived cell lines are described below.
4.2.1 Limiting dilution cloning
Limiting dilution cloning (LDC) is a procedure whereby cells are plated at an appropriately justified low cell density. Some wells will be devoid of cells. This is achieved by preparing a set of increasingly greater dilutions of the non-clonal starting population and visually verifying the number of cells initially deposited per well. Clones that grow out are screened for desirable growth metrics as well as product titer. Clones that appear to be good performers can be expanded through sequentially larger wells until sufficient cells are generated to establish a cell bank. Two rounds of LDC are recommended if manufacturers want to establish a clonal cell line, particularly in the absence of additional supporting technology, to ensure monoclonality (e.g., imaging). The appropriate number of rounds (e.g., 2 rounds) at an appropriate seeding density of LDC provide an approximately 99% probability that the cell line will be monoclonal. However, it is a time consuming process and can take up to 12 months to complete.
4.2.2 Single-round ldc with imaging
The supplemental evidence for the single-cell derivation of the cell line can be based on the use of a qualified imaging system, acquiring images of the single cell at day 0 and on subsequent days to follow the outgrowth of the clones. After LDC, the stained cells are deposited into imaging-quality well plates where day 0 images are acquired in both fluorescence and brightfield imaging. Fluorescence imaging may also be used if necessary. Image verification is reviewed at day 0. Fluorescence and brightfield images are evaluated for quality—based on criteria including image focus and clarity, cell shape, lack of debris or artifacts in the well—as well as for confirmation that the entire well was captured in the image.
4.2.3 Fluorescence-activated cell sorting
This technology can be used alone or in combination with a single round of LDC. The methods used for selection may rely on the detection of fluorescent intracellular reporter(s), cell-surface protein-expression target, or an unrelated marker. The method selection can also be based simply on the sorting of single cells from a preselected population. As with other cloning methods that rely on dilution to separate cells, it is important to verify that only 1 cell was sorted and deposited per well. This could be done by analysts or by an automated imaging machine starting at day 0. Some considerations include verifying that the parameters used for ow are able to distinguish single cells from doublets and that precautions are taken to make sure cell samples are not contaminated with samples previously sorted on the machine (i.e., sanitization). Any biologically derived agent should be tested for adventitious agents. For example, if antibodies are used in the selection process, they should be tested for adventitious agents.
4.2.4 Automated colony picking from semisolid medium
Automated colony-picking machines are increasingly being used for cell line development due to their high throughput capacities. Since the occurrence of high-producing transfectants with desirable growth metrics is relatively rare, screening a larger number of colonies increases the likelihood of identifying good candidates. The process in some cases can be completed in as little as 7 days, with the identification of stable clones by 4 weeks. These systems rely on the viscosity of a semisolid matrix in which the preselected cell population of transfectants is dispersed to separate the individual cells and, subsequently, the colonies and their products from one another. In addition, this semisolid matrix serves to retain product that is produced by the cell colony locally. Production of accumulated recombinant protein is detected by incorporation into a semisolid medium of a fluorescein isothiocyanate (FITC)-conjugated IgG antibody raised against the species of interest. As more recombinant protein is secreted into the medium, a halo of fluorescence builds up around the growing clone of interest. The system is able to image the cells under both white and fluorescent light; thus, both the cell colony that is growing and the amount of fluorescence that is being produced can be monitored. Up to a threshold level, mean exterior fluorescence has been shown to correlate with antibody productivity; however, this relationship was not maintained at levels above the threshold. On the basis of preset thresholds, the machine will automatically pick clones of interest and place them into microtiter plates for expansion. Optimization of the parameters used may be necessary. As with FACS, it is important to verify that the apparatus has been sanitized since its last use to avoid cross-contamination. Also, potential adventitious agents arising from the antibody used in the selection process are a concern; therefore, appropriate testing should be performed. As with other methods, it is important to keep in mind cellular behavior such as clumping. In addition, the plating density can impact the ability of the machine to select only 1 colony. Thus, such machines are often used in combination with a single round of LDC to support assurance of clonality.
