Những vấn đề có thể bạn chưa biết về u thần kinh nội tiết
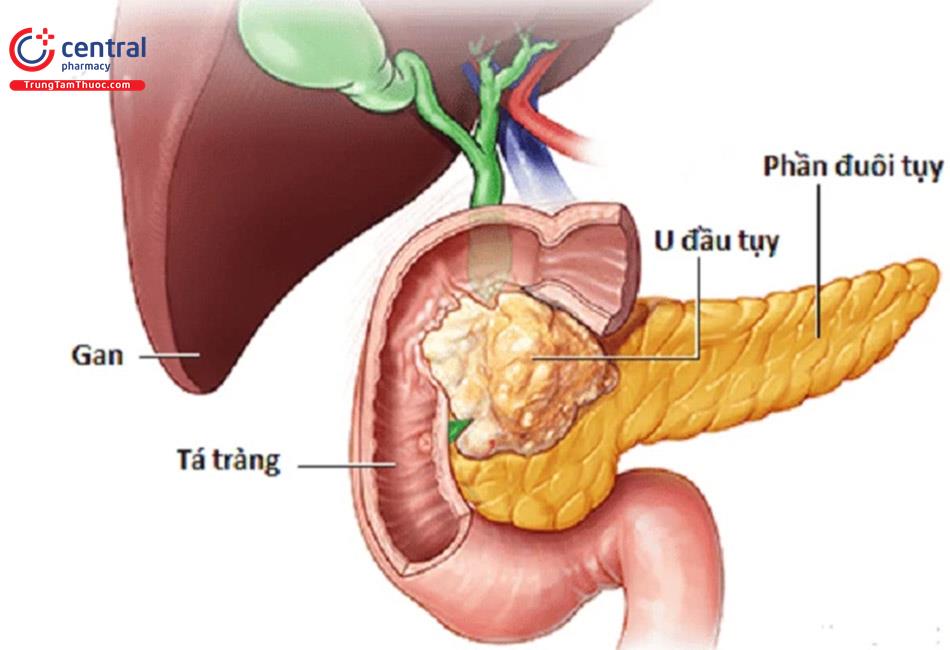
1 NGUYÊN LÝ CHUNG
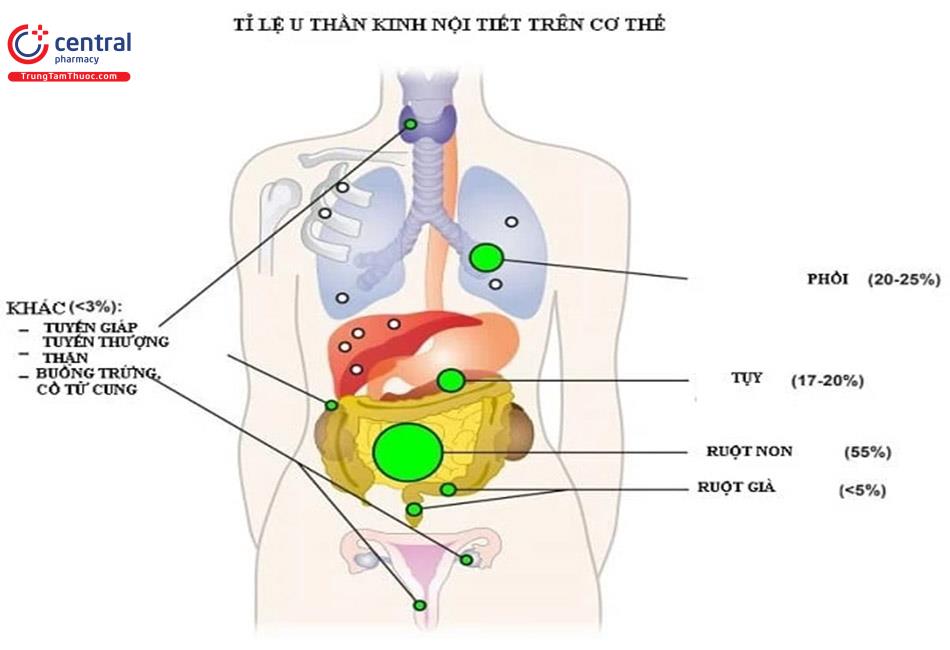
Các u thần kinh nội tiết (NET) là tập hợp không đồng nhất những khối u hiếm gặp xuất phát từ biểu mô nội tiết lan tỏa được phát hiện thấy ở hầu hết các cơ quan trong cơ thể. Phần lớn những khối u này có nguồn gốc từ ruột của bào thai, bao gồm tụy và hệ ruột nhưng cũng có thể từ phổi, buồng trứng và tuyến tiền liệt [1].
Đại đa số các u thần kinh nội tiết biệt hóa cao có đặc điểm mô học của sản xuất hormon nhưng chỉ một số rất ít các trường hợp có dấu hiệu lâm sàng rõ ràng của quá tiết hormon.
Hội chứng Carcinoid ám chỉ một chùm các triệu chứng có liên quan đến sự giải phóng có tính hệ thống các chất giãn mạch và hormon từ NET và nó chỉ chiếm dưới 10% tổng số bệnh nhân u thần kinh nội tiết.
