ASSESSMENT OF SOLID ORAL DRUG PRODUCT PERFORMANCE AND INTERCHANGEABILITY, BIOAVAILABILITY, BIOEQUIVALENCE, AND DISSOLUTION
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
- BACKGROUND
- BIOAVAILABILITY, BIOEQUIVALENCE, AND DISSOLUTION
- BIOEQUIVALENCE
- DISSOLUTION AND IN VITRO PRODUCT PERFORMANCE
- INTERCHANGEABILITY OF DRUG PRODUCTS
- SOLID FORM AND PARTICLE SIZE
- DIFFERENCES IN EXCIPIENTS
- MANUFACTURING PROCESS
- BIOWAIVER
- Biowaiver Based on Dosage Form Proportionality
- Biowaiver Based on the BCS
- APPENDIX
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
DOWNLOAD PDF HERE
1 BACKGROUND
This chapter provides recommendations for the in vivo and in vitro assessment of solid oral drug product performance. The chapter is intended as a guide for scientists and clinicians seeking to evaluate drug product performance by surrogate procedures correlative and/or antecedent to clinical trials in humans. USP–NF provides quality standards for drug substances, excipients, and finished preparations. A USP–NF monograph for an official substance or preparation includes the article's definition; packaging, storage, and other requirements; and a specification. The specification consists of a series of universal tests (description, identification, impurities, and assay) and specific tests—one or more analytical procedures for each test—and acceptance criteria. Quality standards are important attributes that must be built into the drug product. Meeting USP–NF standards is accepted globally as assurance of high quality and is part of the requirements necessary for approval of a bioequivalent , interchangeable drug product. Drug products must meet certain in vivo and/or in vitro performance standards to be considered therapeutically equivalent (TE) and interchangeable. Drug product performance may be defined as the release of the drug substance from the drug product dosage form, normally leading to systemic exposure or less often to local activity of the drug substance necessary for achieving a desired therapeutic response. Bioavailability (BA) is a measure of systemic exposure. This chapter discusses in vivo and in vitro approaches to determining drug product performance. The focus of the chapter is primarily on the performance of solid oral drug products.
This chapter references two Food and Drug Administration (FDA) guidances: Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations (draft guidance March 2014) and Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA (draft guidance December 2013) (search by document title; http://www.fda.gov/); a European Medicines Agency (EMA) guidance: Guideline on the Investigation of Bioequivalence (2010) (search by document title; http://www.ema.europa.eu/ema/), and a World Health Organization (WHO) document, Annex 7: Multisource (Generic)
Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability, WHO Expert Committee on Specifications for Pharmaceutical Products: Fortieth Report. World Health Organization: Geneva; 2006: Annex 7 (WHO Technical Report Series, No. 937). TRS 992 (2015) (search by document title; http://who.int/en/). FDA guidances are used in the United States, and WHO, FDA, EMA, and national/regional guidelines may be used by national/regional drug regulatory authorities outside the U.S. Following approval, control of the quality of a drug product can be achieved in part by using the private and/or public specification, which can include a performance test. USP provides the general chapters Disintegration 〈701〉, Dissolution 〈711〉, Drug Release 〈724〉, In Vitro and In Vivo Evaluation of Dosage Forms〈1088〉, The Dissolution Procedure: Development and Validation 〈1092〉, and Capsules—Dissolution Testing and Related Quality Attributes〈1094〉, which describe these tests and procedures.
This chapter provides general information about the conduct of bioequivalence (BE) studies as a surrogate measure of in vivo drug product performance and dissolution profile comparisons as a measure of in vitro drug product performance as stated in the guidelines cited in this chapter. The chapter also discusses conditions when an in vivo BE requirement may be waived (biowaiver) for certain drug products and shows how the Biopharmaceutics Classification System (BCS) can be used as a predictor of a drug product's performance. An Appendix to this chapter defines key scientific terminology and provides a comparison between FDA, EMA, and WHO in terms of drug product performance assessment. Other BE guidance may exist outside of that given in this chapter.
2 BIOAVAILABILITY, BIOEQUIVALENCE, AND DISSOLUTION
BA studies focus on determining the process and time frame by which a drug substance is released from the oral dosage form and moves to the site of action [see Guidance for Industry: Bioavailability and Bioequivalence Studies Submitted in NDAs or INDs—General Considerations (FDA draft guidance March 2014) and Guidance for Industry: Bioequivalence Studies with Pharmacokinetic Endpoints for Drugs Submitted Under an ANDA (FDA draft guidance December 2013) (search by document title; http://www.fda.gov/]. BA is an indirect or surrogate measure of the rate and extent to which the drug substance or active moiety is absorbed from a drug product and becomes available at its target sites of action. BA data provide an estimate of systemic drug exposure, including fraction of drug absorbed. The determination of availability of drug substance from drug products that are not intended to be absorbed into the bloodstream is outside the scope of this chapter. Drug products are considered bioequivalent if a test (T) drug product does not show a significant difference in rate and extent of absorption by comparison with a designated reference (R) drug product when administered at the same dose under similar experimental conditions in either a single dose or in multiple doses.
BA and BE generally can be obtained by serially measuring drug substance and/or metabolite concentrations in the systemic circulation over a prescribed period. BE studies can use other approaches when systemic drug concentrations cannot be measured or are not appropriate. For these cases, more indirect approaches to BE determination include acute pharmacodynamic endpoints, clinical endpoints, and in vitro studies that typically involve comparisons of the dissolution profiles of (T) and (R) drug products. In the absence of an in vitro–in vivo correlation (IVIVC) or considerations based on the BCS, in vitro performance studies are not used in place of BA studies. BA and BE information are important in regulatory submissions. BA information broadly addresses the absorption, distribution, metabolism, and excretion of the drug substance. For an innovator product, BE studies establish the performance of the product intended for marketing by comparing the BA of the product as developed for marketing approval to the clinical trial material, the drug product used in safety/efficacy trials. For the development and regulatory approval of a generic drug product, the T must be bioequivalent to the R (usually the brand or innovator drug product that is designated by the applicable regulatory authority).
