Assays to Evaluate Fragment Crystallizable (FC)—Mediated Effector Function
If you find any inaccurate information, please let us know by providing your feedback here


Tóm tắt nội dung
This article is compiled based on the United States Pharmacopeia (USP) – 2025 Edition
Issued and maintained by the United States Pharmacopeial Convention (USP)
1 INTRODUCTION
Therapeutic modalities such as antibodies and fragment crystallizable (Fc)-fusion molecules are often engineered with a specific Immunoglobulin G (IgG) isotype and subtype in order to impart particular functionalities, such as half-life extension or induction of cytotoxic activity. Alternatively, a specific isotype and subtype may be selected to mitigate, or lessen, potential induction of unwanted biological activities. These modalities have defined structure/function domains that impart the therapeutic benefit.
In vitro assays that assess complex Fc domain interactions are critical to the biological characterization of Fc-containing therapeutics. This chapter provides information and points to consider about the more commonly used techniques available in both cell-based and non-cell-based formats for measuring these interactions. Finally, the chapter also discusses critical reagents, the foundation for these various assays, as well as assay validation, data analysis, and reporting strategies, but it is not intended to cover all potentially applicable techniques.
2 BACKGROUND
An example of an IgG1 monoclonal antibody (mAb) is shown in Figure 1. The interactions mediated by each structural domain, including antigen binding via the fragment antigen binding (Fab) domain, Fc gamma receptor (FcγR) binding, complement component 1q (C1q) binding, and neonatal Fc receptor (FcRn) binding, are highlighted. The different glycans that may be present in the Fc domain are also noted.
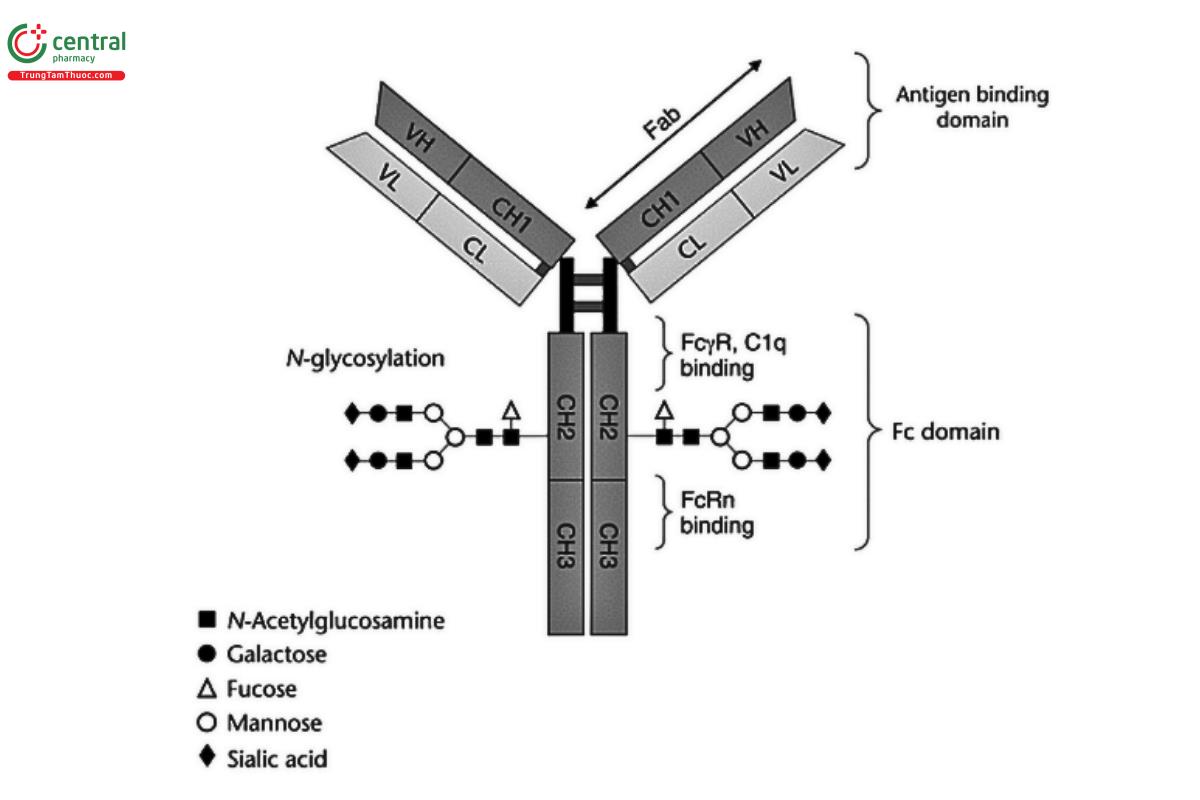
The different Fc-mediated effector function activities may include one or more of the following:
- Antibody-dependent cell-mediated cytotoxicity (ADCC)
- Complement-dependent cytotoxicity (CDC)
- Antibody-dependent cellular phagocytosis (ADCP)
- Fc-dependent apoptosis
Fc effector function activities are mediated via interaction of the Fab domain with the target antigen, and the interaction of the Fc domain with complement or various FcγR expressed on cells of hematopoietic origin (e.g., natural killer, macrophage, dendritic, monocyte, and neutrophil). In some cases, the targeted killing or destruction of cells is part of the mechanism of action (MOA) of the therapeutic and has been linked to its clinical efficacy. In other cases, the unintentional killing of normal cells via these mechanisms can be an undesirable consequence of the therapeutic with potential impact on safety. In addition, the interactions of the Fc-domain with FcRn have implications for the circulating half-life.
Fc receptors (FcRs) represent a large class of glycoproteins in the immunoglobulin superfamily. The FcγR subclass is most involved in interactions with Fc-containing therapeutics. The Fc–FcγR interaction is complex due to the fact that there are multiple subtypes of FcγR with different binding affinities toward the Fc of the various human IgG subtypes: IgG1, IgG2, IgG3, and IgG4. Table 1 shows FcγRs and their relative affinity to each IgG subtype.
Table 1. FcR Class and Affinityᵃ,ᵇ
| Receptor Type | IgG1 | IgG2 | IgG3 | IgG4 |
| FcRn | +++ | +++ | ++ | +++ |
| FcγRI | +++ | – | +++ | ++ |
| FcγRIIa 131H | ++ | + | ++ | + |
| FcγRIIa 131R | ++ | + | ++ | + |
| FcγRIIb | + | +/– | + | + |
| FcγRIIc | + | +/– | + | + |
| FcγRIIIa 158V | ++ | +/– | ++ | + |
| FcγRIIIa 158F | + | +/– | ++ | + |
| FcγRIIIb | + | – | ++ | – |
ᵃ Key: “–” is no measurable affinity; “+/–” is low affinity, barely detectable; “+” to “+++” is increasing affinity.
ᵇ Stewart R, Hammond SA, Oberst M, Wilkinson, RW. The role of Fc gamma receptors in the activity of immunomodulatory antibodies for cancer. J ImmunoTherapy of Cancer, 2014;2(1):1.
It should be noted that affinity measurements are highly dependent on assay format and assay parameters. Consequently, the relative affinities in Table 1 are a compilation of general trends reported from numerous studies and are presented as such for the purpose of making comparisons. Although for most IgG subtypes monomeric IgG interaction with FcγRI is of relatively high affinity while that for other FcγRs is of relatively low affinity, higher-avidity interactions resulting from cross-linking-induced interactions of FcγRs with immune complexes may occur. This is important for the biological function of FcγRs and for the Fc-mediated effector function of IgGs co-bound to target antigen as part of immune complexes, underscoring the importance of cell-based Fc effector function assays in addition to binding assays when potential for known Fc effector function exists. One or more FcγRs on FcγR-expressing hematopoietic cells may be involved in mediating ADCC, ADCP, or Fc-dependent apoptosis. Note also that antigen density and Fc N-glycan composition can impact FcγR-binding interactions and associated effector functions.