4.2.5 Other automated clone selection technology
There are many methods that can be used for clone selection, and other methods may be developed. One example of clone selection methods is to combine high-speed image scanning and quantitation with high-speed laser manipulation, which allows for in situ measurement of recombinant protein secretion of single cells, followed by laser-mediated destruction of the low-producing cells in a population. This can enable higher throughput by using 384-well plates instead of 96-well plates. Image documentation of a clone begins on day 0, immediately after plating, with the aid of a fluorescent viable cell dye and continues throughout the entire expansion process. Multicolor live cell imaging is also possible through the simultaneous use of fluorescence detection reagents that associate specifically with either the cell clones or the expressed recombinant protein. The sample wells are coated with capture matrix to mediate in situ capture of expressed recombinant protein as most of the recombinant proteins are secreted out of the cell. By measuring the fluorescent intensity of the respective detection reagents, each cell in the same well is ranked based on the amount of secreted recombinant protein. Other criteria such as cell growth rate, area, and proximity to other cells also affect the ranking. The highest-ranking cell in a well is identified as the most suitable high-producer cell clone. Subsequently, a laser beam is directed to the lower-ranking cell clones in the same well to induce photomechanical lysis. This allows the high-producing cell remaining in the well to grow and then be transferred to a larger well for expansion, clone characterization, and cell banking. The platform could be used alone or in combination with a single round of LDC. See Table 2 for a comparison of the above methods.
Table 2
Method Comparison | Advantages | Disadvantages |
Limiting dilution cloning | Statistically powered with two rounds Least expensive | One round insufficient to provide monoclonality assurance; requires additional supplemental data Labor and time intensive Requires accurate initial cell counting |
Single-round LDC with imaging | Day 0 image conformation of single cell in well provides high level of monoclonality assurance | Requires expensive equipment Requires equipment qualification Requires highly trained operators |
Fluorescence-activated cell sorting | Single cell deposition High throughput Can be combined with imaging to provide monoclonality assurance Can be used to isolate high producing cells | Requires expensive equipment Requires equipment qualification/validation Requires highly trained operators |
Automated clone picking from semisolid medium | Identification of isolated individual cells Can be coupled with LDC Can be used to identify high producing cells | Use of semisolid media may result in stacked cells in z-axis May require an additional round of LDC Does not allow imaging of single cells Requires expensive equipment Requires equipment qualification Requires highly trained operators |
Other automated clone selection technology | Day 0 image confirmation of single cell in well provides high level of monoclonality assurance | Requires expensive equipment Requires equipment qualification Requires highly trained operators |
| High-throughput image scanning of high-producing cells with elimination of low-producing cells |
|
4.3 Assurance of Clonality
It is important to appropriately document and provide data to support a claim of clonality. The analysis may include: Mathematical probability models, as well as detailed descriptions of the methodologies used and demonstrations of how they achieve single-cell isolation (e.g., experiments with mixed cell lines that have been cultured together)
Images, especially day 0 and over time; genetic data; and consistent process data and product quality
When probability calculations are included, they should be done so prospectively. Back calculation from the number of wells in which the colonies grew does not take into account the number of wells in which cells failed to grow, resulting in an overestimate of monoclonality.
4.3.1 Imagers
Imagers that can document the contents of an entire microplate well2 from day 0 through colony growth can be powerful tools. An image of a single expanded colony would not be enough evidence of monoclonality, but a time lapse showing the development of the colony from a single cell would provide supportive evidence. In this case, it would be important that the entire well was captured to prove that only 1 cell was in the well from the beginning. Furthermore, day 0 fluorescence imaging as applicable allows for the detection of living cells and for differentiation from cell-like artifacts that can be observed in the brightfield image.
4.3.2 Genetic support
For clones with random (non-site-specic) integration, genetic information supporting the location of the integration site provides evidence that the cell line was clonal. Information about the sites of integration can help to identify the number and location of genomic integration sites. Integration site identification does not prove that the cell line was clonally derived.
Change to read:
5 CELL BANK CHARACTERIZATION
Depending on the type of cells used, the biological production processes for cell banking can be variable, which has important consequences for the safety and efficacy of the resulting product. In order to introduce these cell-derived products into routine clinical use, the manufacturer must ensure that robust cell banking procedures are developed on the basis of process understanding and characterization, which should result in consistent production and quality of cell banks. Process understanding and consistency are critical because slight changes in cell growth conditions can occasionally lead to changes (e.g., in glycosylation) that can have major adverse effects later on (e.g., changes in immunogenicity), which may be a critical attribute for the expressed protein or polysaccharide. Validated and/or appropriately qualified-for-use test methods for characterization of the cell bank (e.g., host cell quality, purity, freedom from adventitious agents, in-process testing) are quality recommendations for any MCB or WCB.