The ICH guideline document titled Specifications: Test Procedures and Acceptance Criteria for New Drug Substances and New Drug
Products: Chemical Substances Q6A (2000) (search by document title; http://www.fda.gov/) provides approaches for setting acceptance criteria for drug product performance. These approaches rely on dissolution or disintegration based on clinically acceptable batches, as does FDA's approach. BE studies focus on the performance of the drug product and usually involve comparisons of two drug products: T and R.
The required studies and the determination of BE are the province of regulatory agencies. In the United States, R is termed the RLD and is so noted in FDA's Approved Drug Products with Therapeutic Equivalence Evaluations, 37th Edition [Orange Book (2017) (http://www.accessdata.fda.gov/scripts/cder/ob/)]. To assist countries and regions where the R product may not always be readily identifiable, WHO has prepared a document titled Annex 11: Guidance on the Selection of Comparator Pharmaceutical Products for Equivalence Assessment of Interchangeable Multisource (Generic) Products (2002) (search by document title; http://www.who.int/en/). In the WHO document, R is termed the “comparator pharmaceutical product” (CPP). When a country or region has a clearly defined set of CPPs, the task becomes one of requiring that a manufacturer demonstrate, to the satisfaction of its regulatory authority, that its product is pharmaceutically equivalent (PE) and bioequivalent to the corresponding CPP.
3 BIOEQUIVALENCE
An interchangeable drug product must be PE. The EMA and WHO documents allow pharmaceutical alternatives to be considered TE and interchangeable if they are bioequivalent. Further, generic products must be shown to be bioequivalent to be considered TE to the R product .
For the product to be considered PE, it must have the same drug substance, same strength, same dosage form, same route of administration, and same labeling as the R product. Several methods exist to assess and document BE. These include the following:
1. Comparative pharmacokinetic studies in humans. In these studies, the drug substance and/or its metabolite(s) are measured as a function of time in accessible biological fluid such as blood, plasma, serum, or urine to obtain pharmacokinetic measures such as area under the plasma drug concentration versus time curve (AUC) and maximum concentration (C ) that are refiective of systemic exposure. Other pharmacokinetic parameters such as time to peak concentration (T ) may be informative for safety and efficacy assessment.
BE studies are designed to compare the in vivo performance of 1) a generic product with an R product or 2) a new formulation of an R product with the originally approved product. Generally the design is a two-period, two-sequence, single-dose, crossover randomized study. The number of subjects should be statistically justified and NLT 12. The necessary statistical power of the study will determine the number of subjects >12. During the study, blood samples are collected at sufficient intervals for assessing C , AUC, and other parameters. Blood samples are analyzed using appropriately validated bioanalytical methodology with standard pharmacokinetic measures and statistical approaches. The statistical method for testing pharmacokinetic BE is based on the determination of the 90% confidence interval around the geometric mean ratio of the log-transformed population means (T/R) for AUC and C by carrying out two one-sided tests at the 5% level of significance.
2. Other options. In addition, comparative pharmacodynamic studies in humans and comparative clinical trials can be used to document or supplement BE assessment. In vivo documentation of equivalence is especially important for the following: drugs with a narrow therapeutic range; documented evidence of BE problems; modified-release pharmaceutical products designed to act by systemic absorption; and fixed-dose combination products with systemic action when at least one of the drug substances requires an in vivo study.
3.1 Immediate-Release Drug Products
Single-dose, crossover BE studies are carried out at the highest dose comparing T and R products under fasting conditions. A parallel study design can be used for drugs that have a very long elimination half-life (t ). Sampling truncation at 72 h may be allowable by regulatory agencies. Lower strength(s) of the dosage form can be given a biowaiver based on linear pharmacokinetics, dosage form proportionality, and dissolution profile similarity. Food-effect studies are required as described within scale-up and postapproval changes-immediate release (SUPAC-IR). Food effect studies are required for all T immediate-release oral products except where: 1) the T and R products are both rapidly dissolving with similar dissolution profiles and containing highly soluble and highly permeable drug substances; or 2) where the labeling of the R product states that the product must be taken on an empty stomach; or 3) where the R product labeling does not make a statement about food effect on absorption or administration [see FDA Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies (2002); (search by document title; http://www.fda.gov/)].
3.2 Modified-Release Drug Products
BE studies for modified-release dosage forms are carried out as single-dose, crossover studies under fasting and fed conditions at the highest dose to compare T and R products. A single-dose study is more sensitive than multiple-dose, steady-state studies in assessing in vivo drug product performance, particularly with regard to the phenomenon of dose dumping, i.e., the rapid and unintended premature release of the drug substance from an extended-release product into the gastrointestinal tract. Lower strengths of an extended-release dosage form may not require an in vivo study based on use of the same drug-releasing mechanism, dosage form proportionality, and similar dissolution profile. The FDA Guidance for Industry: SUPAC-MR: Modified Release Solid Oral Dosage Forms Scale-Up and Postapproval Changes:
Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation (search by document title; http://www.fda.gov/) provides guidance on the need for BE studies when changes are made to the formulation. BE studies under fed conditions are required for all generic orally administered modified-release products [see FDA Guidance for Industry: Food-Effect Bioavailability and Fed Bioequivalence Studies (2002)].