Các u thần kinh nội tiết thường được phát hiện muộn và hầu hết đã có di căn tại thời điểm được chẩn đoán. Điều trị bằng nhiều phương pháp, bao gồm phẫu thuật loại bỏ khối u và điều trị nội khoa các triệu chứng, nếu có.
==> Bạn đọc có thể tham khảo thêm: Tổng Quan Hội Chứng Bệnh Lý U Tân Sinh Đa Tuyến Nội Tiết
1.1 Phân loại
1.1.1 Lịch sử
Thuật ngữ carcinoid (karzinoide) được Oberndorfer sử dụng lần đầu vào năm 1907 để mô tả các khối u ở ruột có mô bệnh học giống như ung thư nhưng không ác tính bằng. Sau đó vài năm, người ta nhận thấy các khối u này có những đặc điểm liên quan đến nội tiết và đồng thời phát hiện có các khối u tương tự nhưng nằm ngoài đường tiêu hóa.
Ngày nay chúng ta biết rõ những khối u này có nguồn gốc biểu mô nhưng có các đặc điểm của cả tế bào biểu mô và tế bào thần kinh. Sơ đồ phân loại hiện tại sử dụng thuật ngữ “Nội tiết thần kinh” thay cho carcinoid. Tuy nhiên “carcinoid” vẫn được tiếp tục sử dụng thay thế cho nhau trong y văn với những u nội tiết thần kinh biệt hóa cao.
1.1.2 Phân loại giải phẫu bệnh học các u thần kinh nội tiết
Các tiêu chuẩn giải phẫu bệnh học theo Tổ chức Y tế Thế giới (WHO) khuyến cáo sử dụng khung các khối u biểu mô khác, có nhấn mạnh đến mức độ biệt hóa và bậc tế bào [2]. Tiêu chuẩn của khối u thần kinh nội tiết tiêu hóa ruột tụy (gastroenteropancreatic - GEP) được trình bày trong Bảng 38.1 [2]. Những khối u xuất phát từ các mô khác, ví dụ phổi, có định nghĩa hơi khác biệt.
Các khối u thần kinh nội tiết biệt hóa cao có mô học tương tự với mô thần kinh nội tiết không tăng sinh như các tế bào tròn nhỏ với nhân và bào tương đồng nhất. Đặc điểm của nó là các marker thần kinh nội tiết chromogranin A (CgA) và Synaptophysin bắt màu mạnh khi nhuộm miễn dịch. Trên kính hiển vi điện tử thấy các hạt tiết có màng bao bọc chứa nhiều loại amin và hormon sinh học.
Các khối u thần kinh nội tiết biệt hóa kém có đặc điểm là có ít hạt tiết và các marker thần kinh nội tiết không hoặc bắt màu kém khi nhuộm miễn dịch. Nó sẽ phân bào nhanh hơn và gây tử vong nhiều hơn so với u biệt hóa cao.
1.1.3 Dịch tễ học
Theo dữ liệu từ Khảo sát dịch tễ và kết quả điều trị cuối (SEER), tỷ lệ mới mắc u thần kinh nội tiết là 5/100.000 người [3]. Con số ước tính này có thể không đầy đủ vì bỏ sót các trường hợp lành tính hoặc không xâm lấn, vốn không được báo cáo trong khảo sát.
Các yếu tố nguy cơ bao gồm hội chứng khối u di truyền (được thảo luận sau), giới nữ và người Mỹ gốc Phi [4,5].
Tỷ lệ mới mắc u thần kinh nội tiết ở Mỹ và trên toàn thế giới đã tăng trong vòng 30 năm qua. Nhiều nghiên cứu ở các quốc gia châu Á thấy tỷ lệ mắc khối u nguyên phát khác biệt so với các quốc gia phương Tây [6-8].
Tỷ lệ tăng nhiều nhất trong đa số các nghiên cứu có kiểm tra khối u trực tràng, nó phản ánh tỷ lệ mới mắc được phát hiện tình cờ trong các chương trình sàng lọc bằng nội soi.
| Bảng 38.1. Phân độ khối u thần kinh nội tiết ở dạ dày ruột - tuy (GEP - NET) | |||
| Biệt hoá | Độ | Tỷ lệ tăng sinh | Tên |
| Biệt hóa cao | G1 (độ thấp) | < 2 mitoses/10 HPF và < 3% Ki67 index. | U thần kinh nội tiết |
| G2 (độ trung gian) | 2 - 20 mitoses/10 HPF hoặc 3 - 20% Ki67 index. | U thần kinh nội tiết | |
| Biệt hoá thấp | G3 (độ cao) | > 20 mitoses/10 HPF hoặc > 20% Ki67 index. | Ung thư thần kinh nội tiết (typ tế bào lớn hoặc typ tế bào nhỏ). |
HPF: high-power field Phóng theo Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: A review of nomenclature, grading, and staging systems. Pancreas 2010;39:707-712. | |||
1.2 Cơ chế bệnh sinh
1.2.1 Di truyền phân tử
Đa số các u thần kinh nội tiết là tản phát nhưng có khoảng một phần ba các u thần kinh nội tiết tụy xuất hiện ở những bệnh nhân có đột biến dòng mầm gây ra hội chứng MEN1, von Hipple-Lindau hoặc hội chứng xơ cứng củ. Chẩn đoán u thần kinh nội tiết tụy nhất thiết phải đánh giá các hội chứng này.
