Thẩm định quy trình phân tích: 3 loại quy trình và chỉ tiêu thẩm định

Thẩm định phương pháp phân tích là nhằm chứng minh qui trình đó có phù hợp với mục đích ứng dụng không. Thẩm định phương pháp phân tích là một khâu quan trọng, là cần thiết trong nghiên cứu phát triển thuốc mới. Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc thông tin về thẩm định quy trình phân tích.
1 Thẩm định phương pháp phân tích là gì?
1.1 Khái niệm
Thẩm định phương pháp phân tích là nhằm chứng minh qui trình đó có phù hợp với mục đích ứng dụng không. Thẩm định phương pháp phân tích là một khâu quan trọng, là cần thiết trong nghiên cứu phát triển thuốc mới, là dữ liệu bắt buộc phải có khi lập hồ sơ đăng ký thuốc, là bước khởi đầu cho các nghiên cứu liên quan tới nồng độ thuốc trong cơ thể như dược động học, sinh khả dụng, tương đương sinh học,... của thuốc.
Thẩm định phương pháp phân tích là quá trình xác định bằng nghiên cứu trong phòng thí nghiệm những đặc điểm đặc trưng của phương pháp để đảm bảo phương pháp đó đạt yêu cầu với các ứng dụng phân tích thực tế. Các đặc điểm phân tích đặc trưng cần được thẩm định bao gồm: độ chọn lọc/ độ đặc hiệu, độ tuyến tính - đường chuẩn, độ đúng, độ chính xác, giới hạn định lượng, giới hạn phát hiện, khoảng xác định. Đối với mẫu thử sinh học ngoài các chỉ tiêu trên, còn thẩm định các chỉ tiêu khác như giới hạn định lượng dưới; ảnh hưởng của nền mẫu (yêu cầu đối với phương pháp LC - MS); độ nhiễm chéo; tỷ lệ thu hồi; độ đúng, độ chính xác khi pha loãng mẫu; độ ổn định của dung dịch chuẩn, độ ổn định của mẫu sinh học ở những điều kiện bảo quản khác nhau.
Tầm quan trọng của thẩm định phương pháp phân tích thuốc trong nguyên liệu, thành phẩm và dịch sinh học đã được các nhà sản xuất dược phẩm nhận thức rõ ràng và đã được văn bản hóa trong nhiều hướng dẫn như ICH (International Conference on Harmonisation), EMA, US - FDA, ASEAN, ... Ở Việt Nam, năm 2018, trong Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc, Bộ Y tế đã có hướng dẫn chung về thẩm định phương pháp phân tích. Nội dung chương này trình bày thẩm định phương pháp phân tích bằng kỹ thuật sắc ký áp dụng đối với chất tổng hợp, không áp dụng với chế phẩm sinh học. Giới hạn chấp nhận của các chỉ tiêu thẩm định được đề xuất tuân thủ theo các hướng dẫn trên. Ngoài ra, các phụ lục 1 - 2 minh họa qui trình thẩm định chỉ tiêu định tính, định lượng, xác định tạp chất liên quan của thuốc thành phẩm bằng HPLC, phụ lục 3 trình bày ví dụ xác định mô hình đường chuẩn của qui trình thẩm định phương pháp phân tích thuốc trong dịch sinh học bằng HPLC-UV.
1.2 Các loại qui trình phân tích cần thẩm định
Thẩm định qui trình phân tích liên quan đến 3 loại qui trình chung sau đây:
- Định tính;
- Tạp chất: Bao gồm định lượng hàm lượng tạp chất và thử giới hạn tạp chất;
- Định lượng.
Tùy thuộc vào mục đích của qui trình phân tích mà yêu cầu các chỉ tiêu cần được đánh giá khác nhau.
1.3 Các chỉ tiêu thẩm định
- Độ chọn lọc - độ đặc hiệu: Thể hiện khả năng phương pháp phân tích có thể xác định (định tính - định lượng) chất cần phân tích, không bị nhầm lẫn bởi các thành phần khác có trong mẫu.
+ Độ chọn lọc (selectivity) của một phương pháp phân tích là khả năng phân biệt các chất trong mẫu thử là hỗn hợp nhiều thành phần.
+ Độ đặc hiệu (specificity) của một phương pháp phân tích là khả năng xác định một cách chắc chắn sự có mặt chất phân tích khi có các thành phần khác. Tính đặc hiệu thường được xác định trong phép phân tích mà chỉ có một thông số đáp ứng như hoạt tính phóng xạ trong phép phân tích phóng xạ miễn dịch
Khi xác định chất phân tích có mặt của chất có liên quan như chất phân hủy, chất chuyển hóa, tạp chất, tá dược thì thuật ngữ độ chọn lọc và độ đặc hiệu được dùng giống nhau. Tuy nhiên, tổ chức quốc tế có uy tín như IUPAC, AOAC ưu tiên dùng thuật ngữ “độ chọn lọc”, còn “độ đặc hiệu” dùng cho những phương pháp hoàn toàn có tính chọn lọc. Yêu cầu của thẩm định độ chọn lọc đối phép thử định tính là đánh giá sự có mặt của chất phân tích, phép thử định lượng là đảm bảo kết quả chính xác về hà lượng hay hoạt lực, còn định lượng tạp là phương pháp phân tích cho phép xác định chính xác hàm lượng tạp chất.
- Đường chuẩn - khoảng tuyến tính: Biểu diễn mối quan hệ giữa đáp ứng của píc (diện tích hay tỷ lệ diện tích) và nồng độ thuốc. Khoảng tuyến tính là khoảng nồng độ có mối tương quan tuyến tính với các đáp ứng phân tích.
- Độ đúng (accuracy): Giá trị phản ánh độ sát gần của kết quả phân tích với giá trị thực của mẫu đã biết.
- Độ chính xác (precision): Là mức độ chụm giữa các kết quả riêng biệt khi lặp lại qui trình phân tích nhiều lần trên cùng một mẫu thử đồng nhất, được biểu thị bằng giá trị độ lệch chuẩn tương đối RSD (%) hoặc hệ số biến sai CV (%) Độ chính xác có mức:
+ Độ lặp lại. (Repeatability): là độ chụm của kết quả khi lặp lại qui trình phân tích nhiều lần bởi một người phân tích, một thời điểm, cùng thiết bị, hóa chất.
+ Độ chính xác trung gian (Intermediate Precision) là độ chụm của kết quả khi lặp lại qui trình phân tích nhiều lần khi thay đổi các yếu tố ngẫu nhiên ảnh hưởng đến độ chính xác của qui trình phân tích. Đó là ngày phân tích, kiểm nghiệm viên, thiết bị,... Thực tế không cần phải nghiên cứu những ảnh hưởng này một cách riêng rẽ, mà có thể sử dụng thiết kế thực nghiệm tối ưu để đánh giá.
+ Độ tái lặp (Reproducibility): Độ tái lặp được xác định bằng cách so sánh kết quả giữa các phòng thí nghiệm. Độ tái lập được tiến hành đánh giá trong trường hợp tiêu chuẩn hoá qui trình phân tích ví dụ như đối với các qui trình trong dược điển.
- Giới hạn phát hiện, giới hạn định lượng
+ Giới hạn phát hiện (Limit of detection: LOD) là nồng độ thấp nhất của chất phân tích có thể xác định được nhưng không cần thiết phải định lượng được trong điều kiện thí nghiệm cụ thể.
+ Giới hạn định lượng (Limit of quantitation: LOQ) là nồng độ thấp nhất trong mẫu thử có thể định lượng được với tính đúng và tính chính xác chấp nhận được.
- Khoảng xác định là khoảng nồng độ có tính tuyến tính, độ đúng và độ chính xác chấp nhận được khi áp dụng để định lượng.
- Độ thô (Robustness): khả năng duy trì của qui trình phân tích không bị ảnh hưởng bởi những biến đổi nhỏ nhưng có tính chủ định trong các thông số của phương pháp và chỉ ra mức tin cậy của qui trình trong điều kiện sử dụng bình thường.
- Tỷ lệ thu hồi (của qui trình xử lý mẫu) là tỷ lệ hoạt chất thu được sau khi mẫu được chiết tách theo qui trình đã chọn so với mẫu có cùng nồng độ không được xử lý qua chiết tách, được pha trong nền mẫu đã được chiết.
- Độ ổn định của mẫu phân tích trong dịch sinh học cần được đánh giá trong quá trình xử lý và bảo quản mẫu. Mẫu phân tích phải đảm bảo ổn định trong thời gian dự kiến đủ cho phân tích hết mẫu thử.
- Giới hạn định lượng dưới (Lower Limit of Quantitation: LLOQ) là nồng độ thấp nhất của đường chuẩn có thể xác định được với độ đúng và độ chính xác cho phép.
- Ảnh hưởng của nền mẫu (Matrix Effect: ME) là ảnh hưởng của tất cả các thành phần khác ngoài chất phân tích có trong mẫu.
2 Thẩm định quy trình phân tích thuốc dạng nguyên liệu và thành phẩm
Các chỉ tiêu cần phải thẩm định đối với qui trình phân tích thuốc dạng nguyên liệu và thành phẩm tùy thuộc vào chỉ tiêu chất lượng đánh giá (Bảng 5.1).
Bảng 5.1. Các chỉ tiêu thẩm định phương pháp phân tích thuốc dạng nguyên liệu và thành phẩm
Các chỉ tiêu | Định tính | Xác định tạp chất | Định lượng: -Độ hòa tan -Hàm lượng/hoạt lực | |
Định lượng | Thử giới hạn | |||
Độ đúng | - | + | - | + |
Độ chính xác -Độ lặp lại -Độ chính xác trug gian |
- - |
+ + (1) |
- - |
+ + (1) |
-Độ chọn lọc đặc hiệu (2) | + | + | + | + |
Giới hạn phát hiện (LOD) | - | - (3) | + | - |
Giới hạn định lượng (LOQ) | - | + | - | - |
Tính tuyến tính | - | + | - | + |
Khoảng xác định | - | + | - | + |
Ghi chú:
Dấu - : Không cần phải đánh giá
Dấu +: Cần phải đánh giá
(1): Trong trường hợp đã tiến hành kiểm tra độ tái lặp thì độ chính xác trung gian không cần phải xem xét
(2): Một qui trình phân tích kém đặc hiệu có thể được bổ trợ bằng một hay nhiều qui trình phân tích hỗ trợ khác trong các
(3): Có thể cần trong một số trường hợp.