3.3 Orally Administered Drug Products, Not for Systemic Effect
Some oral drug products are intended for local activity. Mesalamine and Cholestyramine are examples of drugs that are intended for local activity. For these types of drugs, systemic absorption from the gastrointestinal tract is minimal; thus a comparative clinical trial is required while a systemic drug exposure profile also may be required. In some cases, in vitro studies may be appropriate, such as studies including comparison of cholestyramine binding to bile salts.
3.4 Bioequivalence Studies
Objective: The objective of a BE study is to measure and compare formulation performance between two or more drug products. Drug availability from T and R products should not be statistically different when the drug is administered to patients or subjects at the same molar dose under similar experimental conditions.
Design: The design of a BE study depends on the objectives of the study, the ability to analyze the drug substance (and/or metabolites where appropriate) in biological fluids, the pharmacodynamics of the drug substance, the route of drug administration, and the nature of the drug and drug product. Pharmacokinetic parameters, pharmacodynamic parameters, clinical observations, and/or in vitro studies may be used to determine drug BA from a drug product.
Some possible BE study designs include the following:
1. Single-dose, two-way crossover study under fasted conditions
2. Single-dose, two-way crossover study under fed conditions
3. Single-dose, parallel study under fasted conditions
4. Single-dose, replicate design
5. Single-dose, partial replicate design
6. Multiple-dose, two-way crossover study, fasted conditions
7. Pharmacodynamic or clinical endpoint study
8. In vitro dissolution profile comparisons
The standard BE study is a crossover design (e.g., Latin square crossover design) in which each subject receives the T drug product and the R drug product on separate occasions. Studies are usually evaluated by a single-dose, two-period, two-treatment, two-sequence, open-label, randomized crossover design comparing equal doses of the T and R products in fasted or fed adult healthy subjects. A multiple-dose study may be required for some extended-release drug products. A washout is scheduled between the two periods to allow the subjects to completely eliminate the drug absorbed from the first dose before administration of the second dose of drug product. If the predose concentration is ≤5% of the C value in that subject, the subject's data without any adjustments can be included in all pharmacokinetic measurements and calculations. Samples of an accessible biologic fluid such as blood are used to characterize the drug concentration versus time profile. During the fasting study, subjects are fasted at least 10 h. A predose (0 time) blood sample is taken. The drug product is given with 240 mL (8 fluid ounces) of water. No food is allowed for at least 4 h post dose. The T and R drug products are administered at the same time of day to avoid diurnal effects. Blood sampling is performed periodically after administration of drug product according to protocol. A food intervention or food effect study is conducted with standard meal conditions that are expected to provide the greatest effects on gastrointestinal physiology so that systemic drug availability is maximally affected. In addition, the high lipid content of the meal may affect the rate of drug release from the product, in situ. A high-fat (approximately 50% of total caloric content of the meal) and high-calorie (approximately 800–1000 calories) meal is recommended as a test meal for food-effect BA and fed BE studies. This test meal should derive approximately 150, 250, and 500–600 calories from protein, carbohydrate, and fat, respectively. The drug product is given with 240 mL (8 fluid ounces) of water after ingestion of the standard meal. Subjects should consume identical meals within the same interval before administration of the T or R drug products.
Analysis of samples: Samples, usually plasma, are analyzed for the drug substance and, on occasion, active metabolite concentrations by a validated bioanalytical method.
Pharmacokinetic parameters: Pharmacokinetic parameters are obtained from the resulting concentration–time curves. Two major pharmacokinetic parameters are used to assess the rate and extent of systemic drug absorption. AUC reflects the extent of drug absorption, and the Cmax reflects the rate of drug absorption. Other pharmacokinetic parameters may include the Tmax, the elimination rate constant (k), elimination half-life (t1/2), lag time (Tlag), and others.
3.5 Statistical Analysis
Pharmacokinetic parameters are analyzed statistically to determine whether the T and R products yield comparable values. Because BE studies may use small sample sizes, log transformation of the data allows the frequency distribution of the data to be more normalized so that parametric statistical analyses may be performed (see FDA's Guidance for Industry: Statistical Approaches to Establishing Bioequivalence (2001); search by document title; http://www.fda.gov/).
Parametric (normal-theory) general linear model procedures are recommended for the analysis of pharmacokinetic data derived from in vivo BE studies. An analysis of variance (ANOVA) should be performed on the pharmacokinetic parameters AUC and C using appropriate statistical programs and models. For example, for a conventional two-treatment, two-period, two-sequence (2 × 2) randomized crossover study design, the statistical model often includes factors accounting for the following sources of variation:
- Sequence (sometimes called Group or Order)
- Subjects, nested in sequences
- Period (or Phase)
- Treatment (sometimes called Drug or Formulation)
The sequence effect should be tested using the [subject (sequence)] mean square from the ANOVA as an error term. All other main effects should be tested against the residual error (error mean square) from the ANOVA. The least-squares means (LSMEANS) statement should be used to calculate LSMEANS for treatments. Estimates should be obtained for the adjusted differences between treatment means and the standard error associated with these differences.
The statistical assumptions underlying the ANOVA are as follows:
- Randomization of samples
- Homogeneity of variances
- Additivity (linearity) of the statistical model
- Independence and normality of residuals
In BE studies, these assumptions can be interpreted as follows:
- The subjects chosen for the study should be randomly assigned to the sequences of the study.
- The variances associated with the two treatments, as well as between the sequence groups, should be equal or at least comparable.
- The main effects of the statistical model, such as subject, sequence, period, and treatment effect for a standard 2 × 2 crossover study, should be additive. There should be no interactions between these effects.
- The residuals of the model should be independently and normally distributed.