Giải trình tự toàn bộ exome cho thấy các khối u thần kinh nội tiết có ít đột biến soma hơn hầu hết các khối u đặc khác. Các nghiên cứu gần đây gợi ý rằng các đột biến dòng mầm vốn ít được để ý trước đây như MUTYH, CHEK1 và BRCA2 được biểu lộ quá mức trong u thần kinh nội tiết tụy [9].
Các đột biến hoạt hóa mục tiêu cơ học của con đường rapamycin (mTOR) thường có ở các khối u thần kinh nội tiết ở tất cả các vị trí khởi phát. Các con đường phổ biến khác nhắm đến mục tiêu bao gồm chết theo chương trình (DAXX), biến đổi nhiễm sắc (ATRX) trong u thần kinh nội tiết tụy, các con đường tiền sinh ung thư SRC và tín hiệu TGF-B trong u thần kinh nội tiết ruột non [10,11].
1.2.2 Sinh hóa

Các u thần kinh nội tiết chứa các hạt tiết thần kinh có khả năng tổng hợp, dự trữ, giải phóng nhiều loại hormon kinh điển và các chất khác nhu serotonin, histamin, prostaglandin, kallikrein, bradykinin, chất P và nhiều chất khác nữa [12].
Phần lớn các u thần kinh nội tiết là không tiết (không chức năng), nghĩa là sẽ không có bằng chứng lâm sàng về việc quá tiết hormon mặc dù có bằng chứng mô hóa học (histochemical) của sản xuất hormon trưởng thành.
Serotonin (5-hydroxytryptamine (5-HT]) chịu trách nhiệm chính cho các triệu chứng kinh điển của hội chứng carcinoid, bao gồm tiêu chảy, co thắt phế quản và bệnh tim carcinoid. Nguyên nhân gây bốc hỏa không rõ ràng nhưng có sự đóng góp của nhiều chất gồm prostaglandins, kinins, chất P và histamin.
Các bước chính trong chuyển hóa 5-HT bao gồm chuyển tryptophan trong thức ăn thành 5-HT nhờ các tác dụng tiếp nối của tyrosine hydroxylase (TPH) và aromatic L Amino acid decarboyxlase (AAAD) và sự phân hủy 5-HT huyết thanh thành 5-hydroxyindoleacetic acid (5-HIAA) bởi monoamine oxidase.
Một số u thần kinh nội tiết ở Đường tiêu hóa trên không biểu lộ AAAD. Những u này không sản xuất serotonin và do đó không gây ra hội chứng carcinoid kinh điển ngay cả khi có di căn rộng.
Phần lớn các u thần kinh nội tiết, bao gồm cả các u không chức năng, có biểu lộ thụ thể của Somatostatin (SSTRs). Một số phân nhóm, ví dụ u tiết Insulin và u thần kinh nội tiết biệt hóa kém, ít khả năng có biểu lộ mật độ cao SSTR, nó làm giảm hiệu quả của thuốc đồng vận somatostatin (somatostatin analogs - SSAs) trong thăm dò hình ảnh và điều trị cho các phân nhóm này.
2 CHẨN ĐOÁN
2.1 Biểu hiện lâm sàng
Vì đa số các u thần kinh nội tiết là không tiết và tiến triển chậm nên thường được phát hiện tình cờ trong khi phẫu thuật, nội soi hoặc thăm dò hình ảnh vì những chỉ định khác.
Biểu hiện phổ biến nhất là đau bụng, các biểu hiện ít gặp hơn là tắc ruột hoặc chảy máu trực tràng [13].
U thần kinh nội tiết ở ruột non thường di căn đến hạch mạc treo, gây xơ và co thắt mạc treo, với các biểu hiện như là thiếu máu cục bộ hoặc tắc ruột từng đợt.
2.2 Hội chứng Carcinoid và các hormon khác
Hội chứng carcinoid cổ điển (nóng bừng mặt, tiêu chảy, thở khò khè, bệnh tim phải) chỉ gặp ở 5 - 10% các trường hợp u thần kinh nội tiết (Bảng 38.2) [14-17]. Hội chứng này gặp nhiều hơn ở các u tại ruột non so với ở dạ dày - thực quản và đại tràng.
Phổi và gan chuyển hóa nhiều chất do u thần kinh nội tiết tiết ra, do đó sẽ ngăn giải phóng các chất này vào hệ tuần hoàn cho tới khi có di căn. Điều này giải thích tại sao những bệnh nhân u thần kinh nội tiết chỉ có hội chứng carcinoid điển hình khi đã có di căn gan. U thần kinh nội tiết ở phế quản và buồng trứng có thể gây hội chứng carcinoid sớm hơn vì nó tiết trực tiếp các chất vào hệ tuần hoàn.