5.1 Cell Bank Qualification and Characterization
Cell banks used in the manufacturing process must undergo extensive safety testing to demonstrate identity, stability, and purity. See Table 3 for a description of microbial cell bank characterization and testing possibilities. See Table 4 for a description of cell bank characterization of a mammalian cell line (CHO) and testing possibilities. Use of each test should be agreed upon with the applicable health authority.
Cell bank qualification is defined as any testing deemed appropriate to demonstrate that the bank is t for its intended use and the environmental conditions were appropriate. Testing to qualify the mammalian cell bank includes testing to demonstrate freedom from adventitious agents such as bacteria, fungi, mycoplasmas, and viruses and to confirm species identity. Testing is also required to determine if cells produce retroviruses or retrovirus particles.
Table 3. Microbial Cell Bank Characterizationa
Assay Type | MCB | WCB | EOPb |
Specification Analysis | |||
Microbial purity | + | + | — |
Phage contamination | + | + | — |
DNA sequencing | + | + | — |
Strain identity | + | − | — |
Characterization Tests | |||
Viability | + | + | + |
Microbial purityc | — | — | + |
DNA sequence | — | — | + |
Plasmid copy number | + | + | + |
Plasmid frequency or loss of plasmidd | + | + | + |
Restriction enzyme mapping | + | + | + |
Phage contamination | — | — | + |
Nucleic acid fingerprinting | + | + | + |
Nucleic acid profiling | + | + | + |
a ICH Q5A was under revision during development of this chapter. The table is aligned with the current R1 version of ICH Q5A. For more information see current version of ICH Q5A.
b EOP, end of production.
c Purity needs to be performed on EOP if not done in MCB and WCB.
d This test may be more appropriate to perform on the end-of-growth sample.
Table 4. Biological Safety Characterization of Mammalian Cell Banks (CHO Cells)a
Assay Type | MCB | WCB | EOPb |
Microbial Contaminants | |||
Sterility: USP/European Pharmacopoeia/ICH requirements | + FI | + | + |
Mycoplasma: European Pharmacopoeia 2.6.7 (15)/Mycoplasma Tests 〈63〉 | + F | + | + |
Adventitious Virus Detectionc | |||
In vitro assayd,e | + | −f (ERR 1-May-2023) | + (ERR 1-May-2023) |
Antibody production testsg,h | + | — | − |
Other species specific testsi | If applicable | If applicable | If applicable |
In vivo assayj | + | —e | + |
Bovine and porcine virus in vitro assay (16–17)k | − | − | − |
Detection of Retroviruses and Other Endogenous Elements | |||
Electron microscopy | + | — | + |
Infectivity | + | − | + |
Microbial Contaminants | |||
Reverse transcriptase assayl | + | − | + |
Cell Line Identity Tests | |||
Nucleic acid fingerprinting or other | + | + | + |
a ICH Q5A was under revision during development of this chapter. The table is aligned with the current R1 version of ICH Q5A. For more information see current version of ICH Q5A.
b EOP, end of production.
c The strategy for testing extraneous agents should be developed based on a risk assessment following the principles of viral contamination risk (see Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin 〈1050〉).
d The nature of the assay and choice of cells used are governed by the species of origin of the cell bank and the type of virus that may be present based on the handling of the cells. The assay should include a human and/or a non-human primate cell line susceptible to human viruses. Both cytopathic and hemadsorbing viruses should be sought.
e The strategy should include suitable tests that are able to detect different families of extraneous agents that may infect the source materials, including cell substrates and raw materials of animal or plant origin. The strategy also considers the capacity of the manufacturing process to remove or inactivate viruses (see Design, Evaluation, and Characterization of Viral Clearance Procedures 〈1050.1〉).
f Performed on cells generated from the first WCB at the limit of in vitro cell age; for subsequent WCBs, a single in vitro and in vivo test can be done either directly on the WCB or on the cells at the limit of in vitro cell age (LIVCA). On the basis of a risk assessment, these tests may not be required.
g Species-specic viruses present in rodent cell lines may be detected by mouse, rat, or hamster antibody production tests (MAP or HAP). h Alternative validated method (e.g., NAT) agreed upon with the regulatory authority may be used.
i Selection of viruses to be screened should take into account cell lines that are known to have been infected by such agents and the source from which the cell line was derived [e.g., mouse minute virus (MMV), Vesivirus].
j Includes adult and suckling mice and embryonated eggs. Additional animal species may be used depending on the nature and source of the cell lines being tested.
k When bovine and porcine raw materials are used, these tests should be performed. Bovine and porcine virus in vitro assay can be omitted for MCB, WCB, and EOP if the parental cell line used to generate MCB had been tested negative for bovine and porcine viruses and no animal-derived raw material were used in transfection and subsequent steps leading to the generation of MCB and WCB as well as in the production of EOP.
l Not required if infectivity assay is positive.