If these assumptions are not met, additional steps should be taken prior to the ANOVA, including data transformation to improve the fit of the assumptions or use of a nonparametric statistical test in place of ANOVA. However, the normality and constant variance assumptions in the ANOVA model are known to be relatively robust (i.e., a small or moderate departure from one or both of these assumptions will not have a significant effect on the final result). The rationale for log transformation is provided in FDA's Guidance for Industry: Statistical Approaches to Establishing Bioequivalence (2001) [search by document title; see FDA http://www.fda.gov/].
The two one-sided tests procedure: A testing procedure termed the two one-sided tests procedure is used to determine the comparability of geometric mean values for pharmacokinetic parameters measured after administration of the T and R products.1 The two one-sided tests procedure determines whether T is not importantly less than R and whether R is not importantly less than T. Other statistical approaches to the determination of a clinically meaningful difference may be used. Most often, 20% defines a clinically meaningful difference. The statistical procedure involves the calculation of a confidence interval for the ratio (or difference) between T and R pharmacokinetic variable averages. The limits of the observed confidence interval must fall within a predetermined range for the ratio (or difference) of the product averages. Point estimate mean ratios (T/R) derived from the log-transformed AUC and Cmax data must be between 80% and 125%. Because data are log transformed, T/R = 80/100 = 80% and R/T = 100/80 = 125%. In addition, the 90% confidence intervals for the geometric mean ratios (T/R) for AUC and Cmax must be between 80.00% and 125.00%. The regulatory requirements for the range of 90% confidence intervals for Cmax are different in countries outside of the United States.
Bio-inequivalence: The failure to demonstrate BE may be due to a performance failure of the T product or to an inadequate study design. The failure to demonstrate BE because of an inadequate study design can be due to improper sampling in which: 1) the sampling time for Cmax was not properly obtained, or 2) the number of samples taken did not adequately describe the plasma drug concentration versus time profile. Often, with highly variable drugs (e.g., % coefficient of variation [CV] of Cmax or AUC > 30%), where failure to demonstrate BE was observed, the study was not powered adequately due to too few subjects used.
Presentation of data: The drug concentration in biological fluid at each sampling time point should be furnished untransformed for all the subjects who participated in the study. The derived pharmacokinetic parameters also should be furnished untransformed. The mean, the standard deviation, and the CV for each variable should be computed and tabulated in the final report.
To facilitate BE comparisons, pharmacokinetic parameters for each individual should be displayed in parallel for the formulations tested. In particular, for AUC and Cmax, the difference (T − R), the ratio (T/R), and the log of ratio (log T/R or ln T/R) between the T and R values should be tabulated side-by-side for all the subjects. For each subject, the summary tables should indicate in which sequence (T then R, or R then T) the subject received the product. Histograms showing the frequency distribution of the difference and ln ratio (or log ratio) for the major pharmacokinetic parameters (AUC and Cmax) are useful in the submission. NMT 20% of the total AUC (zero to infinity) should be obtained by an extrapolated terminal elimination.
In addition to the arithmetic mean for the T and R products, the geometric means (antilog of the means of the logs), means of the logs, and standard deviations of the logs should be calculated for AUC and Cmax. All means, including arithmetic mean, geometric mean, and means of the logs, as well as standard deviations and CVs, should be included in the report.
4 DISSOLUTION AND IN VITRO PRODUCT PERFORMANCE
As noted for an official preparation, USP monographs provide a public specification that includes a list of tests, references to analytical procedures, and acceptance criteria. Most solid oral dosage forms, including oral suspensions, require a dissolution or drug release test.
Drug dissolution and drug release testing are described in 〈711〉 and 〈724〉. These public specifications are used for quality control tests and for market approval. The USP dissolution test in the monograph is related to BA and BE only when closely allied with a sound regulatory determination. Without this association, the USP dissolution test should be regarded solely as a quality control test for batch release. FDA guidances are: 1) Guidance for Industry: Dissolution Testing of Immediate Release Solid Oral Dosage Forms (1997) (http://www.fda.gov/; search by document title); 2) Guidance for Industry: Extended Release Oral Dosage Forms: Development, Evaluation, and Application of In Vitro/In Vivo Correlations (1997); and 3) Guidance for Industry: Dissolution Testing and Specification Criteria for Immediate-Release Solid Oral Dosage Forms Containing Biopharmaceutics Classification System Class 1 and 3 Drugs (draft July 2015) (search by document title; http://www.fda.gov/).
4.1 Dissolution and In Vitro BA
Drug dissolution and release tests are very useful during drug product development for identifying critical manufacturing attributes such as the impact of ingredient properties and the impact of the manufacturing process on drug product performance. During product development, optimum dissolution conditions need to be developed to discriminate among drug product formulations and changes in manufacturing processes. After the finished dosage form is approved for marketing, drug dissolution and release tests may be useful in predicting possible changes in performance due to scale-up and postapproval changes. See the following FDA guidances:
Guidance for Industry: Immediate Release Solid Oral Dosage Forms, Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls, In Vitro Dissolution Testing, and In Vivo Bioequivalence Documentation (1995) (search by document title; http://www.fda.gov/) and Guidance for Industry: SUPAC-MR: Modified Release Solid Oral Dosage Forms, Scale-Up and Postapproval Changes: Chemistry, Manufacturing, and Controls; In Vitro Dissolution Testing and In Vivo Bioequivalence Documentation (1997) (search by document title; http://www.fda.gov/). For some oral drug products, in vitro drug dissolution can be related to in vivo performance, such as BA and/or systemic drug exposure. Chapter 〈1088〉 describes various approaches to IVIVC.