Các triệu chứng của hội chứng carcinoid thay đổi về mức độ và thời gian và thường mơ hồ, không đặc hiệu và liên quan đến vị trí u, dẫn đến sự chẫm trễ trong chẩn đoán. Thời gian trung bình từ khi có triệu chứng đến khi được chẩn đoán là hơn 9 năm.
Cơn Carcinoid kịch phát là một thể đe dọa tính mạng của hội chứng carcinoid được kích hoạt bởi các biến cố đặc hiệu như gây mê, phẫu thuật hoặc điều trị hóa chất, những tác động này có thể kích thích giải phóng một lượng quá lớn các chất vận mạch.
Các triệu chứng bao gồm nóng bừng mặt kèm theo thay đổi nhiều về huyết áp và cũng có thể có loạn nhịp, co thắt phế quản và thay đổi ý thức.
Có một số các hội chứng lâm sàng khác hiếm gặp trong u thần kinh nội tiết do tiết quá nhiều các hormon nội tiết trưởng thành (xem Bảng 38.2) [14-17].
2.3 Chẩn đoán cận lâm sàng
2.3.1 Cận lâm sàng

Rất hiếm gặp các u thần kinh nội tiết có tiết, do đó cần nghĩ đến các
nguyên nhân khác hay gây ra các triệu chứng như tiêu chảy hay đỏ bừng mặt trước khi chỉ định các xét nghiệm sinh hóa.
Xét nghiệm sàng lọc hội chứng carcinoid ban đầu hữu ích nhất là đo nồng độ 5-HIAA trong nước tiểu 24 giờ.
Xét nghiệm này có độ nhạy khoảng 70% và độ đặc hiệu gần 90% ở các bệnh nhân hội chứng carcinoid và nồng độ thường tăng rất cao (>100 mg/ngày) [18].
Ở những bệnh nhân không có hội chứng carcinoid lâm sàng, xét nghiệm không có giá trị cho chẩn đoán u thần kinh nội tiết vì nồng độ bình thường hoặc chỉ tăng rất nhẹ. Tăng 5-HIAA cũng có thể gặp trong bệnh Celiac sprue (không hấp thu gluten), bệnh Whipple hoặc sau khi ăn các loại thức ăn có nhiều tryptophan.
Trước khi chỉ định xét nghiệm 5-HIAA niệu, bắt buộc phải kiểm tra các nguyên nhân có thể gây kết quả dương tính giả (Bảng 38.3). Nồng độ 5-HIAA dường như có liên quan chặt chẽ với khối u và có thể được sử dụng như là một marker cho mức độ bệnh và để theo dõi đáp ứng với điều trị ở những bệnh nhân có tăng rõ rệt trước điều trị.
Kết quả đo trực tiếp 5-HT trong máu bị ảnh hưởng bởi thức ăn, thuốc và serotonin giải phóng từ tiểu cầu. Nó không được coi là xét nghiệm chẩn đoán hữu ích tại thời điểm hiện nay.
Một chỉ dấu sinh học phổ biến khác là CgA. Nó là một glycoprotein được tiết ra bởi u thần kinh nội tiết cùng với các hormon khác. Nó là một marker nhạy của u carcinoid nhưng kém đặc hiệu [19].
Kết quả có thể dương tính giả ở người suy gan và suy thận, viêm khớp dạng thấp, viêm đại tràng, chấn thương và stress về thể chất, tăng gastrin máu do giảm tiết clo dịch vị (do dùng thuốc ức chế bơm proton kéo dài, viêm teo dạ dày, hay ứ đọng dịch hang vị) và đa u tủy xương [20]. Các hướng dẫn hiện tại không khuyến cáo sử dụng xét nghiệm CgA để chẩn đoán u thần kinh nội tiết.
Không như 5-HIAA, xét nghiệm CgA trong huyết tương không dựa vào sự tiết serotonin và có thể phát hiện được các khối u không tiết. Nó có thể được sử dụng để theo dõi tiến triển của khối u và đáp ứng với điều trị nếu u thần kinh nội tiết được chẩn đoán bằng các phương pháp khác.