The classical pathway of the complement system involved in CDC is a cascade of complement protein complex formation comprising several different complement proteins, which ultimately form a membrane attack complex that can kill complement-bound target cells. The formation of this complement complex is dependent on the first step in which C1q, a component of the complement, binds to the Fc region of the Fc-containing therapeutic, which in turn is bound to the antigen or other binding partner through the Fab or the binding domain of an alternative mAb. IgG1 and IgG3 subtypes have higher CDC activity than IgG2 and IgG4. A comparison of all four together, in general, reveals that the relative C1q binding activity is determined to be IgG1≈IgG3 > IgG2≈IgG4. (1) It is also important to take into consideration the fact that epitope presentation, antigen density, and Fc N-glycan composition can impact C1q binding and CDC activity.
Recycling of antibodies after binding through the Fc region to FcRn is known to prolong their persistence in the bloodstream. Fc fusion proteins are engineered with the Fc for this purpose. The interaction of the Fc domain with FcRn occurs in a strict pH-dependent manner in which maximum binding is observed at a slightly acidic pH (6.0) and dissociation at neutral pH. Although it is always best practice to maintain assay temperature and pH control in any protein–protein interaction measurement, particular attention should be paid to pH level when assessing FcRn binding to Fc.
Specifically, the Fc-containing protein is pinocytosed at the cell surface and associates with FcRn in the acidic environment of the endosome. It is then recycled back to the cell surface where it is released from the FcRn at the more neutral pH of the blood. Human IgG1, IgG2, and IgG4 bind well to FcRn, whereas most human IgG3 binds poorly. Serum half-life is considerably shortened with lower-affinity binding, and this is one reason why IgG3s and the IgG3 Fc domain are generally not used as protein therapeutics. During studies performed at pH 5.8 and 25° that compared all four IgG subtypes together, the rank order of binding affinity to FcRn was IgG1≈IgG2≈IgG4 > IgG3. In contrast to FcγRs, differential glycosylation patterns in the Fc do not appear to alter FcRn binding based on affinity measurements from assays performed at pH 5.8 and 25°.
3 CELL-BASED FUNCTIONAL ASSAYS
This section describes the most commonly used cell-based assays. It is important to note that the intent is to describe these assays from a chemistry, manufacturing, and control perspective, as opposed to a discovery research perspective. As such, the differentiation between the use of relevant reagents and materials, such as primary cells, versus engineered cell lines that overexpress certain proteins, will be highlighted, where appropriate.
For the purpose of drug release, stability, and comparability studies, it is important to have robust, accurate, precise, and stability-indicating analytical assays that have been appropriately developed and qualified. This process should include an evaluation of the method sensitivity to the post-translational modifications, including glycosylation, which is known to impact Fc function. However, an in-depth discussion of each glycan and its respective impact is complex and beyond the scope of this chapter.
Three common components of cell-based assays where technical considerations are important are: 1) the choice of the target cell, 2) concentrations of the therapeutic antibody, and 3) endpoint detection for cell killing or surrogate marker.
Assessment of lack of effector function for a modality is challenging. Points to consider include:
- IgG1 positive control match for IgG2 and IgG4 modalities
- Physiologically relevant versus engineered cell lines that over-express targets
- Soluble targets bound to cell surface receptors or membrane expressed versions of soluble targets
The most effective option may be to carefully consider assay design to address lack of effector function, potentially including proven assay formats for modalities with demonstrated effector function.
Table 2 summarizes the commonly used cell-based assay formats for each Fc-mediated effector function activity. Each assay is then described in more detail. Because fundamental assay optimization approaches such as design of experiment (DOE), as well as assay qualification or validation strategies, are generally applicable to most bioassays and are covered in Design and Development of Biological Assays 〈1032〉 and Biological Assay Validation 〈1033〉, these topics will not be covered extensively in this chapter.
Table 2. Commonly Used Cell-Based Effector Function Assay Formats
| Activity | Assay Format | Points to Consider |
| Antibody-dependent cell-mediated cytotoxicity (ADCC) | Cytotoxicity: Peripheral blood mononuclear cells (PBMCs), NK cell-based assays using chromium-51 (51Cr) radioactivity or fluorescence-labeled target cells, or release of intracellular enzymes or Adenosine triphosphate (ATP) as readouts |
|
| ||
| ||
| Reporter gene assays: Readout quantifying ADCC signaling pathway activation in engineered effector cells |
| |
| Complement-dependent cytotoxicity (CDC) | Cytotoxicity: Single target cell-based assays using viability indicators, fluorescence-labeled target cells, or release of intracellular enzymes or ATP as readouts |
|
| ||
| ||
| Reporter gene assays: Using target cells constitutively expressing luciferase |
| |
| Antibody-dependent cell-mediated phagocytosis (ADCP) | Phagocytosis: Primary macrophages or monocyte cell lines with flow cytometry assay, live cell digital imaging, use of pH-sensitive dye to confirm actual phagocytosis |
|
| Reporter gene assays: Readout quantifying ADCP signaling pathway activation in engineered effector cells |
| |
| Fc-dependent apoptosis | Apoptosis: Cell-based assay with cross-linking reagents; detection modes include caspase activators, DNA strand breaks via nucleic acid stains, and mitochondrial membrane potential changes by fluorescent probes; fluorescent detection of membrane Phospholipid phosphatidylserine |
|
3.1 Antibody-Dependent Cell-Mediated Cytotoxicity
ADCC is a cell-mediated innate immune mechanism whereby an effector cell of the immune system (e.g., NK cell, monocyte, macrophage, or eosinophil) actively lyses a target cell that has been recognized by target-specific antibodies. ADCC can be important for the efficacy and MOA of some mAb therapeutics. However, for other molecules, ADCC activity may be a potential liability that needs to be assessed; for example, if the target is located on normal cells. As Figure 2 shows, to induce ADCC, the Fab domain of the antibody has to bind antigen on the surface of the target cell while its Fc domain binds to the FcγR on the surface of the effector cell.
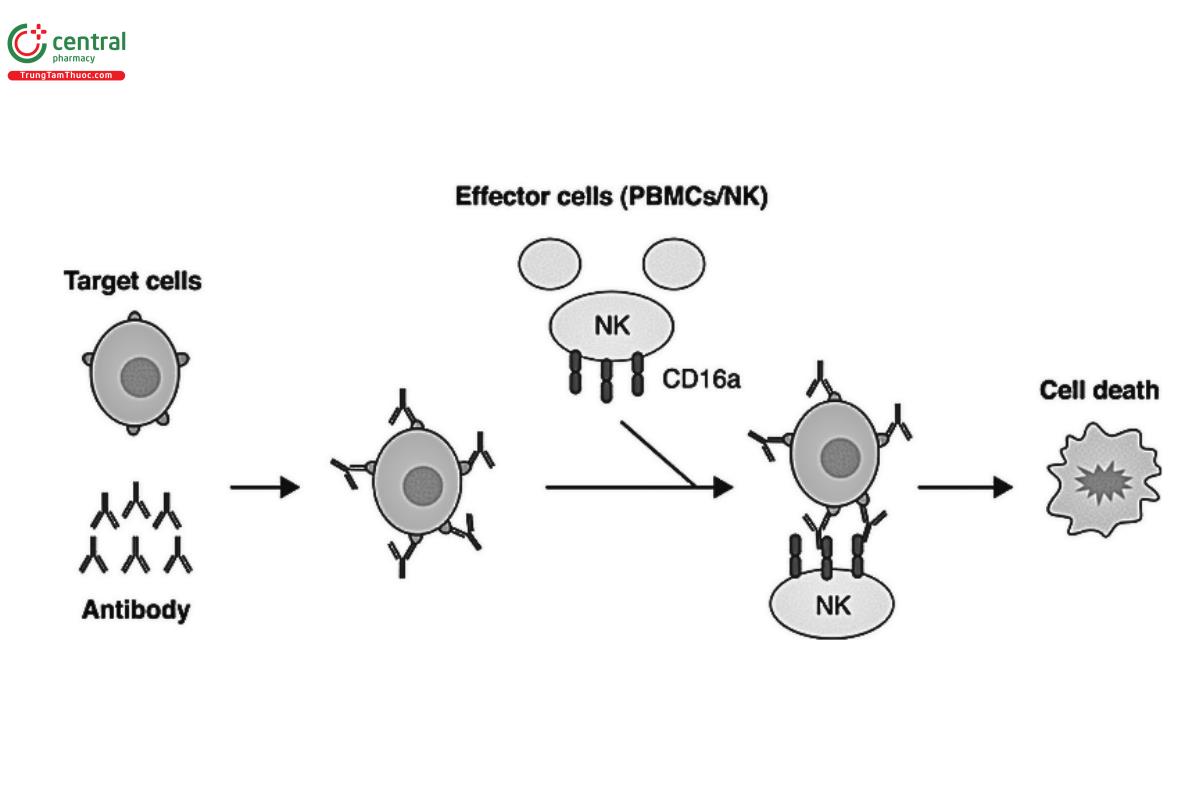
The development of ADCC assays is complicated because two different cell types are involved and many parameters need to be optimized, such as target and effector cell numbers; target-to-effector cell ratio; plating procedure, media, and extracellular matrix; drug concentrations and dilutions; volumes; incubation times; signal reading; and data processing and analysis. In addition, the type of FcγR and genetic polymorphism should be considered as these components can affect the affinity of Fc binding.