5.2 Mammalian Cell Bank Testing
A new cell bank (MCB or WCB) needs to be qualified to demonstrate its suitability for biologic production. The newly prepared cell bank should be characterized extensively using appropriate release tests on an aliquot of the cell bank or on cultures derived from it. The testing on limit of in vitro cell age (LIVCA) cells should be conducted for the initial MCB or WCB. When new WCB are expanded to or beyond the LIVCA, testing should be conducted to assess against the existing MCB. However, extensive characterization and testing is not necessary for a WCB generated from a fully tested MCB. The characterization of the cell bank is dependent on its use, and the specific use should guide decisions about the required recommended testing and whether additional testing might be necessary.
Though rarely banked, primary cells are more likely to contain adventitious agents than banked, well-characterized cell lines. The genetic stability of diploid cell strains should be monitored throughout production.
The tumorigenicity study procedure and suitable animal models are described in the WHO's Technical Report Series No. 978, Annex 3 (13) and the FDA's Guidance for Industry: Characterization and Qualification of Cell Substrates and other Biological Materials Used in the Production of Viral Vaccines for Infections Disease Indications (14). If the cell substrate under evaluation is found to be tumorigenic, it must be evaluated for oncogenicity in suitable animal models (newborn nude mice, newborn hamsters, and newborn rats) using an appropriate positive control.
For an engineered cell line, the inserted GOI should remain intact and the copy number should be determined to support consistent production.
The cell bank should comply with the appropriate test for sterility given in Sterility Tests 〈71〉 and carried out using an appropriate volume from cell cultures. The test should be carried out on 1% of containers with a minimum of 2 containers. The cell bank should also comply with the test for mycoplasma in Mycoplasma Tests 〈63〉 performed on 1 or more containers.
Electron microscopy (EM) of the MCB should be done to detect the presence of any extraneous agents. The MCB should also be assessed by the vitro virus assay with the two endpoints of cytopathic effect and hemadsorption. The end of production (EOP) cells are also characterized to demonstrate absence of extraneous agents that might be present in the MCB.
Additionally, the cell bank should be tested for possible virus contaminants, depending on the origin of the cells, using appropriate specific tests (e.g., mouse, rat, or hamster antibody production tests or a nucleic acid amplification test may be used for specific contaminants). See Viral Safety Evaluation of Biotechnology Products Derived from Cell Lines of Human or Animal Origin 〈1050〉 and Design, Evaluation, and Characterization of Viral Clearance Procedures 〈1050.1〉 for more details.
The MCB should be examined for retroviruses using:
- Infectivity assay
- Transmission electron microscopy (TEM)
- Reverse transcriptase (RT) or other appropriate assays should be performed to detect retroviruses that may be noninfectious if infectivity is not detected and no retrovirus or retrovirus-like particles have been observed by EM
Detection for retroviruses in the MCB is governed by the host species and can include detection methods such as reverse transcriptase activity, infectivity in sensitive cell cultures or electron microscopy. It is recommended to continue to follow advances in new technology to supplement cell bank characterization where appropriate.
5.3 Genetic Characterization
Genetic characterization to support the use of the production cell line for the MCB and WCB and a proposed LIVCA at the EOP stage are essential for any development program. The purpose of genetic characterization is to demonstrate the integrity of the expression construct carrying the GOI throughout the intended commercial manufacturing process. The manufacturing cell culture duration starts with the MCB and/or WCB and continues to the proposed LIVCA for the drug substance production. It is recommended that LIVCA be determined based on the cell age of the EOP cells for a defined duration beyond the routine commercial drug substance manufacturing process. At a minimum, LIVCA should have population doubling levels (PDLs) beyond the typical manufacturing window as appropriate. The additional generations are added to allow for future changes to the manufacturing process and to ensure that the LIVCA is not exceeded in future manufacturing operations. The EOP cells should be harvested from a representative commercial process, either at a pilot or commercial scale.