| Bảng 38.2. Các Hội chứng u thần kinh nội tiết có thiết | |||
| Hội chứng | Các triệu chứng | Tỷ lệ mới mắc | Chẩn đoán |
| Carcinoid | Nóng bừng mặt
| Những người mắc hội chứng carcinoid: 85% | 5-HIAA niệu |
Tiêu chảy
| 30% | ||
Co thắt phế quản
| 10-20% | ||
| Bệnh tim Carcinoid [14] | Phổ biến nhất là hở van ba lá và hẹp van động mạch phổi. Các triệu chứng của suy tim phải, bao gồm phù chân và cổ trướng. | 4-70% | Siêu âm tim |
| Bệnh xơ hóa | Xơ hóa sau phúc mạc, tắc nghẽn ruột/niệu đạo, bệnh Peyronie, xơ phổi | 30 - 60% trong số này có triệu chứng ở bụng | Không có xét nghiệm sinh hoá điển hình |
| Pellagra | Viêm lưỡi, viêm miệng, viêm da, lú lẫn Giảm Albumin máu Do chuyển đổi tryptophan tiền chất để tổng hợp serotonin | Tới 20% [15] | N1 methylnico- tinamide niệu |
| U tiết insulin | Xem lại chương 33 | ||
| U tiết Gastrin | |||
| U tiết Glucagon [16] | Triệu chứng phổ biến Gầy sút, đái tháo đường, ban đỏ hoại tử di chuyển Triệu chứng khác Tiêu chảy tăng tiết, huyết khối tĩnh mạch sâu, các triệu chứng tâm thần kinh. | Khoảng 1 - 10/10 triệu | Glucagon huyết thanh tăng điển hình > 500 pg/mL. |
| U tiết VIP/ hội chứng WDHA/ hội chứng Verner Morrison [17] | Tiêu chảy phân nước, hạ kali máu và giảm tiết acid dich vi. | ~1/10 triệu người | VIP huyết thanh > 75 pg/mL ≥ 2 lần. Tiêu chảy có khoảng trống thẩm thấu thấp. |
| U tiết Somatostatin | Paracrine ức chế giải phóng hormon dẫn đến đái tháo đường, sỏi mật, tiêu chảy/ỉa phân mỡ. | ~1/40 triệu người | Somatostatin huyết tương lúc đói > 30 pg/mL. |
| VIP, vasoactive intestinal peptide: Peptid hoạt tính trong ruột | |||
| Bảng 38.3. Các nguyên nhân gây sai sót kết quả xét nghiệm 5-HIAA niệu | |
| Dương tính giả | Âm tính giả |
Thức ăn
Thuốc
| Thuốc |
2.3.2 Hình ảnh học
Khi đã chắc chắn có chẩn đoán sinh hóa hội chứng carcinoid, cần tìm vị trí khối u.
Chụp CT ổ bụng là xét nghiệm chẩn đoán được lựa chọn để xếp giai đoạn khối u, vì nó phát hiện được khối u nguyên phát và các hạch mạc treo to.
- Chụp CT thường quy thường không phát hiện được các khối u nhỏ ở hỗng tràng, hồi tràng và ruột thừa do kích thước nhỏ. Chụp CT mạch ruột (enterography) được chứng minh là phương pháp nhạy để phát hiện các khối u ở ruột non khi mà chụp CT thường quy không chẩn đoán được [21].
- Các u thần kinh nội tiết di căn thường có hình ảnh CT đặc trưng ở mạc treo với một khối xơ tập trung nhô ra hình cánh sao. Có thể có vôi hóa ở trung tâm.
- Nên cân nhắc chụp CT gan 3 pha vì gan là vị trí di căn phổ biến nhất.
Chẩn đoán hình ảnh liên quan đến Somatostatin
- U thần kinh nội tiết thường có biểu lộ (thụ thể) SSTRs, điều này rất hữu ích cho thăm dò hình ảnh và điều trị.
- Đồng phân Somatostatin (SSA) gắn "In (Indium) được sử dụng để xa hình từ nhiều năm. Độ nhạy ước tính trong phát hiện các khối u carcinoid là từ 65 - 100%. Độ đặc hiệu bị hạn chế do chất đánh dấu bị hấp thu bởi cả các khối u không phải u thần kinh nội tiết, u hạt và trong các bệnh tự miễn.
- Các chất phóng xạ để chụp PET mới được phát triển có độ nhạy vượt xa xạ hình hay chụp CT thường quy. Cặp kết hợp giữa một chất radionuclide chelator (DOTA) với các peptide ngắn có nguồn gốc từ octreotide. Một thử nghiệm lớn trên 131 bệnh nhân nghi ngờ bị u thần kinh nội tiết cho thấy “Ga-DOTATATE- phát hiện được tổn thương ở 95% số bệnh nhân (so với 45% của chụp CT thường và 31% khi làm xạ hình) [22].
Các phương pháp khác như chụp cộng hưởng từ (nhạy với phát hiện bệnh lý ngoài gan) hay siêu âm nội soi/siêu âm trong mổ thường được dành cho những bệnh nhân nghi ngờ u carcinoid mà không thể định vị được u bằng chụp CT hay chụp chức năng (functional imaging).
Chụp CT ngực có thể phát hiện được các khối u carcinoid ở phế quản, siêu âm tim có thể giúp đánh giá mức độ nặng của bệnh tim carcinoid.
3 ĐIỀU TRỊ
Các điều trị chính ở bệnh nhân u carcinoid gồm kiểm soát triệu chứng, kiểm soát các thông số sinh hóa (làm giảm hoặc bình thường hóa nồng độ 5-HIAA), kiểm soát khối u mục tiêu và cải thiện chất lượng cuộc sống.
3.1 Các thuốc điều trị
3.1.1 Kiểm soát triệu chứng liên quan đến hormon
Đồng phân somatostatin (SSA) là liệu phát điều trị triệu chứng chính ở những bệnh nhân có hội chứng carcinoid để kiểm soát các triệu chứng liên quan đến hormon tiết ra. Nó cũng có thể ngăn tiến triển của bệnh tim carcinoid. Khởi đầu có thể cho Octreotide tác dụng ngắn và nếu dung nạp tốt sẽ chuyển sang dạng tác dụng kéo dài. Liều thông thường của depot octreotide LAR là tiêm bắp 20 đến 30 mg mỗi tháng một lần. Lantreotide, một dạng SSA tác dụng kéo dài khác, có hiệu quả lâm sàng tương đương với octreotide LAR (xem phần dưới).