4.2 Dissolution and In Vitro Equivalence
An appropriate dissolution test is a powerful in vitro physiochemical test that measures drug product quality and performance for a variety of dosage forms, such as solid oral dosage forms, transdermal dosage forms, suspensions, and certain semisolid dosage forms. The USP tests for finished dosage forms can be divided into two types: 1) drug product quality tests, and 2) drug product performance tests. Product quality tests are intended to assess attributes such as assay and content uniformity; product performance tests are designed to assess product performance, and in many cases relate to dissolution. For details regarding the performance of a dissolution/drug release test, see〈711〉, 〈724〉, 〈1088〉, 〈1092〉, and Semisolid Drug Products—Performance Tests 〈1724〉.
The in vitro dissolution test was initially developed as a quality control tool to ensure drug product quality and batch-to-batch consistency. The test procedures for conducting dissolution/drug release tests are described in 〈711〉 and 〈724〉. The development of the BCS brings new understanding and power to the dissolution test. The BCS classifies the drug substance according to its aqueous solubility, as well as its permeability through a biomembrane, such as the intestinal mucosal cells. The dissolution rate of the drug substance from the dosage form is important in substantiating biowaivers based on the BCS.
4.3 Dissolution Profile Comparisons
In vitro drug dissolution and release testing can be related to in vivo drug performance, such as BA. The comparisons of dissolution profiles are gaining importance as a means of documenting comparative BA studies—that is, BE. A biowaiver is the replacement or waiver of in vivo BE studies by an in vitro test.
A model independent mathematical approach is used to compare the dissolution profiles of two products: 1) to compare the dissolution profile between the T product and R product in biowaiver considerations; 2) to compare the dissolution profile between the two strengths of products from a given manufacturer; and 3) for SUPAC after the product is approved. For comparing the dissolution profile, the similarity factor f2 should be computed using the equation:
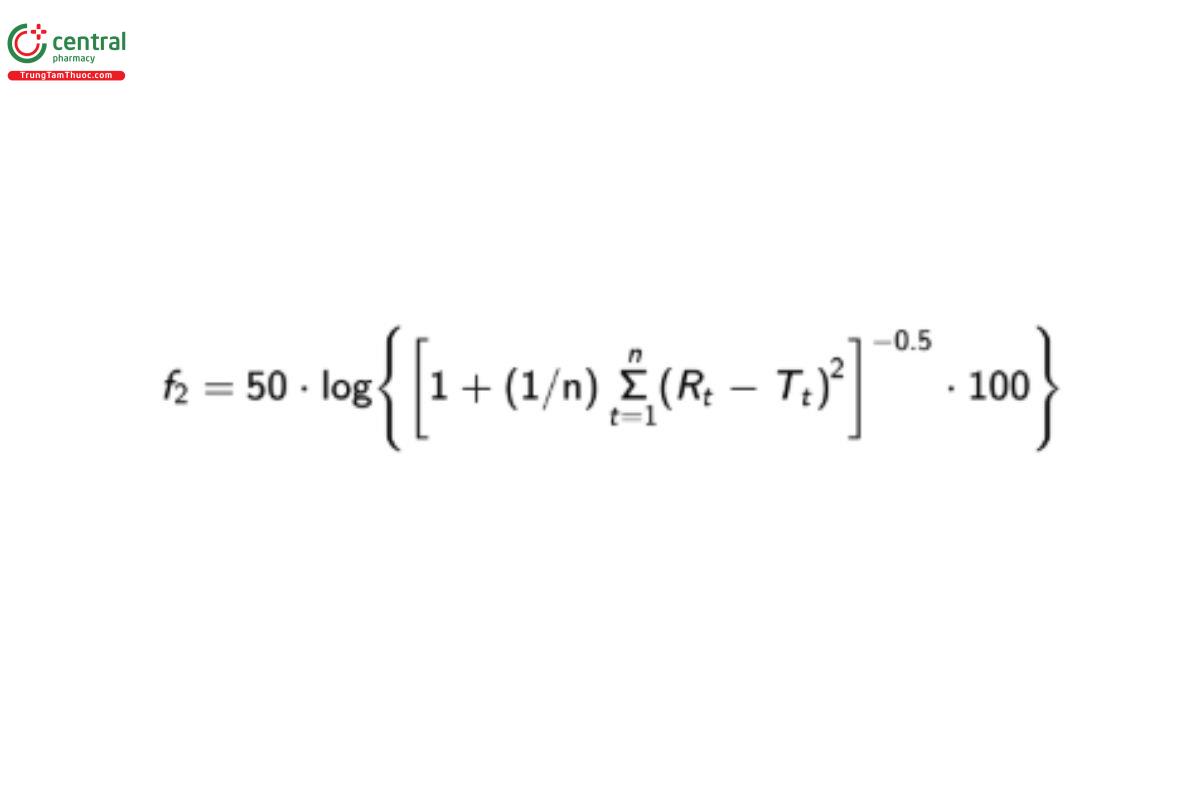
where Rt and Tt are the cumulative percentage of the drug dissolved at each of the selected n time points of the R and T product, respectively. An f2 value of 50 or greater (50–100) ensures dissolution profile similarity and thus equivalence of the performance of the two products. FDA guidance recommends that, at a minimum three points, NMT 1 point exceeding 85% of the label claim dissolved, should be used for similarity profile comparison. For products that dissolve very rapidly (≥85% dissolution in 15 min), a profile comparison is not necessary.
Different guidances have slightly different considerations in the application of dissolution profile comparisons that are given in Table A-1.
5 INTERCHANGEABILITY OF DRUG PRODUCTS
The interchangeability of drug products is a major concern for physicians, pharmacists, and others who prescribe, dispense, or purchase them. Interchangeability of drug products is a regulatory decision requiring a determination of therapeutic equivalence. The FDA determines therapeutic equivalence of products in the U.S. Because the formulation and method of manufacture of the drug product can affect its BA and stability, the manufacturer must demonstrate that the T drug product is bioequivalent and TE to the R. The factors that can affect the drug product performance and therefore interchangeability of the finished drug product include:
- Solid form and particle size of the drug substance
- Differences in excipients in the formulation
- Differences in the manufacturing process
6 SOLID FORM AND PARTICLE SIZE
As a result of the synthetic route and method of purification, the drug substance may be present with different particle size or solid form.