Traditional ADCC assays use PBMCs or isolated NK cells from human donors to induce chromium-51 (⁵¹Cr) release from preloaded target cells after lysis. Although such effector cells are the most physiologically relevant, they also represent an expensive, variable, and unreliable material source. Importantly, donor-to-donor differences due primarily to FcγR polymorphism can significantly affect the precision and accuracy of the results. Consequently, cells are generally pooled from multiple donors.
The use of a radioactive readout in traditional ADCC methods also has been shown to have both advantages and disadvantages. Chromium is non-toxic to cells, and because only target cells are loaded with it, there is no background from effector cells. However, the use of radioactivity is associated with safety and waste concerns. In general, the traditional ADCC assays are challenging to perform and transfer.
To avoid radioactivity, target cells can be preloaded with fluorescent dyes such as calcein O,O′-diacetate tetrakis(acetoxymethyl) ester (calcein AM), carboxyfluorescein succinimidyl ester (CFSE), 2′,7′-bis-(2-carboxyethyl)-5-(and-6)-carboxyfluorescein (BCECF), or europium. Note that these reagents may be toxic to cells and the sensitivity to these dyes may vary significantly between different cell types. The cell penetration of dyes and the ability to retain the dyes intracellularly also may be cell type-dependent.
The advantage of prelabeling cells is that only target cell death is measured with no background from effector cells, which usually provides higher signal-to-background ratios, especially for fluorescent dyes with amplification. The general drawback is the extra time and manipulation required for the loading and washing steps. However, it can be mitigated by implementing thaw-and-use cell banks prelabeled with the fluorescent dye.
Alternatively, the release of intracellular enzymes such as lactate dehydrogenase (LDH) or other markers such as ATP with luminescent, fluorescent, or colorimetric readouts can be used to assess ADCC. However, signals from effector cells or spontaneous death of target cells increases the assay background. Use of a target cell line constitutively expressing a marker, such as luciferase, is an attractive option as well, because it permits a homogeneous assay format.
Engineered effector cell lines can be employed instead of primary cells collected from human donors. The use of engineered cells significantly decreases assay variability and provides a reliable and unlimited source of a critical reagent, which enables high throughput; this makes these assays more amenable to execution for product release and stability testing. Although these types of cells have been accepted for release and stability purposes, it is important to understand how the data generated using engineered cell lines compare to data from the more biologically relevant primary cells.
The final results from an ADCC assay can be presented in terms of potency relative to a reference standard. However, the calculation of percentage cytotoxicity should also be performed during assay development and qualification in order to understand what percentage of the cell population is actually being killed. The calculation is done according to the formula:
% Cytotoxicity = [(Experimental lysis − Spontaneous lysis)/(Maximal lysis − Spontaneous lysis)] × 100
“Spontaneous lysis” in this case is the lysis that occurs in the absence of antibody but in the presence of effector cells.
The use of reporter gene assays as a format is also an option; for example, those assays that use engineered effector cells expressing FcγRIIIa and a reporter gene with ADCC-specific, MOA-relevant, response elements such as nuclear factor of activated T cells (NFAT). The reporter gene assay reflects the early pathway activation of ADCC as a surrogate for cell death. Pathway activation in these assays can only occur when antibody is bound to both target and effector cells. The assays are usually faster and more precise and have higher signal-to-background ratio, making them very suitable for demonstrating consistency of manufacturing and for batch release of the therapeutic. The assays also allow for an assessment of relative potency, but not cytotoxicity. The reporter gene assays are useful for characterization and comparability needs where ADCC is a presumed MOA, especially because accuracy and precision are better than with ADCC assays using primary effector cells.
3.2 Complement-Dependent Cytotoxicity
CDC represents another mechanism by which a therapeutic monoclonal antibody can destroy target cells. As Figure 3 illustrates, CDC is initiated by the binding of the Fab domain of the antibody to the cell-associated target, and binding of the Fc portion to a plasma C1q protein, which triggers the complement cascade, leading to the formation of the membrane attack complex (MAC) (C5b to C9) at the surface of the target cell, resulting in the killing of the target cell. Given the importance of this activity, a robust and quantitative CDC assay can be an important analytical tool to guide drug process and product development as part of a complete analytical control strategy. Different IgG isotypes have different potential to elicit CDC, based on a number of factors including the inherent affinity to C1q protein. In general, the level of CDC is high for human IgG1 and IgG3 and low for IgG2 and IgG4. The ability of an antibody to mediate CDC is also dependent on the location of the epitope on the target receptor. Binding to a specific epitope can impact the conformational display of the antibody, thereby impacting its interaction with C1q protein. In addition to specific epitopes, the density of the target antigen can influence the level and extent of the CDC activity. In general, cell lines with higher target antigen densities tend to be more sensitive to CDC activity than cell lines with lower target antigen densities.
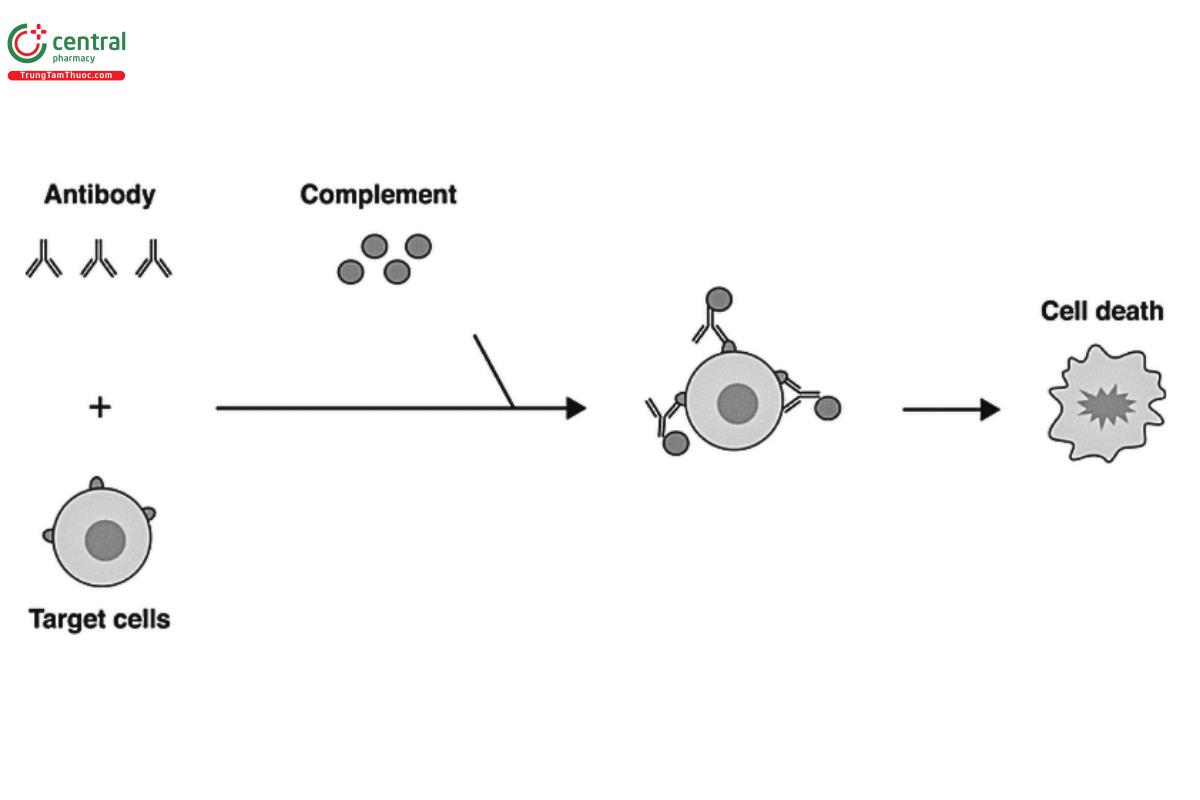
The selected target cell may be a cell that is physiologically representative for the disease indication, or alternatively, a cell line that recombinantly expresses target antigen. However, it might be beneficial to ascertain that target levels in an engineered cell line are representative of target levels in physiologically relevant cells, which can be achieved with techniques such as flow cytometry.