2.1 Độ chọn lọc
Xác định độ chọn lọc yêu cầu được thực hiện khi thẩm định các phép thử định tính, xác định tạp chất và định lượng. Qui trình dùng để xác định độ chọn lọc phụ thuộc vào mục tiêu đã định của qui trình phân tích.
2.1.1 Đối với phép định tính, định lượng
Chuẩn bị các mẫu:
- Mẫu trắng: Dung môi pha động, dung môi hòa tan mẫu, pha loãng mẫu (nếu có),...
- Dung dịch mẫu placebo (đối với thuốc thành phẩm): Chuẩn bị theo qui trình.
- Dung dịch chuẩn: Chuẩn bị theo qui trình.
- Dung dịch thử: Chuẩn bị theo qui trình,
Đánh giá kết quả:
- Thời gian lưu của píc chính trên sắc ký đồ dung dịch thử trùng với thời gian lưu píc chính dung dịch chuẩn
- Sắc ký đồ của mẫu trắng, dung dịch mẫu placebo không xuất hiện píc ở trong khoảng thời gian lưu tương ứng với thời gian lưu của chất chuẩn. Nếu có đáp ứng píc thường yêu cầu phải không lớn hơn 1,0% so với đáp ứng píc của mẫu chuẩn.
- Píc của hoạt chất cần phân tích trong sắc ký đồ dung dịch thử phải tinh khiết.
Hệ số chồng phổ UV của píc hoạt chất cần phân tích thu được trong sắc ký đồ và phổ UV của píc tương ứng trong sắc ký đồ dung dịch chuẩn xấp xỉ 1,0.
2.1.2 Thử tạp chất
Những tạp chất sẵn có
Thêm vào mẫu thử một lượng thích hợp các tạp chất và chứng minh rằng từng tạp chất riêng biệt này được tách riêng rẽ ra khỏi nhau và/hoặc ra khỏi các thành phần khác có trong mẫu.
Những tạp chất không có sẵn
So sánh kết quả phân tích của mẫu thử có chứa tạp chất hoặc các sản phẩm phân huỷ bằng qui trình phân tích đã xây dựng với qui trình chính thống khác.
Có thể so sánh với kết quả của mẫu được xử lý ở các điều kiện khắc nghiệt như: ánh sáng, nhiệt độ, độ ẩm, acid, kiềm, chất oxi hoá, chất khử.
Ví dụ:
Thử tạp chất có sẵn
Chuẩn bị mẫu chuẩn và tạp.
Sắc ký lần lượt mẫu trắng - > mẫu chuẩn -> mẫu tạp - > mẫu chuẩn thêm tạp.
Đánh giá kết quả:
Xác định độ phân giải Rs giữa 2 píc liền kề, thông thường yêu cầu Rs ít nhất không được nhỏ hơn 1,5.
Ví dụ cụ thể về đánh giá ảnh hưởng của các tác nhận đẩy nhanh quá trình phân hủy khi bảo quản được trình bày trong Phụ lục 2.
Trong một số trường hợp đánh giá tỷ lệ đỉnh - hõm (p/v). Giá trị này phải nằm trong giới hạn qui định đối với mỗi trường hợp cụ thể.
2.2 Khoảng tuyến tính
Thực hiện trực tiếp trên mẫu chuẩn bằng cách pha loãng dung dịch chuẩn gốc hoặc cân riêng biệt chuẩn để pha ứng với mỗi điểm nồng độ. Chuẩn bị ít nhất 5 dung dịch chuẩn, có nồng độ tùy thuộc qui trình thẩm định.
Trong các nghiên cứu thẩm định phương pháp định lượng, khoảng nồng độ thường lựa chọn từ 50 - 150% của nồng độ thử đối với mỗi qui trình
Đối với phương pháp định lượng tạp chất tiến hành cùng với định lượng hàm 100% của định lượng lượng tạp chất tiến 100% của định lượng dược chất) hoặc đánh giá bằng tích thì khoảng nồng độ phải từ LOQ hoặc giới hạn báo cáo/bỏ qua (Reporting/disregard limit) đến 120% của chuẩn định lượng dược chất. Trong đó, nồng độ tại LOQ phải nhỏ hơn giới hạn báo cáo/bỏ qua.
Đánh giá kết quả
- Qui trình định lượng: Hệ số tương quan tuyến tính (r) phải ≥ 0,998 và % hệ số chắn tại nồng độ 100% phải không lớn hơn 2,0%.
- Qui trình thử độ hòa tan: Nếu thiết kế thẩm định như 1 qui trình định lượng thì giới hạn như phép định lượng. Còn nếu thiết kế có tính đến yếu tố đặc tính của phép thử (thử trên thiết bị thử độ hòa tan hoặc dung dịch chuẩn được pha trong nền mẫu,...) thì hệ số tương quan tuyến tính (r) phải không nhỏ hơn 0,995; % hệ số chẵn tại nồng độ 100% phải không lớn hơn 3,0%.
- Qui trình định lượng tạp chất: Hệ số tương quan tuyến tính (r) phải không nhỏ hơn 0,99; % hệ số chắn tại nồng độ giới hạn phải không lớn hơn 10,0%.
Trường hợp nằm ngoài khoảng qui định, phải có sự giải thích phù hợp.
2.3 Độ đúng
Độ đúng cần được thiết lập trong khoảng xác định của qui trình phân tích.
2.3.1 Định lượng
2.3.1.1 Nguyên liệu
Thực hiện đối với chất phân tích đã biết rõ độ tinh khiết (ví dụ chất đối chiếu).
2.3.1.2 Thành phẩm thuốc
Có thể xác định bằng một trong các phương pháp sau:
- Phương pháp mẫu tự tạo: Thêm một lượng đã biết chất đối chiếu vào hỗn hợp nền mẫu tự tạo chứa các thành phần của thành phẩm thuốc nhưng không có dược chất.
- Phương pháp thêm chuẩn: Trong trường hợp không có đầy đủ các thành phần để làm mẫu tự tạo thì có thể chấp nhận cho thêm một lượng đã biết của chất cần phân tích vào chế phẩm.
2.3.1.3 Chuẩn bị mẫu
Đối với mỗi qui trình phân tích, khi thẩm định độ đúng thường chuẩn bị ở 3 mức nồng độ:
- Định lượng: 80%, 100%, 120% của nồng độ thử
- Độ đồng đều hàm lượng: 70%, 100% và 130% nồng độ thủ trừ trường hợp do bản chất của dạng bào chế thì cần khoảng nồng độ rộng hơn.
-Thử độ hoà tan: nhỏ hơn 20% mức giới hạn độ hòa tan trong tiêu chuẩn chất lượng, 100% và 120% so với nhãn.
2.3.1.4 Định lượng
Xác định hàm lượng hoạt chất có trong các mẫu thẩm định bằng cách sử dụng dung dịch chuẩn được chuẩn bị theo chỉ dẫn trong qui trình phân tích hoặc phương trình hồi quy tuyến tính được tiến hành song song.
2.3.1.5 Đánh giá kết quả
Xác định độ đúng (Tỷ lệ thu hồi) theo công thức:
Độ đúng (%) = (Lượng hoạt chất tìm lại / Lượng hoạt chất thêm vào) × 100%
- Định lượng: Tỷ lệ thu hồi 98 - 102% ở mỗi mức nồng độ, RSD không lớn hơn 2,0% ở mỗi mức nồng độ.
- Thử độ hòa tan: Nếu thiết kế thẩm định như 1 qui trình định lượng thì giới hạn như phép định lượng. Còn nếu thiết kế có tính đến yếu tố đặc tính của phép thử thì tỷ lệ thu hồi cho phép 95 - 105% ở mỗi mức nồng độ, RSD không lớn hơn 5,0% ở mỗi mức nồng độ.
Trường hợp nằm ngoài khoảng này, phải có sự giải thích phù hợp.
2.3.2 Tạp chất (định lượng)
Độ đúng phải được tiến hành trên các mẫu thử (nguyên liệu hoặc thành phẩm thuốc) đã được thêm một lượng tạp chuẩn đã biết. Trong trường hợp không có tạp và/hoặc sản phẩm phân huỷ chuẩn thì có thể chấp nhận so sánh kết quả thu được với một qui trình độc lập. Hệ số đáp ứng của hoạt chất cũng có thể được sử dụng.
2.3.2.1 Chuẩn bị mẫu
Tùy từng quy trình, chuẩn bị mẫu có khoảng nồng độ khác nhau, nhưng phải đảm bảo khoảng xác định phủ được nồng độ tạp ở mức phải báo cáo đến 120% nồng độ tạp ở mức giới hạn theo tiêu chuẩn.
2.3.2.2 Định lượng
Xác định hàm lượng tạp chất có trong các mẫu thẩm định bằng cách sử dụng dung dịch chuẩn được chuẩn bị theo chỉ dẫn trong qui trình phân tích hoặc phương trình hồi quy tuyến tính được tiến hành song song.
2.3.2.3 Đánh giá kết quả
- Nếu thiết kế thẩm định qui trình xác định tạp chất xung quanh mức giới hạn (± 20%) thì tỷ lệ thu hồi 90 - 110% và RSD không lớn hơn 10,0% ở mỗi mức nồng độ.
- Nếu xác định tạp chất tiến hành cùng với định lượng hàm lượng dược chất thì qui định về tỷ lệ thu hồi và RSD tùy thuộc nồng độ chất phân tích có thể tham khảo giới hạn chấp nhận theo AOAC (Bảng 5.2). Thông thường cho phép tỷ lệ thu hồi 90 - 110% và RSD ≤ 10,0% ở mỗi mức nồng độ. Riêng mức LOQ tỷ lệ thu hồi cho phép từ 85 - 115% và RSD không lớn hơn 15,0%.
Trường hợp nằm ngoài khoảng qui định, phải có sự giải thích phù hợp.