Các tác dụng phụ bao gồm buồn nôn, đau bụng, nôn và tiêu chảy nhưng thường tự hết trong vòng một vài ngày sau tiêm.
Sỏi mật và bùn mật có thể là biến chứng lâu dài, xảy ra ở gần 50% số bệnh nhân do giảm co bóp và làm trống túi mật sau ăn. Điều trị dự phòng bằng ursodeoxycholic acid có thể giúp làm giảm biến chứng này, cắt túi mật đôi khi được thực hiện cùng lúc với cắt khối u nguyên phát.
Hiện tượng nhờn thuốc thường gặp sau khoảng 12 tháng, có thể khắc phục bằng liều cao hơn hoặc dùng thêm interferon-a.
Telotristat ethyl, chất ức chế tryptophan hydroxylase có tác dụng ngăn chuyển ban đầu từ tryptophan thành 5-HT, mới được chấp thuận như là điều trị hỗ trợ cho SSA ở những bệnh nhân điều trị SSA nhưng triệu chứng giảm ít. Nghiên cứu pha III thấy thêm telotristat ethyl vào SSA làm giảm tần suất đại tiện và nồng độ 5-HIAA niệu, so với Placebo [23].
Điều trị đặc hiệu với các triệu chứng khác của hội chứng carcinoid ngoài SSA thường có tác dụng hữu ích. Các thuốc Prednisone, phenoxybenzamin và chlorpromazin đã được chứng minh hiệu quả ở những bệnh nhân bị nóng bừng mặt và tiêu chảy nặng. Các thuốc ức chế histamin có tác dụng tốt ở bệnh nhân carcinoid dạ dày có tiết histamin.
3.1.2 Kiểm soát phát triển của khối u
Với các khối u biệt hóa kém, hóa trị liệu gây độc tế bào là phương pháp chính, vì những khối u này đáp ứng kém với điều trị đích. Phác đồ chuẩn gồm thuốc platinum và Etoposide [1].
Với những bệnh nhân có u biệt hóa cao và bệnh không có triệu chứng, quyết định điều trị được cá thể hóa và những khối u nhỏ, ổn định có thể chỉ cần theo dõi.
Với những bệnh nhân có u tiến triển nặng hoặc có triệu chứng, có nhiều lựa chọn điều trị.
- Các thử nghiệm ban đầu với octreotide LAR (PROMID) và lanreotide (CLARINET) cho thấy kéo dài thời gian sống thêm không tiến triển (PFS) ở những u thần kinh nội tiết tụy và ruột non biệt hóa cao đã có di căn [24,15]. Cân nhắc SSA như là điều trị đầu tay cho những bệnh nhân có di căn, có triệu chứng hoặc u biệt hóa cao tiến triển.
- Everolimus, một chất ức chế con đường mTOR, khi dùng đơn trị liệu hoặc thêm vào SSA trong thử nghiệm RADIANT- 4, đã được chứng minh kéo dài thời gian sống thêm không tiến triển (PFS) ở các u thần kinh nội tiết có di căn [26]. Thuốc được chấp thuận ở Mỹ cho điều trị u thần kinh nội tiết tiến triển, không tiết. Các biến cố ngoại ý ở mức 3/4 gồm viêm niêm mạc miệng, tiêu chảy, nhiễm khuẩn, thiếu máu và tăng đường máu.
- SSA gắn chất phóng xạ, còn gọi là liệu pháp nucleotide phóng xạ thụ thể peptide (PRRT) đã được sử dụng ở châu u và đang được FDA xem xét điều trị các u thần kinh nội tiết dương tính với thụ thể somatostatin. Thử nghiệm NETTER-1 của Lu-DOTATATE cho thấy tăng thời gian sống thêm không tiến triển và tỷ lệ đáp ứng ở những bệnh nhân phối hợp PRRT với octreotide liều thấp so với octreotide liều cao đơn thuần [27]. Phân tích sơ bộ thấy PRRT làm tăng thời gian sống thêm chung nhưng kết quả này cần được khẳng định qua phân tích chung cuộc theo kế hoạch. Tác dụng phụ ức chế tủy xương gặp ở khoảng 10% số bệnh nhân.
- Nhiều chất ức chế phân tử nhỏ, bao gồm Sunitinib, Pazopanib và Bevacizumab được chứng minh có hiệu quả vừa phải trong các thử nghiệm pha II và III.
3.2 Điều trị phẫu thuật
Một số ít bệnh nhân có biểu hiện bệnh khư trú tại chỗ và dường như chưa có di căn, điều trị đầu tay là phẫu thuật cắt bỏ nhằm chữa khỏi bệnh.