The choice of complement protein is also an important consideration for the development of the CDC assay. Different sources of complement such as rabbit, primate, or human have been used. It is important to note that some cancer cell lines express receptors that inhibit complement activation, thereby masking the potential activity of the therapeutic. This again underscores the importance of the choice of the target cell line as well as the choice of the complement.
The choices of cell killing readout for the development of the CDC assay are numerous and can affect the sensitivity, precision, and robustness of the assay. The choices are similar to those mentioned in the ADCC assay in Antibody-Dependent Cell-Mediated Cytotoxicity. Cell killing also can be assessed by measuring the activity of released enzymes upon cell death, such as LDH or adenylate kinase. This method has the advantage of not preloading the cells and is based on loss of membrane integrity. However, the assay also can be variable because of added steps and manipulations of performing the assay. An alternative assay format is to measure ATP in an assay with luminescent, fluorescent, or colorimetric readouts. Luminescence-based assays measuring ATP levels in particular have become more popular because cell killing can be measured by adding the detection reagent without the need for further manipulation, and the assays are sensitive. In addition, like the ADCC assay in Antibody-Dependent Cell-Mediated Cytotoxicity, a cell line constitutively expressing a marker, such as luciferase, as a surrogate measure of viable cell number also may be employed.
3.3 Antibody-Dependent Cellular Phagocytosis
ADCP is an Fc-dependent, cell-mediated, and innate immunity mechanism in which antibody-opsonized target cells, or in some cases, specific fibrillar proteins, engage the FcγR on the surface of immune effector cells to induce phagocytosis. This causes the internalization and degradation of the target (see Figure 4).
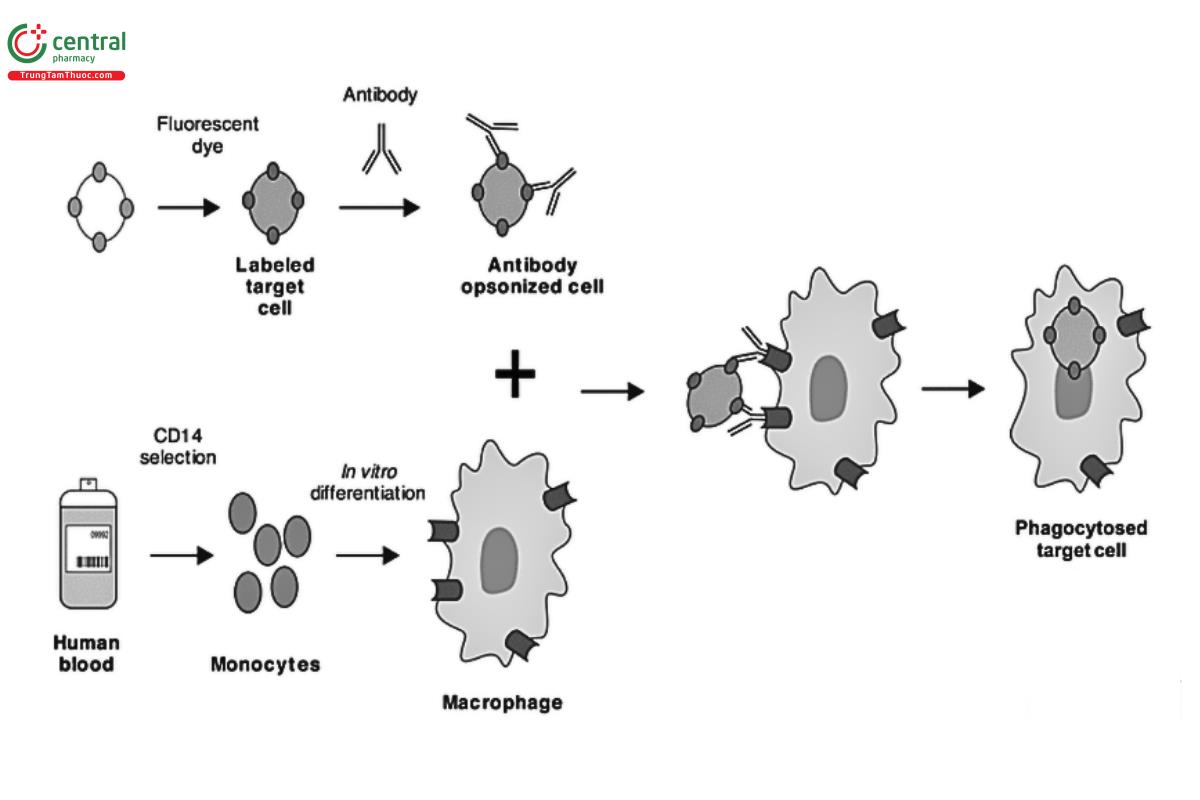
Effector cells capable of mediating ADCP include macrophages, monocytes, neutrophils, and microglia. ADCP assays are generally more complex than ADCC or CDC assays. Part of this complexity is driven by the immune cell types capable of eliciting ADCP. In contrast to ADCC mediated by NK cells expressing FcγRIIIa, the macrophage, monocyte, or neutrophil effector cells involved in ADCP express more than one type of FcγR. The FcγRs that are present and the ratio of their cell surface expression can vary with both cell type and cytokine induction, although FcγRIIa often appears to be the predominant FcγR expressed. In addition, distinct IgG subtypes have different potential to elicit ADCP through the various FcγRs. Fcγ receptor polymorphisms can also affect ADCP activity, notably for IgG2 interactions with FcγRIIa, although also for IgG1 interactions with FcγRIIIa on immune cells capable of ADCP. Furthermore, Fc engineering that enhances effector function may alter the potential contribution in vivo of particular effector cells and their FcγRs on ADCP activity (also true for ADCC). Consideration of these complexities should be used to drive decisions on in vitro ADCP assays to use during antibody drug development.
Traditionally, ADCP assays use primary macrophages as physiologically relevant effector cells. Because macrophages are not a circulating immune cell type, human primary macrophages for ADCP assays must be prepared from blood donor monocytes and differentiated in culture for a week. It is preferable to use differentiation treatments that lead to macrophages representing the desired subtype (i.e., the subtype that most closely represents the specific in vivo situation).
Monocytes also may be used as effector cells to evaluate ADCP activity, but they are less effective at ADCP.
The complexity of primary effector cell preparation, particularly for macrophages, has potential for introducing assay variability. Rigorous QC and development of a robust process of cell preparation, including cell differentiation, are essential. Flow cytometry can be used to evaluate specific markers that represent the desired cells so that their representation in the population is understood and can be used for “go/no go” decisions on use. Expression of specific FcγRs can be confirmed using flow cytometry and quantified using commercially available kits. Flow Cytometry 〈1027〉 provides a thorough review of best practices for flow cytometry and includes references to functional methods for measuring apoptosis, cell viability, and intracellular cytokine expression. Pooling of ADCP effector cells from different donors is not possible because of major histocompatibility (MHC) restrictions, resulting in greater overall potential for assay run-to-run variability.