TT | Hàm lượng (%) | Tỷ lệ chất | Đơn vị | Độ thu hồi | RSDr (%) | RSDR (%) |
|---|---|---|---|---|---|---|
1 | 100 | 1 | 100 % | 98-101 | 1 | 2 |
2 | ≥ 10 | 10-1 | 10 % | 95-102 | 1,5 | 3 |
3 | ≥ 1 | 10-2 | 1 % | 92-105 | 2 | 4 |
4 | ≥ 0,1 | 10-3 | 0,1 % | 90-108 | 3 | 6 |
5 | 0,01 | 10-4 | 100 ppm | 85-110 | 4 | 8 |
6 | 0,001 | 10-5 | 10 ppm | 80-115 | 6 | 12 |
7 | 0,0001 | 10-6 | 1 ppm | 75-120 | 8 | 16 |
8 | 0,000001 | 10-8 | 10 ppb | 70-125 | 15 | 32 |
2.4 Độ chính xác
Độ chính xác được thực hiện trên mẫu thử hoặc mẫu tự tạo. Ưu tiên sử dụng đối tượng thẩm định là thuốc thành phẩm.
2.4.1 Độ lặp lại. (Repeatability)
2.4.1.1 Chuẩn bị mẫu
Có thể thực hiện theo 1 trong 2 cách sau:
- Thực hiện lặp lại qui trình phân tích tối thiểu 9 lần ở 3 mức nồng độ tương tự như xác định độ đúng bằng phương pháp mẫu tự tạo.
- Thực hiện lặp lại qui trình phân tích tối thiểu 6 lần ở mức nồng độ 100%.
2.4.1.2 Định lượng
Xác định hàm lượng hoạt chất có trong các mẫu thẩm định bằng cách sử dụng dung dịch chuẩn được chuẩn bị theo chỉ dẫn trong qui trình phân tích hoặc phương trình hồi quy tuyến tính được tiến hành song song
2.4.1.3 Đánh giá kết quả
- Định lượng: RSD không lớn hơn 2,0% ở mỗi mức nồng độ.
- Thử độ hòa tan: Nếu thiết kế thẩm định như 1 qui trình định lượng thì giới hạn như phép định lượng. Còn nếu thiết kế có tính đến yếu tố đặc tính của phép thử thì RSD không lớn hơn 5,0% ở mỗi mức nồng độ
- Định lượng tạp chất:
+ Nếu thiết kế thẩm định xung quanh mức giới hạn (± 20%) thì RSD phải không lớn hơn 10,0% ở mỗi mức nồng độ.
+ Nếu tạp được xác định cùng với qui trình định lượng thì qui định về RSD tùy thuộc nồng độ chất phân tích theo qui định của AOAC (Bảng 5.2). Thông thường cho phép RSD không lớn hơn 10,0% ở mỗi mức nồng độ, riêng mức LOQ không lớn hơn 15%.
Trường hợp nằm ngoài khoảng qui định, phải có sự giải thích phù hợp.
2.4.2 Độ chính xác trung gian
Thực hiện tương tự như đối với độ lặp lại nhưng thay đổi điều kiện phân tích như người phân tích khác, ngày phân tích, thiết bị,...
Gộp kết quả thẩm định của 2 lô số liệu.
Đánh giá kết quả:
- Qui trình định lượng: RSD không lớn hơn 3,0% ở mỗi mức nồng độ.
- Qui trình thử độ hòa tan: Nếu kết quả trung bình của 2 lô số liệu lớn hơn hoặc bằng 85% thì cho phép RSD không lớn hơn 5%, còn trung bình của 2 lô số liệu nhỏ hơn 85% thì yêu cầu RSD không lớn hơn 10%.
- Qui trình định lượng tạp chất: Tương tự như độ lặp lại
Trường hợp nằm ngoài khoảng qui định, phải có sự giải thích phù hợp.
Khi thực hiện thẩm định độ chính xác chỉ tiêu độ hòa tan hoặc độ đồng đều hàm lượng của thuốc thành phẩm cần loại yếu tố ảnh hưởng tới độ chụm từ mẫu thử bằng cách lựa chọn các đơn vị mẫu có khối lượng lệch nhau không quá 2% (đơn vị bé nhất và đơn vị lớn nhất) trong qui trình thẩm định dùng mẫu chế phẩm.
2.5 Khoảng xác định
Khoảng xác định là khoảng nồng độ đáp ứng yêu cầu về tính tuyến tính, độ đúng và độ chính xác và được áp dụng để định lượng mẫu thử chứa chất phân tích với hàm lượng nằm trong khoảng nồng độ đó.
2.6 Giới hạn phát hiện và giới hạn định lượng
Có thể xác định LOD và LOQ bằng một trong các phương pháp sau:
2.6.1 Dựa vào tỉ lệ đáp ứng so với nhiễu
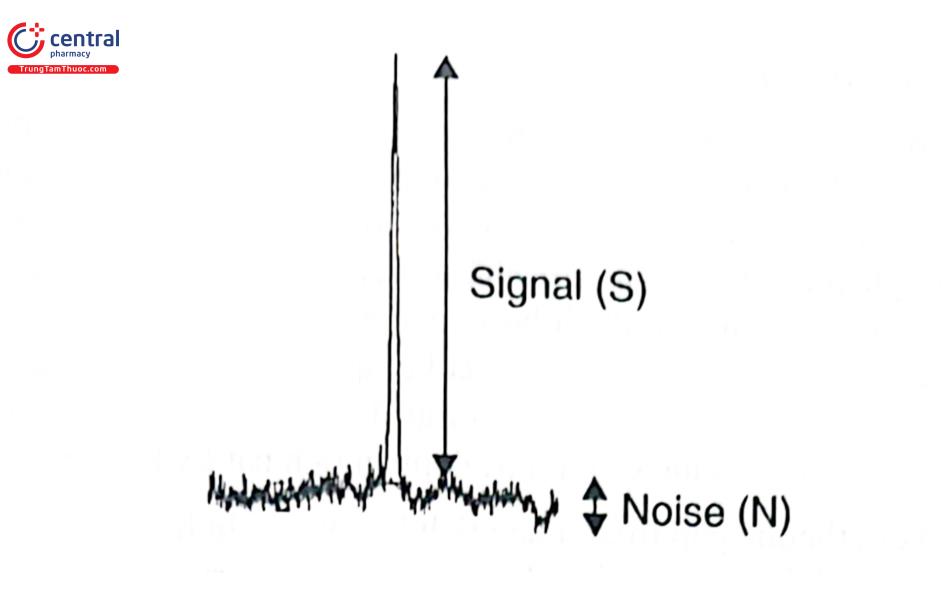
Chuẩn bị dung dịch mẫu thử từ mẫu thử đã biết nồng độ hoặc bằng cách thêm chuẩn vào nền mẫu placebo ở mức độ thấp. Xác định tỉ lệ đáp ứng (S) trên nhiễu (N) của đáp ứng đo được trên mẫu thử với đáp ứng của mẫu trắng, từ đó tính được nồng độ tối thiểu của chất phân tích có thể phát hiện được hoặc định lượng được.
- Nồng độ tại đó có S/N = 2 - 3 là LOD của qui trình.
- Nồng độ tại đó có S/N ~ 10 là LOQ của qui trình.
2.6.2 Dựa vào độ lệch chuẩn của đáp ứng và độ dốc
- Giới hạn phát hiện có thể được tính như sau: LOD = 3,3 ꝍ/S
- Giới hạn định lượng có thể được tính như sau: LOD=10 ꝍ/S
Trong đó:
- ꝍ là độ lệch chuẩn của đáp ứng
- S là độ dốc của đường chuẩn
Độ dốc S có thể được tính dựa vào đường chuẩn của chất phân tích. Có thể xác định ꝍ theo nhiều cách khác nhau, như:
- Dựa vào độ lệch chuẩn của mẫu trắng: Tiến hành lặp lại một số lượng thích hợp phép phân tích trên mẫu trắng (thường là 10 lần, tối thiểu 6 lần), đo đáp ứng nền và tính độ lệch chuẩn của các đáp ứng này.
- Dựa vào đường chuẩn: Dựa vào đường chuẩn đặc trưng của mẫu thử có chứa chất phân tích có nồng độ nằm trong khoảng giới hạn phát hiện. Số dư độ lệch chuẩn của đường hồi quy hoặc độ lệch chuẩn của giá trị giao điểm với trục tung của đường hồi quy có thể được sử dụng như là độ lệch chuẩn.
2.6.3 Dựa vào độ lệch chuẩn của đáp ứng và giá trị trung bình
Dựa vào độ lệch chuẩn khi phân tích trên nền mẫu tự tạo. Chuẩn bị dung dịch mẫu thử bằng cách thêm chuẩn vào nền mẫu placebo ở mức nồng độ thấp. Làm lặp lại tối thiểu 10 lần.
2.6.3.1 Tính toán kết quả
Tính giá trị trung bình x và độ lệch chuẩn SD của đáp ứng phân tích (diện tích píc).
LOD = 3 × SD
LOQ = 10 × SD
Yêu cầu: 4 ≤ R ≤ 10 với R = x /LOD.
2.7 Độ thô (Robustness)
Đánh giá độ thô cần được xem xét trong giai đoạn phát triển phương pháp và tuỳ thuộc vào loại qui trình phân tích đang nghiên cứu. Độ thô chỉ ra được mức độ tin cậy của phương pháp khi có những thay đổi nhỏ có chủ định của các thông số của phương pháp. Nếu những phép đo nhạy cảm với những thay đổi điều kiện phân tích, thì điều kiện phân tích cần được kiểm soát thích hợp hoặc chỉ dẫn những điểm cần lưu ý trong quá trình phân tích. Kết quả đánh giá độ thô là kết quả đánh giá dãy các thông số phản ánh tính thích hợp của hệ thống (ví dụ phép thử độ phân giải) phải được thiết lập để đảm bảo duy trì được tính hiệu lực của qui trình phân tích bất kỳ khi nào sử dụng.
Những biến đổi thường gặp trong phân tích là tính ổn định của các dung dịch phân tích, thời gian chiết, ảnh hưởng của điều kiện sắc ký. Đối với HPLC là sự thay đổi pH pha động, thành phần trong pha động, cột tách khác nhau (do nhà cung cấp và /hoặc lô khác nhau), chương trình nhiệt tốc độ dòng khí.