Different solid forms, crystalline or amorphous, can dissolve at different rates, and therefore the extent of absorption and BA of these substances from the same dosage form, e.g., oral tablet, may be different. For example, crystalline structures are more thermodynamically stable than amorphous forms and as a consequence may dissolve more slowly. Different polymorph, hydrate, or salts of the drug substance may also have different physical chemical properties. These differing forms of the drug may have different aqueous solubilities or may dissolve at different rates. Particle size or particle size distribution of the drug may also affect the rate of dissolution. A drug substance having a particle size distribution with a large number of very fine particles may dissolve faster and lead to systemic absorption that is more rapid than for a particle size distribution with larger particles.
7 DIFFERENCES IN EXCIPIENTS
A pharmaceutical dosage form typically consists of excipients as well as the drug substance(s). An excipient is any component, other than the drug substance(s), intentionally added to the formulation of a dosage form. Excipients, which may have no pharmacodynamic activity, play a critical role in manufacturing, stability, and performance. Excipients are manufactured to comply with compendial standards that address quality. However, the physical and chemical properties of the excipients affect the performance of a finished dosage form at least as much as the physical and chemical properties of the drug substance.
8 MANUFACTURING PROCESS
Variation in the manufacturing process can have a dramatic effect on drug product performance. Blending, sieving, compression force, precompression or granulation, and coating are all manufacturing operations that can impact product performance. Interchangeability can be affected by the manufacturing operation that is as important a consideration as the control of the drug substance form and the properties of the excipients used in the formulation.
9 BIOWAIVER
The term “biowaiver” is applied to a regulatory approval process when the application (dossier) is approved on the basis of evidence of equivalence other than an in vivo BE test. For solid oral dosage forms, the evidence of equivalence is determined on the basis of an in vitro dissolution profile comparison between the T and the R product.
10 Biowaiver Based on Dosage Form Proportionality
When a single-dose fasting BE study is conducted on the designated (usually highest) strength of the drug product, the requirement for the conduct of additional in vivo BE studies on the lower strengths of the same product can be waived, provided that the lower strength: 1) is in the same dosage form; 2) is proportionally similar in its active and inactive ingredients; 3) has the same drug release mechanism (for extended-release products); 4) meets an appropriate in vitro dissolution profile comparison criterion (e.g., f ≥50); and 5) both lower and higher strengths are within the linear pharmacokinetic range.
11 Biowaiver Based on the BCS
BCS provides a regulatory platform for replacing certain BE studies with in vitro dissolution tests. The BCS takes into account three major factors that govern the rate and extent of drug absorption from immediate-release dosage forms. These are aqueous solubility and intestinal permeability of the drug substance in conjunction with the dissolution of the pharmaceutical dosage form. These properties are assessed and categorized as being low or high for the purpose of classification. On the basis of the solubility and permeability of the dosage form, the drug substance is categorized in different classes:
- Class 1: high solubility, high permeability
- Class 2: low solubility, high permeability
- Class 3: high solubility, low permeability
- Class 4: low solubility, low permeability
Solubility classification of a drug substance as low or high depends on the dose of the drug product. FDA defines a drug substance as being highly soluble “when the highest dose strength is soluble in 250 mL or less of aqueous media over the pH range of 1.0–6.8” (See FDA Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System (December 2017) (search by document title; http://www.fda.gov/). However, EMA uses the highest single dose administered, and WHO uses the highest therapeutic dose approved by a competent authority, typically de fined by the labeling for the innovator product.
The classification of drug substance permeability as high or low differs among the FDA, WHO, and EMA. FDA states that a drug substance is highly permeable where the extent of absorption is 90% or more of an administered dose in the absence of evidence of instability in the gastrointestinal tract. WHO considers a drug substance (API) highly permeable when the extent of absorption is 85% or more of an administered dose. EMA finds complete absorption to be established where the measured extent of absorption is ≥85% and goes on to say that complete absorption is generally related to high permeability. FDA, WHO, and EMA all use mass balance or comparison to an intravenous reference dose (EMA: absolute BA) in support of the claim of extent of absorption.
The FDA requirements for BCS-based biowaiver apply to immediate-release drug products when the drug substance is BCS Class 1 and the R and T products are rapidly dissolving (NLT 85% in NMT 30 min) or BCS Class 3 where R and T are very rapidly dissolving (NLT 85% in NMT 15 min) and contain the same excipients. T and R products should be PE and exhibit similar dissolution profiles. The FDA biowaiver excludes immediate-release products that have a narrow therapeutic index or are designed to be absorbed in the oral cavity. The EMA requirements for BCS-based biowaiver are applied to immediate-release solid oral drug products considered not to have a narrow therapeutic index. Both BCS Class 1 and 3 compounds can be considered. The T and R must have the identical drug substance, but different salts are acceptable as long as both belong within BCS Class 1. For BCS Class 1 drug substances, in vitro dissolution is either very rapid (i.e., >85% in 15 min) or similarly rapid (i.e., 85% in <30 min and similar dissolution profiles). For BCS Class 3 drug substances, a biowaiver would require very rapid in vitro dissolution for both T and R. Excipients that might affect BA are the same and other excipients are similar qualitatively and in similar amounts.