Several established cell lines, such as monocytic THP-1, pro-monocytic U937, and BV-2 microglia, are used as effector cells for ADCP assay development. Additionally, cell lines such as Chinese hamster ovary (CHO) and Jurkat cells expressing FcγRIIa or other FcR have been used successfully as surrogates for traditional ADCP assays.
An appropriate target cell selection is equally important. The stability of antigen expression level across cell passage can be monitored. Target cell size and shape can also influence ADCP activity. Different physiologically relevant target cells should be analyzed for suitability or bridging to data using engineered cells line(s), as part of assay development. When the target is extracellular fibrils, the preparation of fibrils needs thorough development as well.
ADCP assays typically involve the tracking of fluorescently labeled target cells by either flow cytometry or confocal microscopy. As such, ADCP is more complex than the ADCC assay. For flow cytometry, effector and target cells used in the ADCP assay are made distinguishable through differential fluorescent dye labeling of cell markers specific for each cell type. Two or more fluorochromes can be used simultaneously in flow cytometry if their peak emission wavelengths are not close to each other and are thus detected by different detectors. The amount of fluorescent signal is proportional to the number of fluorochrome-containing molecules associated with specific cells. The ADCP assay involves reaction of effector and target cells incubated with a dose range of antibody. Different conditions can be evaluated including effector:target cell ratio, assay duration, and antibody dose range. An antibody isotype control should be included as a negative control.
A subsequent wash step is performed and then flow cytometry is used to evaluate percentage of target cells phagocytosed. A uniform population of effector cells, such as an established cell line, simplifies the assay significantly. For example, percentage of labeled cells that are doubly stained compared with total target cells that are singly stained could represent the percentage of target cells phagocytosed. Analysis involves identification and quantification of the different cells as well as gating, which is setting a numerical or graphical boundary to analyze a subset of data. The subset could be target cells alone or cells positive for specific markers of both effector cells and target cells.
Forward and side scatter of cells is often used as an initial gating process to remove debris from the analysis before focusing on gating based on specific, fluorescently labeled markers.
As an alternative to using target cells in ADCP assays, some investigators have used fluorescent beads coated with antigen or fluorescently labeled fibrils. These beads or fibrils are incubated with antibodies while the immune complexes are incubated with effector cells. Effector cells then can be analyzed for fluorescent bead or fibril uptake by using flow cytometry, and ADCP activity also can be evaluated; however, antigen-coated beads may not accurately reflect target cells. It is important to understand how the data generated from this approach compare to data from physiologically relevant target cells.
Because flow cytometry using specific labeling of external markers of target and effector cells actually represents evidence of cell surface association rather than proof of phagocytosis, it is advisable to confirm phagocytosis. This can be done by using a pH-sensitive fluorescent dye, available commercially, to label target cells and thereby detect their presence in the acidic environment of the phagocyte lysosomes. Alternatively, target cell engulfment can be confirmed with confocal fluorescence microscopy or microscopy-based live-cell imaging, including time lapse, to evaluate ADCP by live cells. Effector cells also can be treated with trypsin for removal of bound but non-internalized fluorescence. Live-cell digital imaging equipment and associated software have advanced to a level capable of quantification of ADCP dose responses; this is in addition to qualitative assessment using fluorescent, pH-sensitive or other dyes, and provides a viable, high-quality alternative to flow cytometry analysis. Some additional options of these systems include capability for environmental control for cells in assay plates in the chambers where the cells are digitally imaged. This process also allows cell responses to be evaluated at different times during the assay reaction. For this type of analysis, appropriate balancing of imaging and cell needs is important.
Reporter gene assay formats are also an option (e.g., assays that use engineered effector cells expressing a relevant FcγR and a reporter gene with ADCP-specific, MOA-relevant, response elements such as NFAT). The reporter gene assay reflects the early pathway activation of ADCP as a surrogate for cellular phagocytosis. Pathway activation in these assays can only occur when antibodies are bound to both target and effector cells. Different engineered effector cells expressing FcγRIIa, FcγRIIIa, or FcγRI allow evaluation of the potential impact of factors such as manufacturing changes on different FcγRs involved in ADCP. The assays are usually faster, more precise and accurate, and have higher signal-to-background ratio, making them very suitable for demonstrating consistency of manufacturing and for characterization, for example. They allow for an assessment of relative potency, but not phagocytosis. Comparison to data generated from assays measuring actual cellular phagocytosis is recommended, and because the correlation may differ between particular therapeutics, the correlation should be investigated for each one.
3.4 Fc-Dependent Apoptosis
Apoptosis, or programmed cell death, is the genetically controlled destruction of cells that occurs as a normal part of an organism's growth or development. Apoptosis is characterized by nuclear condensation, cell shrinkage, membrane blebbing, and chromosomal DNA fragmentation. Fc-dependent apoptosis is one of the Fc effector functions in which ligation of certain cell surface molecules by antibodies in the presence of cross-linking antibodies or FcγR-expressing cells initiates signal transduction events that lead to apoptosis of the cells.
Fc-dependent apoptosis has been implicated as a potential MOA for several therapeutic antibodies. As Figure 5 illustrates, the induction of Fc-dependent apoptosis in vivo depends on specific target molecules capable of activating apoptosis pathways and requires cross-linking of the Fc regions by FcγRs on NK cells and other effector cells such as neutrophils and monocytes. Depending on the effector cells involved, Fc-dependent apoptosis may be induced via cross-linking by any of the subclasses of FcγRs (i.e., I, IIa, IIb, IIIa, IIIb). Factors affecting FcγR binding activity, such as antibody isotypes and glycoforms, would also impact Fc-dependent apoptosis. Additionally, the degree of Fc-dependent apoptosis is positively influenced by the expression level of the antigen.
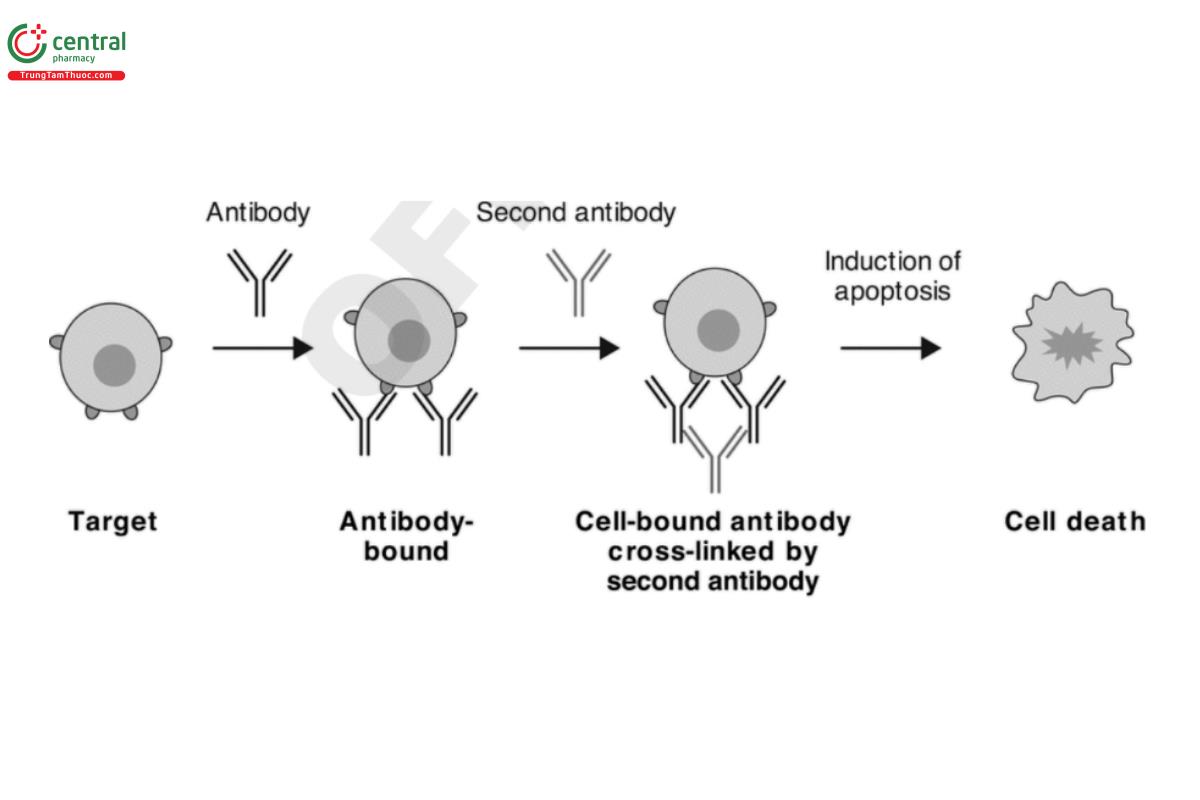
Design of in vitro Fc-dependent apoptosis assays involves selection of relevant target cells and cross-linking agents. Considerations for target cells are similar to those for ADCC and ADCP. Common cross-linking agents include anti-Fc antibodies and protein A. Note that the optimum composition of antibody and cross-linking agents in the assay should be determined empirically. Apoptosis of target cells can be assessed by detection of chromatin condensation, DNA strand breaks or fragmentation with various nucleic acid stains, changes in the permeability of cell membranes or mitochondrial membrane potential by fluorescent probes, translocation of anionic phospholipid Phosphatidylserine with fluorescent Annexin V conjugates, and/or activation of caspase enzymes by caspase activity assays. As some of these tools measure events that may not always lead to apoptosis, their selection should be justified based on knowledge of the apoptosis pathways in the specific target cell.