2.8 Phép thử tính thích hợp của hệ thống
Kiểm tra tính tương thích hệ thống là một phần không thể tách rời trong qui trình phân tích. Đánh giá tính thích hợp của hệ thống là những phép thử thích hợp của toàn hệ thống phân tích được cấu thành bởi các yếu tố bị, hệ thống điện, cách tiến hành phân tích và mẫu thử.
Các thông số của phép thử tính tương thích của hệ thống được thiết lập cho từng qui trình riêng biệt phụ thuộc vào loại qui trình được thẩm định, thành phần mẫu thử đơn hoặc đa thành phần,... Đối với qui trình định lượng thông số bắt buộc đánh giá là giá trị RSD của đáp ứng phân tích để tính kết quả (diện tích píc) phải không lớn hơn 2,0% nếu không có yêu cầu riêng. Ngoài ra, các thông số khác có thể yêu cầu số đĩa lý thuyết, độ phân giải, độ cân xứng píc, ... Còn đối với qui trình xác định tạp chất, thông số độ nhạy hay được yêu cầu đánh giá ngoài các thông số khác.
2.9 Tái thẩm định phương pháp
Phương pháp phải tái thẩm định khi có sự thay đổi trong khâu tổng hợp nguyên liệu dược chất, thay đổi thành phần của thành phẩm hoặc thay đổi qui trình phân tích.
Mức độ thẩm định lại tuỳ thuộc vào bản chất của sự thay đổi
3 Thẩm định phương pháp phân tích thuốc trong dịch sinh học
3.1 Các mức độ thẩm định phương pháp phân tích sinh học
Đối với thẩm định phương pháp phân tích sinh học được chia ra ba mức độ như sau:
3.1.1 Thẩm định đầy đủ (full validation)
Thẩm định đầy đủ rất quan trọng khi:
- Xây dựng và triển khai phương pháp phân tích sinh học lần đầu.
- Đối với thuốc mới.
- Đối với phương pháp định lượng đã phê chuẩn, nếu có mặt sản phẩm chuyển hóa của chất cần phân tích.
3.1.2 Thẩm định một phần (partial validation)
Thẩm định một phần được sử dụng khi có những thay đổi trong phương pháp phân tích sinh học đã được thẩm định. Khi tiến hành, có thể chỉ cần xác định lại độ đúng, độ chính xác hoặc phải làm lại hầu hết các tiêu chí trong thẩm định đầy đủ. Phải các tiêu chí tiến hành thẩm định một phần phương pháp phân tích trong các trường hợp sau:
- Khi chuyển giao phương pháp cho phòng thí nghiệm khác hoặc người khác.
- Thay đổi trong phương pháp luận phân tích (ví dụ: thay đổi bộ phận phát hiện).
- Thay đổi chất chống đông khi lấy dịch sinh học.
- Thay đổi loại mẫu sinh học (ví dụ thay huyết tương bằng nước tiểu).
- Thay đổi trong quá trình xử lý mẫu.
- Thay đổi chủ thể thử nghiệm (ví dụ: huyết tương chuột cống thành huyết tương chuột nhắt)
- Thay đổi khoảng nồng độ cho thích hợp hơn
- Thay đổi thiết bị hoặc phần mềm xử lý kết quả
- Thể tích mẫu giới hạn (ví dụ: nghiên cứu về nhi khoa hoặc loại nền ít gặp).
- Chứng minh độ chọn lọc của phương pháp đối với chất phân tích khi có mặt các chất cản trở.
- Chứng minh độ chọn lọc của phương pháp đối với chất phân tích khi có mặt các chất chuyển hóa xác định.
3.1.3 Thẩm định chéo (cross validation)
Thẩm định chéo là so sánh các thông số thẩm định khi hai hay nhiều phương pháp phân tích được sử dụng để cung cấp dữ liệu cho cùng một nghiên cứu hoặc cho các nghiên cứu khác. Phương pháp dùng làm đối chiếu hay dùng là phương pháp phân tích sinh học gốc đã được thẩm định.
Thẩm định chéo thường được tiến hành để xác lập độ tin cậy giữa các phòng thí nghiệm với nhau khi tiến hành phân tích mẫu trong cùng một nghiên cứu được thực hiện ở nhiều phòng thí nghiệm khác nhau. Thẩm định chéo cũng được xem xét khi các dữ liệu thu được từ nhiều kỹ thuật phân tích khác nhau.
3.2 Các loại mẫu thử trong thẩm định phương pháp phân tích dịch sinh học
- Mẫu trắng (mẫu blank): là dịch sinh học trắng không chứa chất phân tích và/ hoặc chất chuẩn nội.
- Mẫu zero: là mẫu chuẩn bị bằng cách pha chuẩn nội (IS) trong dịch sinh học trắng.
- Mẫu đường chuẩn (Mẫu CC): là mẫu chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích ở các nồng độ xác định khoảng tuyến tính và chuẩn nội trong dịch sinh học trắng.
- Nền mẫu: mẫu dịch sinh học trắng được chiết tách loại tạp theo qui trình.
- Mẫu chuẩn kiểm tra (Quality Control: QC): là những mẫu tự tạo biết trước nồng độ, chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng.
- Mẫu giới hạn định lượng dưới: Mẫu chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng ở nồng độ thấp nhất dự kiến. Giá trị nồng độ LLOQ dự kiến tùy thuộc phạm vi ứng dụng của phương pháp, nếu qui trình phân tích sử dụng để đánh giá tương đương sinh học thì LLOQ lựa chọn nhỏ hơn hoặc bằng 1/20 – 1/30 nồng độ hấp thu cực đại của thuốc.
- Mẫu ULOQ: Mẫu có nồng độ cao nhất của đường chuẩn vẫn đảm bảo tính tuyến tính và píc chất phân tích sắc nét, không bị biến dạng. Giá trị nồng độ ULOQ dự kiến tùy thuộc phạm vi ứng dụng của phương pháp, thông thường ULOQ sẽ lớn gấp 100 - 500 lần LLOQ;
- Mẫu kiểm tra nồng độ thấp (LQC: Low Quality Control): Mẫu có nồng độ thấp của đường chuẩn được chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng ở nồng độ lớn gấp 2 - 3 lần LLOQ;
- Mẫu kiểm tra nồng độ trung bình (MQC: Medium Quality Control): Mẫu có nồng độ trung bình của đường chuẩn được chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng ở nồng độ trong khoảng 40 - 60% ULOQ;
- Mẫu kiểm tra nồng độ cao (HQC: High Quality Control): Mẫu có nồng độ cao của đường chuẩn được chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng ở nồng độ trong khoảng 70 - 90% ULOQ;
- Mẫu kiểm tra pha loãng (DC: Diluted control): Mẫu có nồng độ nằm trong khoảng tuyến tính của đường chuẩn, được chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng ở nồng độ cao gấp 2 - 3 lần nồng độ HỌC, sau đó pha loãng tiếp bằng huyết tương trắng với độ pha loãng theo dự kiến.
Các mẫu tự tạo dùng trong thẩm định phương pháp phân tích có thể được chuẩn bị bằng cách thêm một thể tích nhỏ dung dịch chuẩn vào mẫu sinh học (yêu cầu thể tích thêm vào phải dưới 5% thể tích của mẫu thử để không làm ảnh hưởng đến bản chất mẫu sinh học) hoặc cô khô dung dịch chuẩn dưới luồng khí nitơ và nhiệt độ khoảng 40°C, sau đó hòa tan cắn chất phân tích trong dịch sinh học sử dụng.
3.3 Thực hiện thẩm định phương pháp thuốc trong dịch sinh học
3.3.1 Độ chọn lọc - độ đặc hiệu
Chuẩn bị các mẫu như sau:
- 1 lô mẫu trắng từ huyết tương trắng của ít nhất 6 nguồn khác nhau .
- 1 đường chuẩn (mẫu CC);
- 1 lô mẫu zero gồm ít nhất 6 lần làm lặp lại;
- 2 lô mẫu mỗi mức LLOQ, LQC, MQC, HQC; mỗi mức ít nhất 6 lần làm lặp lại;
3.3.1.1 Đánh giá kết quả
- Tại thời điểm trùng với thời gian lưu của chất phân tích, đáp ứng píc của từng mẫu trắng và mẫu zero phải không được lớn hơn 20% đáp ứng píc của mẫu LLOQ tương ứng;
- Tại thời điểm trùng với thời gian lưu của IS, đáp ứng píc của từng mẫu trắng phải không lớn hơn 5% đáp ứng trung bình của mẫu CC, QC và LLOQ.
Trong thực nghiệm, phép thẩm định chỉ tiêu độ chọn lọc thường được tiến hành song song cùng chỉ tiêu độ đúng để giảm thiểu số mẫu thực nghiệm.
3.3.2 Độ nhiễm chéo
- Chuẩn bị 6 mẫu dịch sinh học trắng, 6 mẫu chứa IS và chất phân tích có nồng độ thấp nhất trên đường chuẩn (LLOQ) và ít nhất 1 mẫu chứa IS và chất phân tích có nồng độ cao nhất trên đường chuẩn (ULOQ).
- Tiến hành xử lý các mẫu trên qui trình
- Tiêm mẫu LLOQ trước, sau đó tiêm xen kẽ từng mẫu trắng sau khi tiêm mẫu ULOQ.
Xác định đáp ứng píc của mẫu trắng và mẫu LLOQ
3.3.2.1 Tiêu chuẩn chấp nhận
- Tại thời điểm trùng thời gian lưu của chất phân tích, đáp ứng píc trung bình của mẫu LLOQ phải gấp ít nhất 5 lần đáp ứng píc của từng mẫu trắng;
- Tại thời điểm trùng thời gian lưu của IS, đáp ứng píc trung bình của mẫu LLOQ phải gấp ít nhất 20 lần đáp ứng píc của từng mẫu trắng.
3.3.3 Đường chuẩn và khoảng tuyến tính
- Tiến hành trên ít nhất 5 đường chuẩn, đường chuẩn bao gồm ít nhất 6 nồng độ của chất chuẩn pha trong cùng một loại mẫu dịch sinh học. Chuẩn bị các mẫu tự tạo theo quy trình.