Genetic characterization is performed on samples taken at the EOP and compared to the MCB. If necessary, WCB could be tested with bridging to the MCB. Based on ICH Q5B, the genetic characterization consists of four aspects:
1. Verification of the GOI coding sequence
2. Assessment of the integration sites
3. Assessment of the integrity of the gross structure of the expression construct, which provides insight into potential insertions and/or deletions of the expression construct
4. Determination of a consistent GOI copy number over the length of time required to perform a biopharmaceutical manufacturing run
For microbial strain-derived cell banks, the GOI is usually carried by extrachromosomal expression systems such as a plasmid expression vector. The nucleotide sequence encoding the product can be directly sequenced from the plasmid isolated from the cell banks without further cloning. For cell banks carrying chromosomal copies of the GOI, the sequence encoding the product can be verified. This is commonly performed by sequencing of the coding regions amplified by the PCR from cDNA. The nucleic acid sequence should be identical, within the limits of detection method, to the sequence determined for the expression construct and should correspond to the expected protein sequence.
The most common methodology for the assessment of gross structure of the expression construct is restriction endonuclease (RE) mapping analysis. New technologies [e.g., next generation DNA sequencing (NGS)] are currently being explored for this purpose. For extrachromosomal expression systems, the RE analysis can be viewed through gel electrophoresis; for chromosomally integrated expression systems, the RE analysis is usually accomplished by Southern blotting or full sequencing by NGS. The RE digested patterns as well as the DNA sequence should be consistent for DNA derived from either the cell banks or the cells at the LIVCA.
Although not required by ICH Q5B, transcript analysis of GOI can be performed if needed to further evaluate the integrity of the GOI expression. A number of techniques can be used for transcript analysis, including Northern blotting, reverse transcription–polymerase chain reaction (RT–PCR) and transcriptome analysis by NGS.
The most common methodology for the evaluation of GOI copy number is quantitative PCR (qPCR). GOI-specic qPCR assays and a normalizer qPCR assay targeting a host genome region are usually applied. In some cases, there is no strong correlation between the stability and integrity of the therapeutic product. The GOI copy number analysis could be considered for the cell bank characterization purposes only. The genetic determination is part of the quality control. New DNA sequencing technology such as NGS may further enhance the capability for genetic characterization.
The impact of a change in cell culture process after the commercial process has been defined should be evaluated to determine if retesting of the cells at LIVCA is necessary. Changes in cell culture scale and/or manufacturing site may not require retesting of cells at LIVCA if the defined cell age covers any increase in population doubling resulting from the change and if cell culture performance meets the predetermined acceptance criteria. However, changes in media components (especially if animal-derived components are used) and growth conditions often result in changes in cell culture proles, so one should consider retesting the integrity of the expression construct at LIVCA to ensure suitability of the cell line for commercial production.
Although a two-tier cell-banking system (MCB and WCB) is commonly established for the commercial manufacturing of a drug substance, the characterization of the expression construct for each WCB is considered unnecessary if the following criteria are met: The MCB has been fully characterized and the expression construct is confirmed to be stable
The cell age of the current and future WCB, as well as the EOP cells derived from the future WCB, will be controlled within the previously determined LIVCA
If the characterization cannot be carried out on the MCB, it should be carried out on each WCB.
5.4 General Considerations for Qualification of Replacement WCBs
Replacement WCBs prepared using procedures equivalent (as described in the license) to those used to generate the previously approved WCB must meet all specified requirements [e.g., certificate of analysis (CoA) testing] but require no further evaluation under a validation protocol. When the new WCB is a “like-for-like” replacement, the WCB can be implemented after meeting the following criteria:
1. The new WCB must meet all cell bank release testing criteria, including tests for freedom from adventitious agents. 2. Prior to at-scale manufacturing, the WCB may be evaluated using either scale-down or industrial-scale cell culture tests from thaw through production culture to confirm cell culture performance. A minimum number of independent thaws should be included in the evaluation.
3. There is an option to use scale-down cell culture evaluation criteria including cell culture process key performance indicators (KPIs) and relevant product attributes and/or CQAs, but at least one full scale with expanded analytical characterization should be evaluated. For example, the KPI assessment may include specific growth rate and nal viabilities for seed and inoculum train passages, nal production culture viability, and nal product titer. Product quality assessments may include purity, size-exclusion chromatography (SEC), and ion exchange chromatography (IEC) assays. The evaluation criteria can be based on 95% confidence/99% probability tolerance intervals (95/99 TIs) generated using representative data available at the time the evaluation is performed (where appropriate). Results outside the evaluation criteria should be justified or further assessed using additional cell culture studies and/or product attribute testing.
4. The new WCB should produce manufacturing-scale material that meets all specified drug substance release testing requirements. A drug substance manufactured from a replacement bank may not need to be on stability protocol, but does require a CoA.