4 BINDING ASSAY FORMATS
Cell-Based Functional Assays provided information on the biological pathways and associated proteins that are critical to measure these mechanisms of action. In contrast, this section is structured to describe the most commonly used binding assay formats and link the various tools used to the biology described in Cell-Based Functional Assays. Cell-based binding assay formats are not used as commonly as non-cell-based assays. A synopsis of the more commonly used formats for cell-based binding assays follows the more detailed descriptions of the non-cell-based binding assays.
Table 3 summarizes the commonly used non-cell-based binding assay formats and cell-based binding formats. Each of these assays is then described in more detail.
Table 3. Commonly Used Binding Assay Formats
| Assay Format | Merits/Limitations | Points to Consider |
| Enzyme-linked immunosorbent assay (ELISA) |
|
|
| Surface plasmon resonance (SPR), biolayer interferometry (BLI) |
|
|
| Kinetic exclusion |
|
|
| Bead-based proximity assay format |
|
|
| Fluorescence resonance energy transfer |
|
|
| Cell-based binding assays |
|
|
For all of these assay formats, the reagents used in the assay are critical to the assay performance. In some cases, specific recombinant proteins will need to be obtained, whereas in other instances, vendor-supplied secondary antibodies, detection reagents, and specialized plates or binding surfaces are required.
Because of the large number of assay formats discussed in the following sections, each one is structured in a manner that introduces the technique and then discusses the advantages, disadvantages, and points to consider.
4.1 Non-Cell-Based Binding Assays
ELISA
The enzyme-linked immunosorbent assay (ELISA) is the most common type of binding assay. This non-cell-based assay format is flexible, and many different formats exist. Immunological Test Methods—Enzyme-Linked Immunosorbent Assay (ELISA) 〈1103〉 provides information on the various formats. Different ELISA assays have been developed and used to evaluate Fc-containing protein binding to C1q and to various FcγRs, and to evaluate pH-dependent binding to FcRn.
Advantages: High throughput, automatable, standard equipment, economical, easily transferred.
Disadvantages: Multiple wash steps, lengthy, requires plate precoating, may be sensitive to the presence of aggregates in the samples depending on the assay format. The presence of aggregates may complicate the interpretation of results, especially for IgG2, IgG4, and FcRn binding. In addition, the ELISA format may have binding limitations, particularly when the binding interactions are weak (e.g., FcγRII and FcγRIII). In these cases, consider cross-linking to improve sensitivity (refer to 〈1103〉).
Points to consider: Critical reagent quality, inventory and concentration, affinity of interaction being measured.
SURFACE PLASMON RESONANCE
Surface plasmon resonance (SPR) technology can be used for label-free, real-time detection and characterization of ligand binding and dissociation in FcR and Fc-containing protein interactions. Association rate (kₐ) and dissociation rate (k_d) constants, and the equilibrium dissociation constant or affinity constant (Kᴅ = k_d/kₐ) can be determined. Typical systems use microfluidics flow cells. One of the interacting proteins is immobilized, either directly or indirectly via capture, as the ligand on a biosensor chip surface within the microfluidics system. Typically, the other protein partner, as an analyte, is then flowed in a solution over the surface of the biosensor chip with immobilized ligand to a desired association level to measure association; then a subsequent dissociation step follows with buffer alone. Association and dissociation of FcR with Fc-containing protein can be measured through change in SPR or SPR response units (RUs), which, in turn, reflect a change in mass at the sensor chip surface reflective of binding.
The technique is sensitive, and different formats and conditions can be used to obtain data under specific conditions. For interactions where a steady state equilibrium between association and dissociation cannot be reached in a reasonable time, but kinetic information exists, a kinetic analysis to obtain kₐ and k_d allows the affinity constant Kᴅ to be determined. When the kinetics of association and dissociation are very fast and a steady state can be reached in a reasonable time, a steady state equilibrium analysis can be used to determine Kᴅ. Different fit models of the binding data are used to determine further information about the binding interaction (e.g., best fit to a 1:1 binding model), as well as to obtain values for the desired binding parameters. A more extensive discussion of SPR as an immunological test method is provided in Immunological Test Methods—Surface Plasmon Resonance 〈1105〉, which provides guidance on practical aspects of methods, controls, and analyses, including those pertinent to obtaining accurate information on kₐ, k_d and Kᴅ, as well as how to avoid pitfalls.
Advantages: No labeling required; provides more precise kinetic data; can measure a relatively wide range of affinities; can automate sample preparation; data analysis is straightforward; and can use with a reference material to express relative Kᴅ.
Disadvantages: Lower throughput, specialized equipment needed, harder to transfer and validate, and requires greater technical expertise than some other non-cell-based methods.
Points to consider: Technology requires special expertise and careful assessment of the quality of the critical reagent quality.
The generation of a valid and useful SPR binding data set for an FcR and Fc-containing protein interaction can be generally described as being dependent on three major factors: the quality of the FcγR or FcRn and Fc-containing reagents, the experimental design format, and data analysis.
More specifically, for FcRn, binding occurs at somewhat acidic pH (approximately pH 6) in the endosome after the Fc-containing therapeutic, such as a monoclonal antibody, is pinocytosed at the cell surface and associates with FcRn when released into the endosome. Typically, in SPR-based FcRn assays, binding and dissociation at pH 6 are analyzed to obtain the equilibrium dissociation constant or
affinity, Kᴅ, at this pH. Additionally, binding at pH 6 and dissociation at a neutral pH, such as pH 7.2–7.4, can be performed to obtain an understanding of dissociation at the neutral pH; dissociation at neutral pH is essential for recycling of the antibody in vivo through release again at the cell surface. Antibody engineering to improve binding to FcRn, as a component potentially contributing to improved pharmacokinetics (PK), may alter the ability of FcRn to release antibody at neutral pH, so evaluating dissociation under neutral pH buffer flow can provide important information in such work. For Fc-containing proteins directed at soluble targets, evaluating FcRn binding of the protein in the presence of its soluble target also should be considered.