- Xác định hệ số trọng số (weighting):
+ Xác định mức độ biến thiên (variance) đáp ứng ở 2 nồng độ LLOQ và ULOQ;
+ Tính Fthực nghiệm theo công thức:
Fthực nghiệm = Phương sai của ULOQ/Phương sai của LLOQ.
+ Xác định giá trị Fbảng bằng cách tra bảng F - distribution table với a = 0,01; df1 = n - 1; df2 = n - 1, n là số đường chuẩn thực nghiệm.
+ Nếu Fthực nghiệm < F - bảng → Không sử dụng hệ số trọng số. Nếu Fthực nghiệm > F - bảng chứng tỏ tập hợp kết quả không đồng nhất → Cần sử dụng hệ số trọng số.
+ Khảo sát các hệ số trọng số: 1, 1/x, 1/x2, 1/x1/2. Xác định phương trình hồi qui tuyến tính theo công thức:

+ Đối với mỗi mô hình trọng số, tính % tìm lại ở mỗi điểm chuẩn, tính sai số dư bằng % tìm lại trừ đi 100% tính tổng sai số dư (Sum % RE) bằng tổng giá trị tuyệt đối của các giá trị sai số dư. Thực hiện bước này cho từng đường chuẩn đã xây dựng.
+ Lựa chọn mô hình nào có tổng sai số dư (Sum % RE) của các đường chuẩn thực nghiệm nhỏ nhất và trên đồ thị phần dư của mô hình đó, dữ liệu thực nghiệm phân bố đều nhất xung quanh trục nồng độ.
- Tính lại nồng độ mỗi điểm chuẩn theo phương trình hồi quy đã xây dựng được, xác định độ đúng so với giá trị thực của từng nồng độ.
3.3.3.1 Tiêu chuẩn chấp nhận
- Độ đúng so với giá trị thực của các nồng độ phải từ 85% - 115%, trừ tại điểm LLOQ được chấp nhận 80% - 120%.
- Có ít nhất 75% số điểm trong dãy chuẩn, trong đó LLOQ và ULOQ phải đạt được tiêu chuẩn trên.
Khi tiến hành phân tích mẫu thực, thang nồng độ chuẩn để xây dựng đường chuẩn có thể được chuẩn bị theo 2 cách: 1) Chuẩn bị ngay trong ngày phân tích, hoặc 2) Chuẩn bị cùng ngày lấy mẫu và bảo quản cùng với mẫu thử. Cách thứ nhất dùng mẫu chuẩn mới sẽ kiểm soát được độ ổn định của mẫu. Cách thứ hai sẽ dễ hơn và chính xác hơn do loại trừ được ảnh hưởng trong quá trình bảo quản lên các mẫu thử,
3.3.4 Giới hạn định lượng dưới
- Chuẩn bị ít nhất 6 mẫu thử có chứa IS và chất phân tích ở nồng độ LLOQ dự kiến.
- Chuẩn bị song song đường chuẩn tương ứng.
- Xác định độ đúng bằng cách so sánh nồng độ tính được từ đường chuẩn với nồng độ thực đã pha. Xác định độ chính xác bằng cách tính giá trị CV giữa kết quả 6 lần phân tích.
- Tiến hành lặp lại chỉ tiêu này trên ít nhất 3 lô phân tích khác
3.3.4.1 Tiêu chuẩn chấp nhận
- Tại thời điểm trùng với thời gian lưu của chất phân tích, đáp ứng trung bình mẫu LLOQ phải gấp ít nhất 5 lần đáp ứng của mẫu zero.
- Độ đúng trung bình từng lô và độ đúng trung bình của ít nhất 3 lô phải đạt từ 80% - 120% so với nồng độ thực.
- Độ chính xác (CV) của từng lô và của ít nhất 3 lỗ phải không lớn hơn 20%.
3.3.5 Độ đúng, độ chính xác
3.3.5.1 Độ đúng và độ chính xác trong ngày
Tiến hành trong cùng 1 điều kiện:
- Lô mẫu QC bao gồm ít nhất 3 khoảng nồng độ thấp, trung bình và cao so với khoảng đường chuẩn. Tuy nhiên, với những nghiên cứu có khoảng đường chuẩn rộng có thể bổ sung thêm một vài nồng độ QC ở khoảng nồng độ phù hợp.
- Chuẩn bị các lô mẫu ở các mức nồng độ LLOQ, LỌC, MỌC và HỌC mỗi lô mẫu gồm ít nhất 6 mẫu độc lập.
- Chuẩn bị một đường chuẩn trong cùng điều kiện.
- Phân tích các mẫu theo phương pháp cần đánh giá. Ghi lại sắc ký đồ và đáp ứng píc.
- Tính lại nồng độ các mẫu QC, LLOQ theo đường chuẩn.
- Xác định độ đúng của phương pháp bằng cách so sánh nồng độ phân tích được so với nồng độ thực đã pha.
- Xác định độ lặp lại trong ngày bằng cách tính giá trị CV giữa các nồng độ phân tích được của mỗi lô mẫu QC và LLOQ được phân tích trong cùng một ngày.
3.3.5.2 Yêu cầu
- Độ đúng trung bình của từng lô mẫu QC phải nằm trong khoảng 85% - 115% nồng độ thực đã pha, trừ tại nồng độ LLOQ cho phép độ đúng trong khoảng 80 - 120%.
- Độ lặp lại giữa các nồng độ phân tích được của mỗi lô mẫu QC có giá trị CV không lớn hơn 15%, trừ tại nồng độ LLOQ: CV không lớn hơn 20%,
3.3.5.3 Độ đúng và độ chính xác giữa khác ngày
- Tiến hành đánh giá độ đúng, độ chính xác của phương pháp nhưng lặp lại trên it nhất 3 lô phân tích khác nhau trong các ngày khác nhau.
- Xác định độ chính xác khác ngày bằng cách tính toán độ lệch CV % giữa các giả trị độ đúng thu được của mỗi mức nồng độ QC và LLOQ trong các ngày đã đánh giá.
3.3.5.4 Yêu cầu
Giá trị CV giữa giá trị độ đúng của các ngày phải không lớn hơn 15%, ngoại trừ tại nồng độ LLOQ giá trị CV không lớn hơn 20%
3.3.5.5 Độ đúng - độ chính xác mở rộng
- Phân tích 1 đường chuẩn và mẫu LQC, MQC và HQC trong lô phân tích.
- Lô phân tích bao gồm 1 đường chuẩn “n” lần lặp lại của mẫu QC ở các mức nồng độ (LQC, MQC và HQC). “n” tương ứng với cỡ lô dự kiến trong quá trình phân tích.
3.3.5.6 Tiêu chuẩn chấp nhận
- Độ đúng trung bình của mỗi mức nồng độ QC phải nằm trong khoảng 85% - 115% nồng độ lý thuyết.
- Độ chính xác (CV) ở mỗi mức nồng độ không quá 15%.
3.3.5.7 Độ đúng, độ chính xác khi tiêm lại mẫu
- Xử lý mẫu đường chuẩn và 6 mẫu của mỗi mức LQC, MQC và HQC.
- Tiêm mẫu QC cùng với đường chuẩn đã được xử lý.
- Tiêm lại các mẫu QC ở trên và tính lại nồng độ các mẫu QC dựa vào đường chuẩn ban đầu.
3.3.5.8 Tiêu chuẩn chấp nhận
- Độ đúng trung bình của mỗi mức nồng độ QC phải nằm trong khoảng 85% - 115% nồng độ thực pha được.
- Độ chính xác (CV) ở mỗi mức nồng độ không quá 15%.
3.3.5.9 Độ đúng, độ chính xác khi pha loãng mẫu (mẫu DC)
- Xác định hệ số pha loãng dự định (x lần).
- Chuẩn bị các mẫu pha loãng trong dịch sinh học tương ứng có nồng độ cao gấp x lần nồng độ mẫu HỌC. Mỗi lô mẫu gồm ít nhất 6 mẫu.
- Tiến hành pha loãng các mẫu DC với dịch sinh học trắng để thu được các mẫu có nồng độ HQC.
- Chuẩn bị một đường chuẩn và các mẫu QC trong cùng điều kiện.
- Phân tích các mẫu trên theo qui trình, ghi lại sắc ký đồ và đáp ứng píc.
- Xác định độ đúng, độ chính xác của các mẫu DC.
3.3.5.10 Tiêu chuẩn chấp nhận
Độ đúng trung bình phải nằm trong khoảng 85% - 115% nồng độ thực đã pha.
- Độ lặp lại giữa các nồng độ phân tích được của mỗi lô mẫu DC có giá trị CV không lớn hơn 15%.
3.3.6 Tỷ lệ thu hồi
- Chuẩn bị các lô mẫu QC ở 3 mức nồng độ LỌC, MỌC and HỌC trong dịch sinh học, mỗi mức nồng độ gồm ít nhất 5 mẫu độc lập. Tiến hành chiết tách và phân tích sắc kỷ theo qui trình. Ghi lại sắc ký đồ và đáp ứng píc. Song song chuẩn bị các lô mẫu QC trong dung dịch nền mẫu. Phân tích định lượng trực tiếp không qua giai đoạn chiết tách. Ghi lại sắc ký đồ và đáp ứng píc.
- Tỷ lệ thu hồi của chuẩn nội và chất phân tích được xác định bằng cách so sánh đáp ứng của IS và chất phân tích trong các mẫu QC pha trong dịch sinh học (có qua chiết tách) với đáp ứng của chất phân tích và IS trong các mẫu QC pha trong dung dịch nền mẫu (không qua chiết tách).
3.3.6.1 Yêu cầu
- Tỷ lệ thu hồi tại các nồng độ khác nhau không được quá ±15%.
- Giá trị CV giữa các đáp ứng của chất phân tích và IS trong các mẫu QC có qua chiết tách ở mỗi nồng độ phải không lớn hơn 15%.
- Giá trị CV giữa các đáp ứng của chất phân tích và IS trong các mẫu QC không chiết tách ở mỗi nồng độ phải không lớn hơn 10%.