The WHO requirements for BCS-based biowaiver apply to BCS Class 1 and 3 drug substances. For BCS Class 1 substances, both T and R products should be rapidly dissolving and demonstrate similar dissolution profiles. For BCS Class 3 drug substances, dissolution should be very rapid in vitro. The same excipient composition for both T and R drug products is required.
Use of the BCS has become a means of documenting BE without the conduct of an in vivo study; see the FDA guidance Guidance for Industry: Waiver of In Vivo Bioavailability and Bioequivalence Studies for Immediate-Release Solid Oral Dosage Forms Based on a Biopharmaceutics Classification System (December 2017) (search by document title; http://www.fda.gov/).
The in vitro dissolution studies are generally carried out by basket method at 100 rpm or by paddle method at 50 rpm [FDA guidance cited immediately above and EMA guidance, Guideline on the Investigation of Bioequivalence (2010)] or 75 rpm [WHO guidance, Annex 7: Multisource (Generic) Pharmaceutical Products: Guidelines on Registration Requirements to Establish Interchangeability, WHO Expert
Committee on Specifications for Pharmaceutical Products: Fortieth Report. WHO: Geneva; 2006: Annex 7 WHO Technical Report Series, No. 937). TRS 992] (search by document title; http://www.who.int/en/) in 900 mL of medium at pH 1.2, 4.5, and 6.8. On the basis of dissolution rate, the pharmaceutical dosage forms are classified as: 1) very rapidly (FDA, EMA and WHO) dissolving, if 85% or more of the labeled content dissolves in 15 min or less, or 2) rapidly dissolving, if 85% or more of the labeled content dissolves in 30 min.
For biowaiver, the dissolution tests should be carried out for both T and R product under the same test conditions. For the generic product to be eligible for biowaiver, the R product should belong to the same BCS class and should meet dissolution profile comparison criteria. On the basis of the BCS classification and dissolution profile comparison, a biowaiver can be considered by regulatory authorities, provided the dissolution profile similarity criteria are met Table A-1).
11.1 Dissolution as a quality control test and versus as an in vitro equivalence test
There is a clear difference between dissolution as a quality control test and dissolution as an in vitro equivalence test. For immediate-release dosage forms, the quality control test involves a single-point dissolution test in only one medium (generally a compendial test). On the other hand, the in vitro equivalence test involves dissolution profile comparison in pH 1.2, 4.5, and 6.8 between the T product and the R product.
12 APPENDIX
12.1 Comparisons Among FDA, EMA, and WHO
Table A-1. Comparison of Approaches to BCS-Based Biowaiver
| FDA | EMA | WHO | |
| Conditions for f2 calculation | Only one measurement should be considered after 85% dissolution of both the products | A minimum of 3 time points (0 excluded) and NMT 1 mean value of >85% dissolved for any of the formulations | A maximum of 1 time point should be considered after 85% dissolution of the reference (comparator product) has been reached. In cases where 85% dissolution cannot be reached, the dissolution should be conducted until an asymptote (plateau) has been reached. |
| Requirements | f2 ≥ 50 | 50 ≤ f2 ≤ 100 | 50 ≤ f2 ≤ 100 |
| Very rapidly dissolving | NLT 85% in 15 min (using the method according to the guideline) | >85% of the labeled amount dissolves in 15 min (using the method according to the guideline) | NLT 85% of the labeled amount dissolves in 15 min (using the method according to the guideline) |
| Rapidly dissolving | ≥85% of the labeled amount dissolves in 30 min (using the method according to the guideline) | ≥85% of the labeled amount dissolves within 30 min (similarly rapid) (using the method according to the guideline) | NLT 85% of the labeled amount dissolves in 30 min (using the method according to the guideline) |
| Biowaiver | BCS Class 1 drug product (T and R) is rapidly dissolving, and T does not contain any excipients that would affect rate and extent of absorption of the drug. BCS Class 3: drug product (T and R) is very rapidly dissolving, and T is qualitatively very similar in composition to R. | BCS Class 1 drugs and either very rapid dissolution or similarly rapid. Excipients that might affect BA are qualitatively and quantitatively the same; BCS Class 3 drugs and very rapid dissolution and excipients that might affect BA are qualitatively and quantitatively the same; and other excipients are qualitatively the same and quantitatively very similar. | BCS Class 1: T and R are very rapidly dissolving or similarly rapidly dissolving drugs; BCS Class 3: T and R are very rapidly dissolving. |
| Apparatus | USP Apparatus 1 USP Apparatus 2 | Basket Paddle | Basket Paddle |
| Dissolution media | 0.1 N hydrochloride or For capsules and tablets with | pH 1.2 (0.1 N hydrochloride or Absolutely no addition of | Buffer pH 1.2 Buffer pH 4.5 (acetate buffer) Buffer pH 6.8 International pharmacopoeia buffers are preferred |
| Volume | 500 mL or less | 900 mL or less | 900 mL or less |
| Temperature | 37 ± 0.5° | 37 ± 1° | 37° |
| Agitation | Apparatus 1: 100 rpm Apparatus 2: 50 rpm (or 75 rpm when appropriately justified) | Basket: 100 rpm Paddle: 50 rpm | Basket: 100 rpm Paddle: 75 rpm |
| Sample number | 12 | 12 | 12 |
| Sampling time | Suficient number of intervals, e.g., 10, 15, 20, 30 min | 10, 15, 20, 30, 45 min | 10, 15, 20, 30, 45, 60 min |
Table A-2. Comparison of FDA, EMA, and WHO Definitions
| Term | FDA | EMA | WHO |
| Pharmaceutical equivalents | Drug products are considered pharmaceutical equivalents if they contain the same active ingredient(s), are of the same dosage form, have the same route of administration, and are identical in strength or concentration. Pharmaceutically equivalent drug products are formulated to contain the same amount of active ingredient in the same dosage form and to meet the same or compendial or other applicable standards (strength, quality, purity, and identity); but they may differ in characteristics such as shape, scoring configuration, release mechanisms, packaging, excipients, expiration time, and, within certain limits, labeling. | Medicinal products are pharmaceutically equivalent if they contain the same amount of the same active substance(s) in the same dosage forms that meet the same or comparable standards. Pharmaceutical equivalence does not necessarily imply bioequivalence as differences in the excipients and/or the manufacturing process can lead to faster or slower dissolution and/or absorption. | Products are pharmaceutical equivalents if they contain the same molar amount of the same API(s) in the same dosage form, if they meet comparable standards, and if they are intended to be administered by the same route. Pharmaceutical equivalence does not necessarily imply TE as differences in the API solid state properties, the excipients and/or the manufacturing process and other variables can lead to differences in product performance. |