Trái ngược với phần lớn các u đặc di căn, phẫu thuật cắt bỏ có thể vẫn được chỉ định cho các NET di căn. Phẫu thuật làm giảm NET nguyên phát hoặc u di căn có thể cải thiện chất lượng sống và làm giảm gánh nặng triệu chứng do hormon tiết ra hoặc triệu chứng chén ép của khối u.
Phẫu thuật giảm tế bào hoặc cắt bỏ có mục tiêu khối di căn gan nên được cân nhắc cho những bệnh nhân NET có triệu chứng và ảnh hưởng nặng trên gan mà có thể cắt bỏ gần hoàn toàn được. Nghiên cứu hồi cứu chùm ca bệnh thấy có giảm gánh nặng triệu chứng và thời gian sống thêm không tiến triển, không thua kém mặc dù tái phát gần như toàn bộ sau 5 đến 10 năm [28,29].
Cơn carcinoid kịch phát trong mổ hiếm gặp nhưng là một biến chứng tiềm tàng nặng có thể được thúc đẩy bởi phẫu thuật hoặc gây mê. Cần điều trị somatostatin analog trước và trong mổ cho những bệnh nhân có hội chứng carcinoid tiết hormon.
4 KIỂM TRA VÀ THEO DÕI
Có rất ít bằng chứng khuyến cáo việc theo dõi bệnh, do đó các hướng dẫn hiện tại chủ yếu dựa trên ý kiến chuyên gia [30].
Các u thần kinh nội tiết (NET) rất nhỏ, bao gồm NET < 2 cm ở ruột thừa và < 1 cm ở trực tràng, thường được điều trị khỏi bằng phẫu thuật và có thể không cần theo dõi chuyên biệt.
Các NET lớn hơn đã được cắt bỏ hoàn toàn nên được theo dõi về hình ảnh và có thể là các xét nghiệm sinh hóa (nếu có tăng hormon trước mổ) trong 3-12 tháng đầu sau mổ, rồi mỗi 1 - 2 năm sau đó.
Các bệnh nhân đã có di căn hoặc còn sót u (sau mổ) cần được theo dõi bằng thăm khám lâm sàng, hình ảnh và xét nghiệm sinh hóa thường xuyên hơn phụ thuộc vào mức độ triệu chứng, điều trị SSA và bằng chứng bệnh tiến triển.
Siêu âm tim định kỳ để phát hiện sớm bệnh tim carcinoid ở những bệnh nhân có hội chứng carcinoid có thể cải thiện tiên lượng nhưng chưa được kiểm chứng qua các thử nghiệm ngẫu nhiên có đối chứng.
5 TIÊN LƯỢNG
Tiên lượng dựa vào vị trí, kích thước, mức độ xâm lấn và mô bệnh học của khối u nguyên phát.
Phân tích dữ liệu SEER cho thấy trong số những bệnh nhân NET biệt hóa cao, thời gian sống thêm trung vị với bệnh khư trú tại chỗ, vùng hay đã di căn lần lượt là 223, 111 và 33 tháng [3].
Các khối u ở ruột thừa hay trực tràng có tiên lượng đặc biệt tốt, với tỷ lệ sống thêm 5 năm là trên 90% nếu là u tại chỗ.
Tiên lượng của các bệnh nhân NET biệt hóa kém còn tồi hơn, thời gian sống thêm trung vị chỉ là 10 tháng.
Tuổi cao là yếu tố tiên lượng tồi. Giới nữ có cải thiện nhỏ nhưng có ý nghĩa về tiên lượng sống thêm so với giới nam.
6 TÀI LIỆU THAM KHẢO
1. Kunz PL. Carcinoid and neuroendo- crine tumors: Building on success. J Clin Oncol 2015;33:1855-1863.
2. Klimstra DS, Modlin IR, Coppola D, Lloyd RV, Suster S. The pathologic classification of neuroendocrine tumors: A review of nomenclature, grading, and staging systems. Pan- creas 2010;39:707–712.
3. Yao JC, Hassan M, Phan A, et al. One hundred years after "car- cinoid": Epidemiology of and prognostic factors for neuroen- docrine tumors in 35,825 cases in the United States. J Clin On- col 2008;26:3063-3072.
4. Broder MS, Cai B, Chang E, Neary MP. Epidemiology of gastroin- testinal neuroendocrine tumors in a US commercially insured pop- ulation. Endocr Pract 2017;23: 1210-1216.
5. Hauso O, Gustafsson BI, Kidd M, et al. Neuroendocrine tumor epide- miology: Contrasting Norway and North America. Cancer 2008; 113: 2655-2664.
6. Ito T, Igarashi H, Nakamura K, et al. Epidemiological trends of pancreatic and gastrointestinal neuroendocrine tumors in Japan: A nationwide survey analysis. J Gastroenterol 2015;50:58-64.
7. Cho MY, Kim JM, Sohn JH, et al. Current trends in the incidence and pathologic diagnosis of gastro- enteropancreatic neuroendocrine tu- mors (GEP-NETs) in Korea 2000- 2009: Multicenter study. Cancer Res Treat 2012;44:157-165.
8. Tsai HJ, Wu CC, Tsai CR, Lin SF, Chen LT, Chang JS. The epidemiology of neuroendocrine tumors in Taiwan: A nation-wide cancer registry-based study. PLOS One 2013;8:e62487.