3.3.7 Độ ổn định
Độ ổn định của chất phân tích phải được khảo sát cả trong dung dịch chuẩn gốc và trong mẫu sinh học, trong điều kiện và thời gian bảo quản dài, trong quá trình xử lý mẫu và sắc ký. Nếu chất phân tích kém ổn định, có thể thêm vào mẫu chất bảo quản hoặc chất chống oxi hóa nào đó mà không cản trở việc phân tích.
Các dung dịch theo dõi độ ổn định được chuẩn bị và bảo quản trong các điều kiện qui định, trong những khoảng thời gian theo dự kiến. Sau thời gian bảo quản, phân tích các dung dịch nghiên cứu độ ổn định song song với chuẩn mới pha tương ứng. Lặp lại quá trình trên 3 lần riêng biệt. Tính đáp ứng píc trung bình.
Độ ổn định (%) = (Trung bình đáp ứng mẫu độ ổn định × Nồng độ mẫu mới pha x 100) / (Trung bình đáp ứng mẫu mới pha × Nồng độ mẫu độ ổn định)
3.3.7.1 Độ ổn định của dung dịch gốc
Dung dịch gốc bao gồm dung dịch chuẩn nội gốc và dung dịch gốc của chất phân tích
Độ ổn định thời gian ngắn ở nhiệt độ phòng
- Dự kiến kế hoạch/ thời gian cần đánh giá
- Chuẩn bị một dung dịch gốc trong dung môi ở nồng độ thích hợp. Chia lượng dung dịch gốc đã pha thành nhiều phần, đủ cho số lần dự kiến đánh giá, mã hóa và ghi nhãn. Bảo quản các mẫu theo qui định ở nhiệt độ phòng trong khoảng thời gian dự kiến đánh giá.
- Ngay sau khi pha, lấy một phần dung dịch gốc, pha loãng đến nồng độ thích hợp, nếu cần. Phân tích theo qui trình, ghi lại sắc ký đồ và đáp ứng píc. Lặp lại quá trình trên 3 lần riêng biệt. Tính đáp ứng píc trung bình.
- Sau từng khoảng thời gian bảo quản như dự kiến, tiến hành tương tự để xác định đáp ứng píc trung bình của dung dịch gốc sau bảo quản.
- So sánh đáp ứng píc trung bình của dung dịch gốc sau thời gian bảo quản với đáp ứng píc trung bình của dung dịch gốc ngay sau khi pha. Cũng có thể tính kết quả của mẫu nghiên cứu độ ổn định dựa vào nồng độ và đáp ứng của mẫu chuẩn ban đầu.
- Xác định thời gian ổn định thời gian ngắn của dung dịch gốc ở nhiệt độ phòng, khi kết quả đáp ứng yêu cầu.
Yêu cầu:
- Dung dịch gốc được coi là ổn định nếu % độ ổn định nằm trong khoảng 85% - 115%.
- Giá trị CV giữa các kết quả định lượng tại từng thời điểm phải không lớn n (đối với phương pháp LC - MS) và không lớn hơn 2% (đối với phương pháp HPLC).
Độ ổn định thời gian dài
Tiến hành và đánh giá kết quả tương tự như nghiên cứu Độ ổn định thời gian ngắn ở nhiệt độ phòng nhưng các mẫu được bảo quản trong điều kiện và khoảng thời gian đánh giá dự kiến. Thông thường, dung dịch chuẩn gốc được bảo quản ở nhiệt độ 2 - 8°C, tránh ánh sáng.
3.3.7.2 Độ ổn định của dung dịch làm việc
Dung dịch làm việc bao gồm dung dịch chuẩn nội làm việc và dung dịch chuẩn làm việc của chất phân tích.
Độ ổn định thời gian ngắn ở nhiệt độ phòng
Tiến hành và đánh giá kết quả tương tự như nghiên cứu Độ ổn định thời gian ngắn ở nhiệt độ phòng đối với dung dịch chuẩn gốc nhưng là thực hiện trên mẫu dung dịch chuẩn làm việc.
Độ ổn định thời gian dài
Tiến hành và đánh giá kết quả tương tự như nghiên cứu Độ ổn định thời gian ngắn ở nhiệt độ phòng đối với dung dịch chuẩn gốc nhưng là thực hiện trên mẫu dung dịch chuẩn làm việc và các mẫu được bảo quản các mẫu trong điều kiện và khoảng thời gian đánh giá dự Thông thường, dung dịch chuẩn làm việc được bảo quản ở nhiệt độ 2 - 8°C, tránh ánh sáng.
3.3.7.3 Độ ổn định chất phân tích trong dịch sinh học
Chất phân tích trong dịch sinh học bị ảnh hưởng bởi điều kiện bảo quản, đặc điểm 1 định của chất phân tích trong một đựng trong đồ bao gói thể chỉ thích hợp cho mẫu đó đồ bao không được ngoại suy độ ổn định đó cho mẫu và đồ bao gói khác. Điều kiện khi đánh giá độ ổn định phải phản ánh đúng điều kiện khi xử lý mẫu và phân tích mẫu thực, Tất cả việc xác định độ ổn định phải dùng một loạt các mẫu được chuẩn bị từ dung dịch gốc của chất phân tích chuẩn mới pha trong mẫu sinh học không có chất phân tích và chất cản trở. Phép thử phải được thực hiện tại ít nhất 2 mức nồng độ là LQC và HQC.
Chuẩn bị các lô mẫu, bảo quản tại các điều kiện định trước. Sau thời gian bảo quản, phân tích các mẫu nghiên cứu độ ổn định. Lặp lại quá trình trên 3 lần riêng biệt. Song song tiến hành phân tích mẫu của đường chuẩn mới pha.
Tính nồng độ các mẫu nghiên cứu độ ổn định theo đường chuẩn, từ đó xác định được độ ổn định của mẫu thử.
% ĐOĐ = (Nồng độ trung bình của mẫu ĐOĐ x100) / Nồng độ thực pha được
Yêu cầu:
- Tại mỗi mức nồng độ mẫu QC, nồng độ trung bình nằm trong khoảng 85% - 115% so với nồng độ thực pha được.
- CV của nồng độ định lượng được ở mỗi mức nồng độ không quá 15%.
3.3.7.4 Độ ổn định sau ít nhất 3 chu kỳ đông rã
- Chuẩn bị hai lô mẫu mỗi loại của LQC và HQC, mỗi lô có ít nhất 05 mẫu LQC và 05 mẫu HQC.
- Phân tích 01 lô mẫu mỗi loại LQC và HỌC ngay sau khi pha cùng đường chuẩn mới pha để xác định nồng độ trung bình của các mẫu QC mới pha.
- Hai lô mẫu LQC và HQC còn lại được bảo quản đông lạnh trong cùng điều kiện bảo quản của mẫu độ ổn định dài ngày. Sau thời gian bảo quản đông lạnh, lấy mẫu ra để ở nhiệt độ phòng đến rã động hoàn toàn, kết thúc một chu kỳ đông - rã. Sau đó cất lại mẫu vào tủ bảo quản và tiếp tục chu kỳ đông rã. Thời gian bảo quản đông lạnh cho mỗi chu kỳ đông rã ít nhất là 12 giờ.
- Sau khi kết thúc chu kỳ đông rã, tiến hành phân tích mẫu theo phương pháp cần đánh giá, xác định nồng độ trung bình của các mẫu QC của mỗi loại dựa vào đường chuẩn mới pha được tiến hành song song
- Tính độ lệch (%) giữa nồng độ trung bình của các mẫu LQC và HQC sau ít nhất 3 chu kỳ đông - rã so với nồng độ trung bình của các mẫu tương ứng mới pha. Đồng thời, tính độ lệch CV giữa các giá trị phân tích được của mỗi khoảng nồng độ QC trước và sau ít nhất ba chu kỳ đông rã.
Nếu chất phân tích không ổn định ở điều kiện bảo quản dự định tiến hành, mẫu đánh giá độ ổn định được để đông ở -70°C sau ít nhất ba lần đông - rã.
3.3.7.5 Độ ổn định ở nhiệt độ phòng trong thời gian
- Dự kiến kế hoạch/ khoảng thời gian cần đánh giá.
- Chuẩn bị các lô mẫu LỌC và HỌC, mỗi lô mẫu gồm ít nhất 5 mẫu LỌC và 5 mẫu HQC. Số lô mẫu QC được tính theo số lần dự kiến đánh giá. Mã hóa và ghi nhãn cho từng lô mẫu. Bảo quản với điều kiện giống bảo quản mẫu độ ổn định dài ngày.
- Khi phân tích, cùng lấy các lô mẫu QC để rã đông và phân tích ngay 1 lô QC xác định nồng độ trung bình ban đầu. Bảo quản các lô mẫu QC còn lại ở nhiệt độ phòng. Sau các khoảng thời gian dự kiến, phân tích để xác định nồng độ trung bình sau bảo quản.
- Tính độ lệch (%) giữa nồng độ trung bình sau từng thời điểm đánh giá so với nồng độ trung bình của mẫu mới pha ở mỗi khoảng nồng độ QC. Đồng thời, tính độ lệch CV giữa các giá trị phân tích được của mỗi khoảng nồng độ đó.
- Xác định thời gian ổn định của mẫu dịch sinh học ở nhiệt độ phòng, khi kết quả của mẫu đáp ứng yêu cầu.
3.3.7.6 Độ ổn định của mẫu dịch sinh học trong thời gian dài
- Dự kiến kế hoạch/ khoảng thời gian cần đánh giá.
- Chuẩn bị các lô mẫu LỌC và HỌC, mỗi lô mẫu gồm ít nhất 5 mẫu LQC và 5 mẫu HQC. Số lô mẫu QC được tính theo số lần dự kiến đánh giá. Mã hóa và ghi nhãn cho từng lô mẫu.
- Phân tích 1 lô mẫu QC ngay sau khi pha cùng đường chuẩn mới pha để xác định nồng độ ban đầu.
- Các lô mẫu còn lại được bảo quản đông lạnh ở nhiệt độ dự kiến. Sau từng khoảng thời gian bảo quản như dự kiến, lấy 1 lô mẫu QC, để rã đông hoàn toàn ở nhiệt độ phòng, phân tích cùng đường chuẩn mới pha để xác định nồng độ sau thời gian bảo quản.