The release of batches derived from the new WCB would be predicated on successfully completing all the above-mentioned criteria. For WCBs prepared using a manufacturing process that differs significantly from the process used to produce previously approved WCBs, the same evaluation procedure described above (criteria 1–3) applies. However, additional evaluations may be performed and could include further KPI assessments and the testing of additional CQAs and non-CQAs. For cell culture-based infectivity assays (in vitro adventitious agent assays), refer to 〈1050〉 and 〈1050.1〉.
5.5 Other General Considerations for Cell Banks
5.5.1 Cell bank cryopreservation
It is critical that cells are characterized and a feasibility assessment is performed to verify that cell performance is achieved when expanded at a defined concentration and duration using a specific technology platform. Containers should be appropriately labeled, and cryolabels must be able to withstand cryopreservation. Cryology considerations for cells are addressed in 〈1044〉. Additional considerations for cell banking are as follows:
- Cells are frozen at early passage (MCB), intermediate passage (WCB), or late passage (EOP).
- Frozen cell volumes can vary from 0.5 to >100 mL depending on the number of cells required upon thawing.
- Cell banks can be stored in cryovials or cryobags.
- Cell density can range from 106 to 108 cells/mL.
- Cell bank storage containers should be filled with an excess to accommodate loss of cells upon thaw.
- For MCB or WCB, cells should be banked at a density that allows for direct inoculation into vessels for expansion upon thawing.
- Mammalian cells should be preserved in GMP-grade cryopreservation solution with appropriate cryoprotectant (e.g., DMSO or glycerol) to support cell recovery consistent with established in-process viability limits.
- Post-bank thaw testing with assessment of cell viability should be performed and documented. A viability limit should be set and justified to demonstrate that the cells survived the cryopreservation process. The viability limit could be set based on historical data.
- Each cell bank vial or bag should be labeled in advance with the appropriate information (e.g., cell bank lot number, tube number, and production date) on liquid nitrogen-resistant cryolabels; use appropriate controls and limits.
If different cell banks are stored in the same facility, appropriate measures and controls (for example segregation or cryolabels) should be in place to prevent errors due to switching and cross contamination. Different freezers or storage areas within freezers should be used to separate the cell banks prior to release testing from the cell banks to be used for production.
5.5.2 Cell bank storage stability and monitoring
A cell bank is considered stable if, upon thaw and subsequent passage, it can consistently meet growth and viability criteria based on historical performance. Bank-specic limits for viability and growth rates can be set after thaw studies have been completed. Cell bank stability can be equally ensured through trending of thaw performance data across a more substantive data set such as WCB vial thaw and/or batch.
Stability limits for mammalian cell banks should be site independent, product specific, cell bank specific, and determined on the basis of thaw performance. Stability limits can also be vial-size specific due to the operational differences between seed strain sources from different-sized vials. Data used to establish final viability and specific growth rate limits should be documented.
Data for MCB stability monitoring should include data from when 1 or more vials of the cryopreserved MCB are thawed for:
- Initial characterization
- Production of clinical or commercial material
- Preparation of a new WCB
Data for WCB stability monitoring can include data from when 1 or more vials of the cryopreserved WCB are thawed for a clinical or commercial production campaign.
Data for WCB stability monitoring may also include data from appropriately documented scale-down or pilot-scale processes, such as those performed during process characterization and validation studies. WCB stability should be periodically monitored unless the bank is no longer in use for commercial production or the product has been removed from the market.
5.5.3 Cell bank transportation
A temperature-cycling study can be done to determine the effects of repetitive temperature fluctuations on frozen-cell stability and shelf life. Chapter 〈1044〉 outlines good storage and distribution practices for cells, cell therapy products, and cell banks to maintain the integrity of frozen cells during movement or transportation.
It is crucial to ensure that the facility maintaining the cell bank provides storage management systems, which should include an inventory of all stored material and routine documentation of entries and withdrawals. Transportation controls should be in place as per 〈1044〉. Distribution of cell bank vials should be handled carefully to ensure that integrity and cell viability are maintained throughout the transfer. Cells can be shipped on dry ice in containers, with appropriate procedures for replenishment of dry ice as needed. Dry shippers (well insulated Dewars that contain a cryoabsorbent material) charged by addition of liquid nitrogen provide an optimal method for the shipping and handling of cell banks.
5.5.4 Raw materials
Control of raw materials is necessary to reduce the risk of contamination due to adventitious agents and possible cross contamination from incoming raw materials. Contamination risk for all raw materials should be assessed; the assessment can then be used to support product exposure history.