FcRn protein should preferably comprise the soluble extracellular domain of the heavy chain (α-chain) of FcRn and associated beta-2 microglobulin (β2M) because the β2M is important for efficient pH-dependent IgG binding and reflects physiological binding more closely. Recombinant FcRn heterodimer is available commercially or can be produced to obtain better control over quality, consistency, and supply. Mammalian cell expression is best to maximize proper protein folding and post-translational modifications. The soluble extracellular domain of FcRn with associated, species-specific β2M that is copurified can be used, or a fusion protein of the FcRn extracellular domain with β2M is also possible. Alternatively, the heavy chain (α-chain) of FcRn can be, and often is, used alone in comparative SPR binding studies of Fc-containing molecules, although IgG binding is decreased compared with that of native FcRn heterodimer.
For FcγRs (other than to FcγRI) binding of Fc-containing proteins such as antibodies is a low-affinity interaction. To obtain suitable results, a higher concentration of analyte than usual may need to be flowed over the chip surface to which the binding partner is bound.
For all FcRs, thorough characterization and high and consistent functional quality of protein is important for an accurate and reproducible assay. For example, it is important in obtaining kₐ in kinetic analysis because concentration is a factor of kₐ and the concentration should reflect that of functionally active protein.
Consideration should be given to whether to immobilize the FcR or the Fc-containing protein to the surface of the SPR chip since both formats are possible. Different biosensor-prepared surfaces are available commercially. Some chip surfaces can slow association of the specific binding partners so that mass-transport limitations or steric hindrance are avoided in order to obtain accurate results. A low density of ligand on the chip surface is desirable for the same reasons. Direct or indirect ligand immobilization is possible. Overall, surface preparation that avoids avidity artifacts and achieves relatively low-level ligand binding under gentle immobilization to avoid mass-transport-limited conditions and to direct simple analyte binding may best reflect conditions on the cell.
Indirect immobilization of ligand is preferred because it is gentler. Examples include amine-immobilized neutravidin to capture biotinylated IgG ligand or immobilized antigen to capture IgG ligand. Direct immobilization formats, such as amine coupling of the ligand directly to the chip, can be considered, but with caution, because this type of direct immobilization through multiple sites throughout the protein has the potential to alter protein structure and binding characteristics. Outputs of SPR analyses that signify achievement of appropriate binding conditions include good fitting to simple binding models and low and randomly scattered residuals around the fitted data. Prior determination of the concentration of active analyte being tested against a specific immobilized partner allows more accurate input of values toward binding parameter determination.
Biolayer interferometry
Biolayer interferometry (BLI) is another label-free, real-time detection technology used to detect intermolecular interactions. The technology is suitable for measuring FcR and Fc-containing protein interactions. It is an optical analytical method that measures interference pattern changes of light wavelengths (optical interferometry) and is dependent on a fiber-optic biosensor. When target molecules bind to or dissociate from the surface of the biosensor to which the binding partner is bound, a spectral shift occurs to a degree proportional to the number of in-solution binding partner molecules bound to the biosensor surface. The change is monitored in real time and can provide kinetic data or relative Kᴅ data on molecular interactions between the two binding partners. BLI also can be used to obtain concentrations of molecules.
Advantages: No labeling; high throughput; can provide kinetic and quantitation data; semi-automatable.
Disadvantages: Specialized equipment and biosensors needed; more difficult to transfer.
Points to consider: Selection of the appropriate biosensor and assay orientation (i.e., which molecule to immobilize) is important. The affinity of FcγR–IgG interactions is often relatively low, requiring that high concentrations of analyte be used. An excess of ligand molecules on the biosensors can lead to data artifacts due to crowding, steric hindrance, avidity, mass-transport effects, and non-specific binding, while a low loading density could result in poor signal-to-noise ratio. The buffer conditions should account for optimal binding yet reduce as much as possible the non-specific binding. Length of the association step should be considered because shorter times may prevent secondary interactions.
Kinetic exclusion assay
The kinetic exclusion assay is a solution equilibrium/exclusion-based method. It is particularly sensitive in determining Kᴅ in the pM to low nM range, but low-affinity interactions are more of a challenge because of sample volume and/or concentration needs. The kinetic exclusion assay can also determine concentrations of interacting binding partners. In the assay, one binding partner is held constant in concentration, the other is titrated into the solution, and the binding is allowed to come to equilibrium. For antibody–antigen interactions, the antibody is normally titrated in. The solution is then rapidly passed into a flow cell. The flow cell contains antigen-coated beads that bind any free antibody, but antigen-bound antibodies in the solution do not have time to dissociate; they are kinetically excluded from binding to the bead. The free antibody bound to the beads is subsequently detected with fluorescently labeled secondary antibodies of appropriate specificity; they are then quantified, giving a measure of free antibody remaining at equilibrium. This value then can be used to determine binding parameters.
Advantages: Can measure high-affinity interactions and a broad range of affinities; measurements can be made with unpurified mixtures and unmodified proteins; can measure concentrations and binding beyond the limits of popular surface-based methods (e.g., very slow dissociation).
Disadvantages: Low to medium throughput; possible non-specific binding to beads; less useful for weaker binding interactions such as some FcγR interactions with antibodies; may not reflect physiological binding if physiological binding is not in solution in vivo; specialized equipment needed.
Points to consider: Whether the binding partners normally bind in solution; orientation of the assay (i.e., which binding partner is captured in the flow cell); whether a binding partner is available only in an unpurified state.
Bead-based proximity assay
Bead-based assay technologies are now widely used to study biomolecular interactions in a microplate format. These technologies are non-radioactive, homogeneous proximity assays. Binding of molecules captured on the beads leads to an energy transfer from one bead to another, ultimately producing a luminescent/fluorescent signal. The technology allows the detection of molecules in a wide range of sample matrices. Almost any sandwich assay can be developed to detect an analyte of interest. An example FcR assay may have the following competitive assay format. An Fc or Fc-containing therapeutic can be biotinylated and coupled to streptavidin donor beads, while a recombinant histidine-tagged FcR (FcR-His) molecule is conjugated to acceptor beads. In the absence of an unlabeled Fc-containing sample, the beads come into close proximity and a signal (e.g., luminescence) is generated. However, when a different Fc-containing moiety (i.e., test article) is added into the reaction, the Fc region competes with the Fc-biotin reagent in solution at pH 6.0 for binding to FcR-His. The assay would measure the dose-dependent decrease in signal when an Fc-containing test article is added to a reaction containing FcR-His and Fc-biotin. Test sample activity would be determined by comparing the test sample response to the response obtained for the reference standard. Results would be reported as percent relative binding values.
Advantages: High throughput; homogeneous; suitable for lower-affinity interaction; high signal-to-noise ratio; easily automatable; transferable.
Disadvantages: Expensive reagents; requires labeling and relatively expensive plate reader; more sensitive to the presence of aggregates in the samples due to avidity effects.
Points to consider: A number of variables should be considered during the development and optimization of the assay. The concentration of the FcR and Fc-containing molecules should be varied to produce the most desirable dose-response curve. Bead-coupling ratios should be optimized based on the amount of each binding partner used in the assay.
Fluorescence resonance energy transfer
Fluorescence resonance energy transfer (FRET) is based on the transfer of energy between two fluorophores, a donor and an acceptor, when in close proximity. Molecular interactions between biomolecules can be assessed by coupling each partner with a fluorescent label and by detecting the level of energy transfer. A donor–acceptor complex can be detected without the need for physical separation from the unbound partners, allowing for fully homogeneous assays that do not require separation steps such as centrifuging, washing, or filtration. However, traditional FRET is affected by fluorescence from sample components such as buffers, proteins, chemical compounds, and cell lysate. This type of background fluorescence is extremely transient and therefore can be eliminated using time-resolved methodologies. Homogeneous time-resolved fluorescence (HTRF) combines standard FRET technology with time-resolved measurement of fluorescence, eliminating short-lived background fluorescence. Introducing a time delay of approximately 50–150 µs between the system excitation and fluorescence measurement allows the signal to be cleared of all non-specific, short-lived emissions. In contrast, HTRF acceptors emit long-lived fluorescence when engaged in a FRET process. Therefore, long-lived emissions signify energy transfer due to the proximity of the labeled biomolecules. Also, FRET is governed by the physics of molecular proximity, which only allows this phenomenon to occur when the distance between the donor and the acceptor is short enough. In practice, FRET systems are characterized by the Förster radius (R₀) distance at which FRET efficiency is 50%. For HTRF, R₀ lies between 50 and 90 Å, depending on the acceptor used.