- Tính độ lệch (%) giữa nồng độ trung bình tại mỗi thời điểm đánh giá so với nồng độ trung bình của mẫu mới pha ở từng khoảng nồng độ QC. Tính độ lệch CV giữa các kết quả định lượng của mỗi khoảng nồng độ đó.
- Xác định thời gian ổn định của mẫu dịch sinh học ở nhiệt độ qui định, khi kết quả của mẫu đáp ứng yêu cầu.
3.3.7.7 Độ ổn định của mẫu sau xử lý (trong auto - sampler)
- Dự kiến kế hoạch/ khoảng thời gian cần đánh giá.
- Chuẩn bị các lô mẫu LQC và HQC, mỗi lô mẫu gồm ít nhất 5 mẫu LQC và 5 mẫu HQC.
- Xử lý toàn bộ mẫu theo qui trình. Tiến hành tiêm sắc ký ngay các mẫu đó song song với đường chuẩn để xác định nồng độ trung bình ban đầu.
- Sau khoảng thời gian bảo quản trong autosampler như dự kiến, tiến hành tiêm lại các mẫu trên để xác định nồng độ sau khoảng thời gian bảo quản.
- Tính độ lệch (%) giữa nồng độ trung bình tại mỗi thời điểm tiêm lại so với nồng độ trung bình của lần tiêm ban đầu ở mỗi khoảng nồng độ QC. Đồng thời, tính độ lệch CV giữa các kết quả phân tích được của mỗi khoảng nồng độ đó.
3.3.7.8 Độ ổn định chất phân tích trong cắn sau bốc hơi dung môi (áp dụng với trường hợp xử lý mẫu bằng phương pháp chiết lỏng - lỏng)
- Dự kiến kế hoạch/ khoảng thời gian cần đánh giá.
- Chuẩn bị các lô mẫu LQC và HQC, mỗi lô mẫu gồm ít nhất 5 mẫu LỌC và 5 mẫu HQC. Số lô mẫu QC được tính theo số lần dự kiến đánh giá. Mã hóa và ghi nhãn cho từng lô mẫu.
- Chiết toàn bộ mẫu theo qui trình đến giai đoạn cắn sau khi loại dung môi. Hòa tan cắn chiết của 1 lô mẫu ngay sau khi thu được cắn và tiến hành sắc ký. Song song thực hiện xây dựng một đường chuẩn để xác định nồng độ trung bình ban đầu.
- Các lô mẫu còn lại được bảo quản ở điều kiện dự kiến. Sau từng khoảng thời gian bảo quản như dự kiến, lấy 1 lộ cắn chiết của mẫu QC để phân tích. Song song thực hiện xây dựng một đường chuẩn để xác định nồng độ trung bình của các lô mẫu.
- Tính độ lệch (%) giữa nồng độ trung bình sau từng thời điểm đánh giá so với nồng độ trung bình của mẫu mới pha ở mỗi khoảng nồng độ QC. Đồng thời, tính độ lệch CV giữa các giá trị phân tích được của mỗi khoảng nồng độ đó.
- Xác định thời gian ổn định của mẫu cắn chiết ở điều kiện bảo quản, khi kết quả của mẫu đáp ứng yêu cầu.
3.3.8 Ảnh hưởng của nền mẫu (Yêu cầu khi sử dụng phương pháp LC - MS)
- Chuẩn bị ít nhất 6 mẫu dịch sinh học trắng có nguồn gốc khác nhau. Tiến hành chiết tách theo qui trình thu được các dung dịch nền mẫu tương ứng.
- Chuẩn bị các mẫu chuẩn có chứa IS và chất phân tích ở 2 mức nồng độ: LQC và HQC trong từng dung dịch nền mẫu.
- Song song chuẩn bị các mẫu chuẩn trong pha động hoặc dung môi thích hợp chứa IS và chất phân tích ở nồng độ tương ứng với nồng độ của các mẫu chuẩn trong nền mẫu. l
- Xác định hệ số MF (matrix factor) theo công thức
MF= A/B
Trong đó:
A: đáp ứng píc của chất phân tích hoặc IS pha trong dung dịch nền mẫu.
B: đáp ứng píc trung bình của chất phân tích hoặc IS pha trong pha động hoặc trong dung môi thích hợp ở cùng nồng độ.
- Xác định tỷ số MFs/is = MFs / MFis
Trong đó: MFs là MF của chất phân tích, MFis là MF của chuẩn nội
3.3.8.1 Tiêu chuẩn chấp nhận
Giá trị CV % của các tỉ số MFs/is ≤ 15%.
4 Kết luận
Thẩm định phương pháp phân tích rất quan trọng và cần thiết để chứng minh tích rất muốn tình phương pháp phân tích đảm bảo độ chọn lọc, có độ đúng, độ chính xác, độ nhạy phù hợp với đối tượng phân tích ở mức nồng độ thử nghiệm. Mỗi đối tượng mẫu thử (nguyên liệu, thành phẩm, dịch sinh học) có những yêu cầu về tiêu chí thẩm định, giới hạn chấp nhận khác nhau do đó khi tiến hành thực hiện thẩm định phương pháp cần xem xét đối tượng mẫu thử, tính chất của chất phân tích, phương pháp phân tích sử dụng, thiết kế thẩm định để đưa ra yêu cầu phù hợp. Một phương pháp phân tích thuốc đã thẩm định đạt yêu cầu cho phép thực hiện ở các phòng thí nghiệm khác nhau với điều kiện phân tích cụ thể qui định sẽ cho kết quả tương tự nhau. Do đó, thẩm định phương pháp có vai trò quan trọng trong kiểm tra, giám sát chất lượng thuốc sản xuất, lưu hành trên thị trường, cũng như theo dõi nồng độ thuốc trong dịch sinh học của người sử dụng phục vụ nghiên cứu phát triển thuốc mới, nghiên cứu Sinh khả dụng và đánh giá tương đương sinh học, phục vụ kiểm soát thuốc trong điều trị, ...
5 Phụ lục
5.1 Phụ lục 1. Thẩm định phương pháp định tính, định lượng thuốc thành phẩm theo Tiêu chuẩn cơ sở (Viên nén bao phim API 250 mg)
5.1.1 Quy trình phân tích
5.1.1.1 Thuốc thử
Theo DĐVN
- Acetonitril HPLC (TT)
- Acid acetic băng HPLC (TT)
- Nước (TT)
5.1.1.2 Điều kiện sắc ký
- Cột: Hypersil BDS C18 (250 x 4,6 mm; 5 um)
- Tốc độ dòng: 1,0 ml/phút.
- Detector UV: 230 nm.
- Thể tích tiêm: 20 ul.
- Nhiệt độ cột: 30°C
- Pha động: Acetonitril - nước - acid acetic băng (50 : 50:0,1)
5.1.1.3 Tiến hành
- Dung môi pha mẫu: Acetonitril - nước (1:1)
- Dung dịch chuẩn: Cân chính xác khoảng 50 mg chuẩn API vào bình định mức 250 ml. Hòa tan và pha loãng vừa đủ thể tích bằng dung môi pha mẫu, lắc đều. Lọc qua màng lọc 0,45 um.
- Dung dịch thử: Lấy 20 viên, cân xác định khối lượng trung bình viên, nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với 250 mg API vào bình định mức 100 ml, thêm 10 ml nước, lắc kỹ cho bột viên phân tán đều, thêm 80 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tẩm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
- Sự phù hợp của hệ thống sắc ký: Tiêm dung dịch chuẩn vào hệ thống sắc ký. Trên sắc đồ thu được, hệ số kéo đuôi của píc API không được quá 2,0; số đĩa lý thuyết không được nhỏ hơn 2000, RSD của diện tích píc giữa 6 lần tiêm lặp lại không được lớn hơn 2,0%.
- Tiến hành sắc ký lần lượt với dung dịch chuẩn và dung dịch thử.
5.1.1.4 Kết quả
Hàm lượng % API (CxHyNzOn) trong viên so với hàm lượng ghi trên nhãn, tính theo khối lượng trung bình viên, tính theo công thức:
X (%) = (ST/SC) x [(Mc x HPLC x (1-HC/100))/250] x (1250/MT) x (MTB/250)
Trong đó:
ST: Diện tích píc API trên sắc đồ dung dịch thử.
Sc: Diện tích píc API trên sắc đồ dung dịch chuẩn
Mc: Khối lượng API chuẩn (mg).
MT: Khối lượng mẫu thử (mg)
MTB: Khối lượng trung bình viên (mg).
250: Hàm lượng API ghi trên nhãn (mg).
HLC: Hàm lượng API chuẩn (%).
HC: Độ ẩm của chuẩn (%).
5.1.2 Đề cương thẩm định
5.1.2.1 Độ chọn lọc
* Thực nghiệm:
- Tiến hành sắc ký các loại mẫu sau đây theo qui trình phân tích: .
+ Dung dịch mẫu placebo: Chuẩn bị theo qui trình, thay mẫu thử bằng placebo
+ Dung dịch chuẩn: Chuẩn bị theo qui trình.
+ Dung dịch thử: Chuẩn bị theo qui trình.
- Ghi lại các sắc ký đồ. Xác định thời gian lưu của hoạt chất cần phân tích; phổ UV của píc hoạt chất cần phân tích trên sắc ký đồ các dung dịch chuẩn, thử.
* Yêu cầu:
- Sắc ký đồ dung dịch thử phải cho píc chính có thời gian lưu tương ứng với thời gian lưu của píc API trên sắc ký đồ dung dịch chuẩn.
- Sắc ký đồ của mẫu placebo không được xuất hiện píc ở trong khoảng thời gian lưu tương ứng với thời gian lưu của píc API. Nếu có, đáp ứng píc phải ≤ 1,0% so với đáp ứng píc của dung dịch chuẩn.
- Phổ UV của píc hoạt chất cần phân tích thu được trên sắc ký đồ dung dịch thử và phổ UV của píc tương ứng trên sắc ký đồ dung dịch chuẩn phải giống nhau.
5.1.2.2 Độ thích hợp của hệ thống
* Thực nghiệm:
- Dung dịch chuẩn: Sử dụng dung dịch chuẩn từ phần thẩm định độ đặc hiệu.