Qualification of raw materials: Raw materials should be qualified and characterized as to their identity, purity, functionality, and safety. Appropriate testing for raw materials should be performed; examples include testing for endotoxins, mycoplasma, adventitious agents, and/or sterility (see Ancillary Materials for Cell, Gene, and Tissue-Engineered Products 〈1043〉). Any raw material used in the manufacture of a cell substrate (including media components) that is obtained from suppliers should be controlled appropriately by the cell substrate manufacturer (see 〈1043〉).
The qualification program should evaluate the source, identity, purity, and safety of the raw material relative to the established specifications for raw materials. Identification and selection of raw materials should be performed on the basis of suitability for use in the intended process and evaluation of toxicity, availability, consistency, traceability, and potential to be sources of contamination. If the raw materials are human derived, the certificate of origin (CoO) and donor infectious disease status should be evaluated. If the raw materials are animal-derived, the herd qualification and country of origin certificates, should be evaluated.
For some human- and animal-derived raw materials used in cell culture medium (e.g., Insulin, transferrin), a validated production process used for elimination of viruses may substitute for virus detection tests, when justified. The evaluation of the suitability for use should encompass any effects on safety, potency, and purity. It should also include assessment of the potential risk of toxicity and adventitious agents and their removal if such raw materials are used in large amounts in manufacturing.
Drug manufacturers should develop a raw materials management system as part of their quality systems that would include a raw materials qualification program. This program should include:
Incoming receipt, segregation, inspection, and quality release of the raw materials prior to use
2. Vendor audits and certification
3. CoA verification testing or identity testing, as applicable
4. CoO
5. Formal procedures and policies for out of specification results
6. Stability testing
7. Archival sample storage
Any raw material supporting the cell bank growth will be an ancillary product; therefore, supplier material should be controlled by the manufacturer. Usually a CoA and/or a risk statement addressing the likelihood of the presence of animal-derived materials [e.g., bovine material, which carries the risk of bovine spongiform encephalopathy (BSE)] should be provided for all media components. In addition, contracts ensuring a steady supply of a material of the proper purity level should be secured.
6 CONCLUSION
The production of cell banks needs to satisfy quality standards for identity, purity, clonality, stability, and the suitability of cells for use in manufacturing. In addition to the appropriate testing for identity, purity, and adventitious agents, the homogeneity, reproducibility, history, cell bank generation and qualification, genetic stability, storage, raw materials, and reagent qualification, as well as good manufacturing practices, must be demonstrated.
7 GLOSSARY
Adventitious agent:
A microorganism [including bacteria, fungi, Mycoplasma/Spiroplasma, Mycobacteria, Rickettsia, viruses, protozoa, parasites, or transmissible spongiform encephalopathy (TSE) agents] that is inadvertently introduced into the production of a biological product (15).
Cell bank:
A collection of appropriate containers, whose contents are of uniform composition, stored under defined conditions. Each container represents an aliquot of a single pool of cells (7).
Cell bank qualification: Any testing deemed appropriate to demonstrate fitness of use of the bank for its intended purpose and the environmental conditions where appropriate.
Cell line:
Type of cell population that originates by serial subculture of a primary cell population that can be banked (7).
Cell substrate:
Microbial cells or cell lines derived from human or animal sources that possess the full potential for generation of the desired biotechnological/biological products for human in vivo or ex vivo use (7).
Clonality:
A group of cells derived from a single progenitor sharing common ancestry (7).
Diploid cell line:
Type of cell population having a nite in vitro lifespan in which the chromosomes are paired (euploid) and are structurally identical to those of the species from which they were derived (ICH Q5D).
End of production cells (EOP cells):
Cells cultured (under conditions comparable to those used in production) from the MCB or WCB to a passage level or population doubling level comparable to or beyond the highest level reached in production (15).
Expression construct:
The expression vector that contains the coding sequence of the recombinant protein and the elements necessary for its expression (6).
Host cells:
See Parental cells below (7).
Genetic integrity: Test to establish that the gene of interest is intact, sequence of coding sequence is correct, and there are no insertions or deletions.
Integration site:
The site where 1 or more copies of the expression construct are integrated into the host cell genome (6).
Limit of in vitro cell age (LIVCA):
Time between thaw of the MCB vial(s) and harvest of the production vessel as measured by elapsed chronological time, by population doubling level of the cells, or by passage level of the cells when subcultivated by a defined procedure for dilution of the culture (6–7).