Advantages: High throughput, homogeneous, fast, wide assay response window, easily automatable, transferable.
Disadvantages: Does not work well for lower-affinity interaction; requires labeling and specialized plate reader.
Points to consider: Distance between binding partners is smaller than that for bead-based proximity assays.
4.2 Cell-Based Binding Assays
This format can be a suitable choice when a recombinant reagent such as a receptor extracellular domain (ECD) is not commercially available or when it is challenging to produce the recombinant reagent in sufficient amounts. Two types of cell-based binding assays can be established, as shown in Table 4. A cell-based binding assay is often preferred for measurement of FcRn binding because of the structure of the FcRn. As such, this assay format is discussed in the context of an FcRn binding assay.
Table 4. Cell-Based Binding Assay Formats
| Type | Platform |
| Direct assay | Conventional — Plate-based or flow cytometry |
| Competitive assay | Nonhomogeneous — Plate-based |
| Competitive assay | Homogeneous — Flow cytometry, HTRF |
Advantages: Arguably more physiologically relevant because receptors are presented in a membrane environment; no need to purify receptors; standard equipment; medium-to-high throughput.
Disadvantages: Need to express receptors and maintain cell lines for a longer time. In general more difficult to automate and transfer than non-cell-based methods. Levels of receptors may lead to avidity artifacts for high-molecular-weight aggregates. Cell detachment can be a challenge; wash steps must be gentle to reduce cell detachment from well bottoms, which may be partially mitigated by use of round-bottom well plates and centrifugation. Potentially stronger plate effects; multiple wash steps; lengthy; requires precoating of plates with cells. In addition, cell licensing fees may be a consideration.
Points to consider: The following points apply for FcRn cell-based assays. A cell-based assay in which the Fc region of IgG binds to the cell surface expressed FcRn complex may be more indicative of physiology where the FcRn is present as a membrane-bound form and the IgG floats freely as a soluble form. Therefore, this model system may be more appropriate to measure Fc-FcRn interactions. Typically, binding of Fc to FcRn takes place under mildly acidic conditions (pH 6–6.5) and releases the IgG at neutral to basic pH (7–7.4).
5 CRITICAL REAGENTS
The validity of testing results for Fc functionality is highly dependent on the quality of the critical reagents used in these assays. Just as the purity and glycan composition can impact the effector function activity of an Fc-containing therapeutic, the composition of the reagent with which they interact is equally important. As highlighted in Cell-Based Functional Assays, it is important that purified FcRs utilized in binding based assays represent the appropriate genetic polymorphisms in that receptor. For cell-based assays, the cells themselves are considered critical reagents. Specifically, it is important to characterize receptor densities and changes to receptor expression on cells used in ADCC, ADCP, and CDC assays because these changes can affect assay sensitivity. Implementation of critical reagent standards is a compelling idea that would permit standardization of the evaluation of the Fc domain across a multitude of therapeutics.
As with most potency assays, Fc-mediated effector function assays require product reference standards that are well-characterized and controlled. Reference standards are generally derived from a representative product batch. It is important to note that glycosylation and glycan patterns need to be well-characterized in reference standards because these attributes affect Fc functionality. It is not uncommon to observe statistically significant differences in the levels of glycan structures when comparing different batches. As such, the choice of a batch as a reference standard becomes important. One approach that can be considered is to select a batch that represents the midpoint of process variability as the reference standard batch.
5.1 Complement
Serum, complement, and cells are important critical reagents for CDC assays. Assays use commercially available, non-heat-treated serum complement fractions. Assays also incorporate sources of complement that include human, rabbit, guinea pig, and mouse. It is important to note that the lot-to-lot variability of complement batches can complicate results. This can be mitigated by the purchase and qualification of a large lot size that can last a few years based on the testing requirements of the lab.
5.2 Recombinant Fc Receptors
Many FcRs are commercially available or can be manufactured in-house or by an external contract manufacturing organization (CMO). The FcR heterodimerizes with the invariant gamma chain for functional activity, or in the case of FcγRII, the gamma chain is contiguous with the FcR region. However, because the gamma chain is not involved in any direct interactions with the Fc region, the FcR component of the heterodimer is sufficient for direct interaction studies. If FcRs are purchased as a catalog item from an external vendor, it is important to ascertain lot-to-lot variability of the materials purchased. This can be mitigated by the purchase of a large lot size that can last several months to years based on the testing requirements of the lab. The host cell line is an important consideration because glycosylation of some FcRs, such as FcγRIIIa, can affect binding to Fc regions.
5.3 Quality of Reagents
The quality and consistency of FcR critical reagents used in Fc function assays should be ensured by using biochemical analytical techniques to assess purity, homogeneity, stability, lot-to-lot variability, and desired functional properties such as pH specificity of binding for FcRn. These analyses help ensure proper protein folding necessary for FcR binding to Fc-containing protein. If the reagents were purchased from vendors, this information is still important to collect and to supplement what is provided by the vendor. It is important to understand the best design for engineered proteins such as FcRn alpha chain and co-expressed β2M. For the Fc-containing therapeutic protein, aggregation and other modifications (e.g., specific Methionine oxidations in the Fc region known to affect FcRn binding or levels of other post-translational modifications such as N-glycans known to affect FcγR binding) also should be assessed prior to assay so that this information can be used to help interpret results.
The quality of critical cell reagents should be ensured through assessment of stability, lot-to-lot variability, and consistent, desired functional properties. Stability of the cell line should be established during several passages to ensure a functional assay and the ability to create cell banks. Stability can be evaluated using flow cytometry to evaluate, for example, receptor expression, and also by evaluating consistency of assay response. If cells require a pretreatment before an assay, then the consistency of response to this pretreatment also needs to be established.
5.4 Qualification of Reagents
For the qualification of reagents, appropriate criteria need to be developed that are based not only on passing the method acceptance criteria but also on consistency and linkage to data generated from previous batches. Careful qualification is essential to preventing drift in the assay response when changing batches of critical reagent. The stability of FcR and FcRn proteins needs to be adequately monitored during long-term storage.
6 ASSAY VALIDATION
The fundamentals of potency assay validation, including key method parameters to be evaluated, are outlined in 〈1033〉. Use of a validated assay is a requirement for assays that support product lot release and stability testing. Characterization assays are not subject to current good manufacturing practices (cGMPs) or good laboratory practices (GLPs). However, potency assay validation principles should be considered to demonstrate the method performance characteristics and the fitness for use of the assay results.
7 DATA ANALYSIS AND REPORTING
Data analysis and the reporting of results are important outputs from each assay. Fundamental aspects of these are described in 〈1032〉, Analysis of Biological Assays 〈1034〉, and Design and Analysis of Biological Assays 〈111〉. For lot release and stability testing, the convention is to use “relative potency” or “percent relative potency” for the reporting of results. With this approach, a test sample response is compared to the response obtained with a reference standard. As described in 〈1032〉 and 〈1034〉, the central tenet of this approach is the assumption that the same active ingredient is present in both materials. Only after parallelism, or similarity, has been established for the sample response relative to the reference standard response is it permissible to compute the relative potency result. For non-cell-based binding assay formats such as SPR, BLI, and kinetic exclusion, a number of strategies can be used for determining activity, such as reporting the absolute affinity constants, Kᴅ values, kinetic measurements, or relative binding.
A key consideration for the biological characterization of Fc effector function is post-translational modifications and their impact on activity. Following the modeling of the response, consideration should be given to the comparison of the dose-response curves for the reference standard and the test sample(s) as well as the impact of certain attributes, such as glycans, on those responses. In some cases, the use of relative potency as a result output may not be warranted.
8 REFERENCES
Noubia Abdiche Y, et al. The neonatal Fc receptor (FcRn) binds independently to both sites of the IgG homodimer with identical affinity.
MAbs 2015;7(2):331–343.
(USP 1-Dec-2020)