9. Scarpa A, Chang DK, Nones K, et al. Whole-genome landscape of pancreatic neuroendocrine tumours. Nature 2017;543:65-71.
10. Jiao Y, Shi C, Edil BH, et al. DAXX/ATRX, MEN1, and mTOR pathway genes are frequently altered in pancreatic neuroen- docrine tumors. Science 2011; 331:1199-1203.
11. Banck MS, Kanwar R, Kulkarni AA, et al. The genomic landscape of small intestinal neuroendocrine tumors. J Clin Invest 2013; 123: 2502-2508.
12. Vinik AI, Chaya C. Clinical presen- tation and diagnosis of neuroendo- crine tumors. Hematol Oncol Clin North Am 2016;30:21-48.
13. Onaitis MW, Kirshbom PM, Hay- ward TZ, et al. Gastrointestinal carcinoids: Characterization by site of origin and hormone produc- tion. Ann Surg 2000; 232:549–556.
14. Bhattacharyya S, Davar J, Dreyfus G, Caplin ME. Carcinoid heart disease. Circulation 2007; 116: 2860-2865.
15. Bell HK, Poston GJ, Vora J, Wilson NJ. Cutaneous mani- festations of the malignant car- cinoid syndrome. Br J Derma- tol 2005;152:71-75.
16. Wermers RA, Fatourechi V, Wynne AG, Kvols LK, Lloyd RV. The glucaconoma syndrome. Clinical and pathologic features in 21 patients. Medicine (Balti- more) 1996;75:53–63.
17. Ghaferi AA, Chojnacki KA, Long WD, Cameron JL, Yeo CJ. Pancre- atic VIPomas: Subject review and one institutional experience. J Gas- trointest Surg 2008; 12:382-393.
18. O'Toole D, Grossman A, Gross D, et al. ENETS consensus guide- lines for the standards of care in neuroendocrine tumors: Biochem- ical markers. Neuroendocrinolo- gy 2009;90:194-202.
19. Campana D, Nori F, Piscitelli L, et al. Chromogranin A: Is it a useful marker of endocrine tumors?. J Clin Oncol 2007;25:1967-1973.
20. Modlin IM, Gustafsson BI, Moss
SF, Pavel M, Tsolakis AV, Kidd M. Chromogranin A-biological func- tion and clinical utility in neuro- endocrine tumor disease. Ann Surg Oncol 2010;17:2427-2443.
21. Hakim FA, Alexander JA, Huprich JE, Grover M, Enders FT. CT-en- terography may identify small bow- el tumors not detected by capsule endoscopy: Eight years experience at Mayo Clinic Rochester. Dig Dis Sci 2011;56:2914-2919.
22. Sandowski SM, Neychev V, Millo C, et al. Prospective study of 68Ga-DOTATATE positron emission tomography/computed to- mography for detecting gastro-en- tero-pancreatic neuroendocrine tu- mors and unknown primary sites. J Clin Oncol 2016;34:588–596.
23. Kulke MH, Hörsch D, Caplin ME, et al. Telotristat ethyl, a tryptophan hydroxylase inhibitor for the treat- ment of carcinoid syndrome. J Clin Oncol 2017;35:14-23.
24. Rinke A, Müller HH, Schade-Brit- tinger C, et al. Placebo-controlled, double-blind, prospective, ran- domized study on the effect of octreotide LAR in the control of tumor growth in patients with metastatic neuroendocrine midgut tumors: A report from the PRO- MID Study Group. J Clin Oncol 2009;27:4656-4663.
25. Caplin ME, Pavel M, C'wikła JB, et al. Lanreotide in metastatic entero- pancreatic neuroendocrine tumors. N Engl J Med 2014;371:224-233.
26. Yao JC, Fazio N, Singh S, et al. Everolimus for the treatment of advanced, non-functional neuroen- docrine tumours of the lung or gas- trointestinal tract (RADIANT-4): A randomised, placebo-controlled, phase 3 study. Lancet 2016; 387:968-977.
27. Strosberg J, El-Haddad G, Wolin E, et al. Phase 3 trial of Lu-Dotatate for midgut neuroendocrine tumors. N Engl J Med 2017;376:125-135.
28. Mayo SC, de Jong MC, Pulitano C, et al. Surgical management of he- patic neuroendocrine tumor metas- tasis: Results from an international multi-institutional analysis. Ann Surg Oncol 2010; 17: 3129-3136.
29. Glazer ES, Tseng JF, Al-Refaie W, et al. Long-term survival after surgical management of neuroen- docrine hepatic metastases. HPB (Oxford) 2010;12:427–433.
30. Kulke MH, Shah MH, Benson AB 3rd, et al. Neuroendocrine tumors, version 1.2015. J Natl Compr Canc Netw 2015;13:78-108.
31. Thomas J.Braranski, MD, PhD; Janet B.McGill, MD, MA, FACE; Julie M.Silverstein, MD và các tác giả khác tham gia biên soạn, Khoa nội tiết chuyển hóa và nghiên cứu