Master cell bank (MCB):
An aliquot of a single pool of cells that generally has been prepared from the selected cell clone under defined conditions, dispensed into multiple containers, and stored under defined conditions (6–7, 16).
Parental cells:
Cells to be manipulated to give rise to a cell substrate or an intermediate cell line. For microbial expression systems, it is typical to also describe the parental cells as the host cell (7).
Passage level:
The number of times that a cell population has been removed from the culture vessel and has undergone a subculture (passage) process (15).
Pilot plant scale:
The production of a recombinant protein by using a procedure fully representative of and simulating that to be applied on a full commercial manufacturing scale. The methods of cell expansion, harvest, and product purification should be identical except for the scale of production (6).
Research cell bank (RCB)/Premaster research cell bank (pmRCB): Cell bank(s) developed as a precursor to the master cell bank.
Recombinant biologic:
Biological substance produced by genetic engineering techniques.
Tumorigenicity:
Process by which immortalized cells form tumors when inoculated into animals (15).
Viral clearance:
Elimination of target virus by removal of viral particles or inactivation of viral infectivity (5; 〈1050〉, 〈1050.1〉). Virus-like particles:
Structures visible by electron microscopy that morphologically appear to be related to known viruses (5).
Working cell bank (WCB):
A collection of cells prepared from aliquots of a homogeneous suspension of cells obtained by culturing the MCB under defined culture conditions (6–7).
8 REFERENCES
1. Food and Drug Administration. US Code of Federal Regulations. Title 21 CFR 610.18 Cultures.
2. Food and Drug Administration. Supplement to the points to consider in the production and testing of new drugs and biologics produced by recombinant DNA technology: nucleic acid characterization and genetic stability, Rockville, MD: Food and Drug Administration; April 1992.
3. Food and Drug Administration. Points to consider in the characterization of cell lines used to produce biologics. Rockville, MD: Food and Drug Administration; July 1993.
4. Food and Drug Administration. Points to consider in the manufacture and testing of monoclonal antibody products for human use. Rockville, MD: Food and Drug Administration; February 1997.
5. International Council for Harmonisation. Q5A(R1) Viral safety evaluation of biotechnology products derived from cell lines of human or animal origin. 1998. https://www.fda.gov/downloads/Drugs/GuidanceComplianceRegulatoryInformation/Guidances/ucm073454.pdf.
6. International Council for Harmonisation. Q5B Quality of biotechnological products: analysis of the expression construct in cells used for the production of rDNA derived protein products. 1995. https://www.ich.org/page/quality-guidelines.
7. International Council for Harmonisation. Q5D Derivation and characterization of cell substrates used for production of biotechnological/biological products. 1997. https://www.ich.org/page/quality-guidelines.
8. International Council for Harmonisation. Q7 Good manufacturing practice guide for active pharmaceutical ingredients. 2000. http://academy.gmp-compliance.org/guidemgr/les/3-1-18.pdf.
9. International Council for Harmonisation. Q9 Quality risk management. 2005.
10. Xu X, Nagarajan H, Lewis NE, et al. The Genomic Sequence of the Chinese Hamster Ovary (CHO) K1 cell line. Nat Biotechnol. 29(8), 735–741; 2011.
11. Berting A, Faucet M, Kreil T. Biotechnology, Bioengineering. Vol. 106, 598–607; July 1, 2010.
12. Frye C, Deshpande R, Estes S, Francissen K, Joly J, Lubiniecki A, et al. Industry view on the relative importance of “clonality” of biopharmaceutical-producing cell lines. Biologicals. 2016;44(2):117–122.
13. European Medicines Agency. Guideline on development, production, characterisation and specification for monoclonal antibodies and related product (3AB4A); July 2016.
14. World Health Organization. WHO Technical Report Series No. 978, Annex-3. Recommendations for the evaluation of animal cell cultures as substrates for the manufacture of biological medicinal products and for the characterization of cell banks; 2013.
15. Food and Drug Administration. Guidance for Industry: Characterization and qualification of cell substrates and other biological starting materials used in the production of viral vaccines for infectious disease indications. Rockville, MD: Food and Drug Administration; February 2010.
16. European Pharmacopoeia (Ph. Eur.). 9th edition. 2.6.7 Mycoplasmas.
17. Food and Drug Administration. US Code of Federal Regulations. Title 9—Animals and animal products; 2000.
(USP 1-May-2023)
1 See ICH and appropriate FDA guidelines for more specific information.
2 For example, CloneSelect Imager.