| Pharmaceutical alternatives | Drug products are considered pharmaceutical alternatives if they contain the same therapeutic moiety but are different salts, esters, or complexes of that moiety or are different dosage forms or strengths. | Medicinal products that contain different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of the active moiety, or which differ in dosage form or strength | Products are pharmaceutical alternative(s) if they contain the same active pharmaceutical moiety or moieties but differ in dosage form (e.g., tablets vs. capsules), strength, and/or chemical form (e.g., different salts, different esters). Pharmaceutical alternatives deliver the same active moiety by the same route of administration but are otherwise not pharmaceutically equivalent. They may or may not be bioequivalent or TE to the comparator product |
| Therapeutic equivalents | Drug products are considered to be therapeutic equivalents only if they are pharmaceutical equivalents and if they can be expected to have the same clinical effect and safety profile when administered to patients under the conditions specified in the labeling. | A medicinal product is TE to another product if it contains the same active substance or therapeutic moiety and, clinically, shows the same efficacy and safety as that product, whose efficacy and safety have been established. In practice, demonstration of BE is generally the most appropriate method of substantiating therapeutic equivalence between medicinal products, which are pharmaceutically equivalent or pharmaceutical alternatives, provided they contain excipients generally recognized as not having an in fluence on safety and efficacy and comply with labeling requirements with respect to excipients. However, in some cases where similar extent of absorption but different rates of absorption are observed, the products can still be judged TE if those differences are not of therapeutic relevance. A clinical study to prove that differences in absorption rate are not therapeutically relevant may be necessary. | Two pharmaceutical products are considered to be TE if they are pharmaceutically equivalent or pharmaceutical alternatives and after administration in the same molar dose, their effects, with respect to both efficacy and safety, are essentially the same when administered to patients by the same route under the conditions specified in the labeling. This can be demonstrated by appropriate equivalence studies, such as pharmacokinetic, pharmacodynamic, clinical, or in vitro studies. |
| BA (bioavailability) | This term means the rate and extent to which the active ingredient or active moiety is absorbed from a drug product and becomes available at the site of action. | The rate and extent to which a substance or its active moiety is delivered from a pharmaceutical form and becomes available in the general circulation (taking into consideration that the substance in the general circulation is in exchange with the substance at the site of action). | The rate and extent to which essentially similar |
| Bioequivalent drug products | This term describes pharmaceutical equivalent or pharmaceutical alternative products that display comparable BA when studied under similar experimental conditions. | Two medicinal products are bioequivalent if they are pharmaceutically equivalent or pharmaceutical alternatives and if their bioavailabilities after administration in the same molar dose are similar to such degree that their effects, with respect to both efficacy and safety, will be essentially the same. | Two pharmaceutical products are bioequivalent if they are pharmaceutically equivalent or pharmaceutical alternatives and their BA, in terms of rate Cmax and tmax and the extent of absorption (area under the curve [AUC]) after administration of the same molar dose under the same conditions, are similar to such a degree that their effects can be expected to be essentially the same. |
| R drug product | An RLD [21 CFR 314.94(a)(3)] means the listed drug identified by FDA as the drug product upon which an applicant relies in seeking approval of its ANDA. | A reference medicinal product is a medicinal product authorized under Article 6, in accordance with the provisions of Article 8 (EC Directive 2001/83/EC) | The comparator pharmaceutical product (CPP) is a pharmaceutical product with which the multisource product is intended to be interchangeable in clinical practice. The comparator product normally will be the innovator product for which efficacy, safety, and quality have been established. The selection of the comparator product usually is made at the national level by the drug regulatory authority. |
| T drug product | A generic product is a product that is therapeutically equivalent to the RLD and is intended to be interchangeable with the innovator product. | A generic medicinal product which has the same qualitative and quantitative composition in active substances and the same pharmaceutical form as the reference medicinal product, and whose BE with the reference medicinal product has been demonstrated by appropriate BA studies. The different salts, esters, ethers, isomers, mixtures of isomers, complexes or derivatives of an active substance shall be considered to be the same active substance, unless they differ significantly in properties with regard to safety and/or efficacy. In such cases, additional information providing proof of the safety and/or efficacy of the various salts, esters, or derivatives of an authorized active substance must be supplied by the applicant. The various immediate-release oral pharmaceutical forms shall be considered to be one and the same pharmaceutical form. | See Multisource pharmaceutical products. |
| Multisource pharmaceutical products | Pharmaceutically equivalent or pharmaceutically alternative products that may or may not be therapeutically equivalent. Multisource pharmaceutical products that are therapeutically equivalent are interchangeable. | ||
| Interchangeable pharmaceutical product | An interchangeable pharmaceutical product is one which is therapeutically equivalent to a comparator product and can be interchanged with the comparator in clinical practice. |
1 Schuirmann DJ. A comparison of the two one-sided tests procedure and the power