- Tiến hành sắc ký dung dịch chuẩn, tiêm lặp lại 6 lần, ghi lại các sắc ký đồ và xác định giá trị thời gian lưu, diện tích píc, hệ số kéo đuôi, số đĩa lý thuyết.
* Yêu cầu:
- Giá trị RSD của thời gian lưu ≤ 1,0%, của diện tích píc API ≤ 2,0%
- Số đĩa lý thuyết: ≥ 2000.
- Hệ số kéo đuôi: ≤ 2,0
5.1.2.3 Độ tuyến tính
* Thực nghiệm:
- Chuẩn bị 05 dung dịch chuẩn, có nồng độ API tương ứng với 50%; 80%; 100%, 120% và 150% nồng độ định lượng.
- Tiến hành sắc ký các dung dịch chuẩn, ghi lại các sắc ký đồ và xác định đáp ứng của píc.
- Xác định phương trình hồi quy tuyến tính, hệ số tương quan tuyến tính giữa tích píc API thu được trên các sắc ký đồ bằng
phương pháp bình phương tối thiểu.
* Yêu cầu:
- Hệ số tương quan tuyến tính (r) phải ≥ 0,998.
- % Hệ số chắn (% Y) tại nồng độ 100% ≤ 2,0%
% Y= (|Hệ số chắn|/ S píc chuẩn 100%) x 100
5.1.2.4 Độ đúng
* Thực nghiệm:
- Xác định độ đúng của phương pháp bằng cách thêm chính xác một lượng chất chuẩn cần phân tích vào các mẫu placebo.
- Chuẩn bị 03 loại mẫu tự tạo bằng cách thêm chính xác một lượng chất chuẩn vào các mẫu placebo. Lượng chất chuẩn thêm vào tương ứng với 3 mức nồng độ 80%, 100% và 120% so với mức hàm lượng ghi nhãn. Tại mỗi mức nồng độ, thực hiện ít nhất 03 mẫu độc lập.
- Phân tích mẫu theo qui trình phân tích. Xác định hoạt chất thu hồi theo trung bình diện tích píc API của dung dịch chuẩn ở phần thử độ đặc hiệu.
- Xác định độ đúng của phương pháp theo công thức:
Độ đúng (Tỷ lệ thu hồi) (%) = (Lượng hoạt chất thu hồi / Lượng hoạt chất thêm vào) x 100%
Yêu cầu:
- Tỷ lệ thu hồi: 98,0% - 102,0%
- RSD tỷ lệ thu hồi: ≤2,0%
5.1.2.5 Độ chính xác
Độ lặp lại
* Thực nghiệm:
- Tiến hành định lượng 06 mẫu thử độc lập.
- Xác định hàm lượng các hoạt chất có trong các mẫu thử theo trung bình diện tích píc API của dung dịch chuẩn ở phần thử độ đặc hiệu.
- Độ lặp lại của phương pháp được xác định bằng giá trị RSD kết quả định lượng hàm lượng hoạt chất có trong các mẫu.
* Yêu cầu:
- Giá trị RSD kết quả định lượng hàm lượng hoạt chất có trong các mẫu ≤2,0%.
Độ chính xác trung gian
* Thực nghiệm:
- Tiến hành như độ lặp lại nhưng khác kiểm nghiệm viên.
- Xác định giá trị trung bình và giá trị bình và giá trị RSD hàm lượng hoạt chất có trong các mẫu do mỗi kiểm nghiệm viên tiến hành.
- Xác định mức độ sai khác kết quả định lượng giữa 2 kiểm nghiệm viên.
* Yêu cầu:
- Giá trị RSD kết quả định lượng của mỗi kiểm nghiệm viên (n=6) ≤ 2,0% và của cả hai kiểm nghiệm viên (n=12) phải ≤ 3,0%.
5.1.2.6 Khoảng xác định
Được suy ra từ kết quả độ đúng, độ chính xác và độ tuyến tính (Từ giá trị nhỏ nhất đến giá trị lớn nhất của độ đúng).
5.1.3 Báo cáo kết quả thẩm định
Nguyên, vật liệu - thiết bị và dụng cụ
* Chất chuẩn:
- API: Chuẩn Viện Kiểm nghiệm thuốc trung ương; Hàm lượng: 99,94%; Độ ẩm: 0,47%; SKS: WS.0218abc.02
* Mẫu thử, mẫu placebo:
- Mẫu thử: Viên nén API 250 mg
- Mẫu placebo: Là mẫu chứa các tá dược bào chế viên được cân và trộn đều theo đúng tỷ lệ trong công thức. Khối lượng tương ứng một viên là 251,4 mg.
* Hóa chất thuốc thử: Acetonitril, Acid acetic băng loại dùng cho HPLC của Merck, Đức.
* Thiết bị phân tích: Máy HPLC Shimadzu SPD - M30A, máy HPLC Agilent 1260 hiệu chuẩn đạt.
5.1.3.1 Kết quả thẩm định
Độ chọn lọc
* Thực nghiệm:
- Dung dịch placebo: Cân 0,2515 g mẫu placebo vào bình định mức 100 ml, thêm 10 ml nước, lắc để bột viên phân tán đều, thêm 80 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 m dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
Dung dịch thử: Cân 0,5018 g bột thuốc vào bình định mức 100 ml, thêm 10 ml nước, lắc để bột viên phân tán đều thêm 80 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Đọc qua màng lọc 0,45 um
Dung dịch chuẩn: Cân 51,18 mg chuẩn API vào bình định mức pha loãng tới vừa đủ thể tích bằng dung môi pha mẫu, lắc đều. Lọc qua màng lọc 0,45 um
* Kết quả:
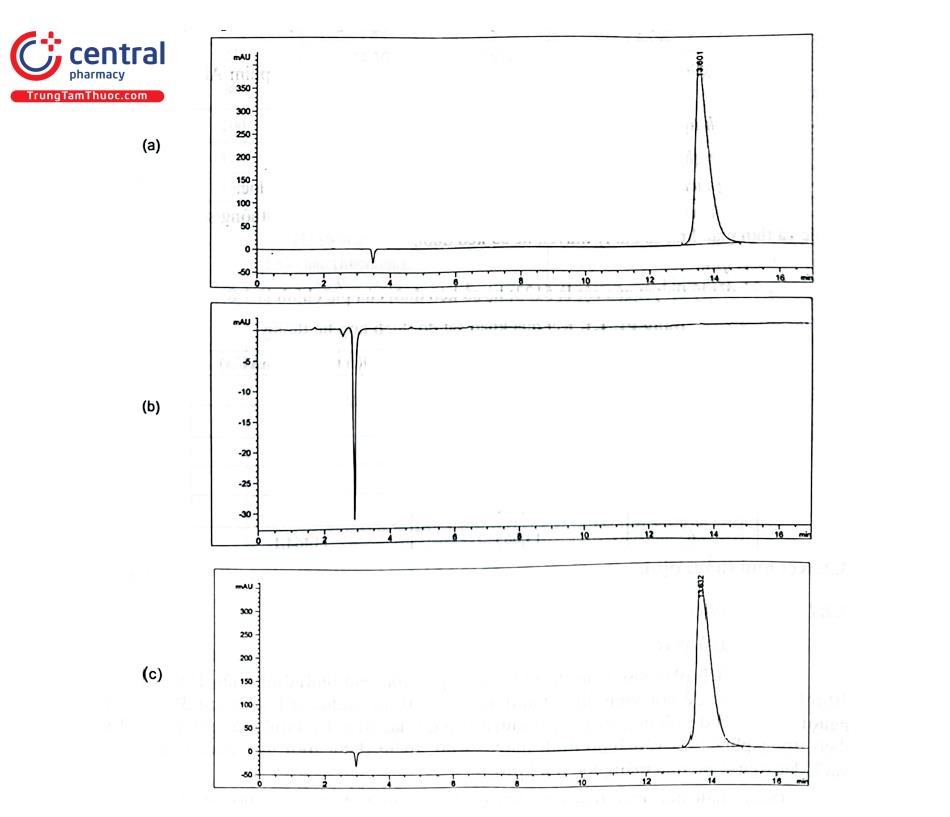
- Trên sắc ký đồ dung dịch placebo không xuất hiện các píc có thời gian lưu tương ứng với thời gian lưu của píc API trên sắc ký đồ dung dịch chuẩn.
- Trên sắc ký đồ mẫu thử cho píc có thời gian lưu tương ứng với thời gian lưu của pic API trên sắc ký đồ mẫu chuẩn (khoảng 13,6 phút). Hệ số match xấp xỉ 1000.
- Phổ UV của píc chính trên sắc ký đồ mẫu thử giống với phổ UV của píc API trên sắc kỷ đồ mẫu chuẩn.
* Nhận xét: Phương pháp định lượng API trong viên nén bao phim API 250 mg là chọn lọc.
Độ thích hợp của hệ thống
* Thực nghiệm:
- Dung dịch chuẩn: Sử dụng dung dịch chuẩn phần thử độ chọn lọc.
Tiêm 6 lần dung dịch chuẩn. Ghi lại các sắc ký đồ, xác định các thông số diện tích píc và thời gian lưu, số đĩa lý thuyết, hệ số kéo đuôi.
* Kết quả:
- Số đĩa lý thuyết của cột là 5123; hệ số kéo đuôi của píc chính là 1,8.
STT | Thời gian lưu (phút) | Diện tích pic (mAU.s) |
|---|---|---|
1 | 13,547 | 11391,7 |
2 | 13,599 | 11374,2 |
3 | 13,618 | 11376,6 |
4 | 13,650 | 11391,4 |
5 | 13,601 | 11415,1 |
6 | 13,665 | 11464,1 |
7 | 13,631 | 11402,2 |
8 | 0,27 | 0,14 |
* Nhận xét: RSDthời gian lưu < 1,0%; RSD diện tích pic < 2,0%; N > 2000; T<2,0=> hệ thống phù hợp để định lượng API trong chế phẩm.
Độ tuyến tính
* Thực nghiệm:
Chuẩn bị các dung dịch chuẩn API trong dung môi pha mẫu có nồng độ API biến thiên trong khoảng 0,10 - 0,30 mg/ml (tương ứng 50 - 150% nồng độ định lượng). Tiêm các dung dịch này vào hệ thống sắc ký, ghi lại sắc ký đồ.
* Kết quả:
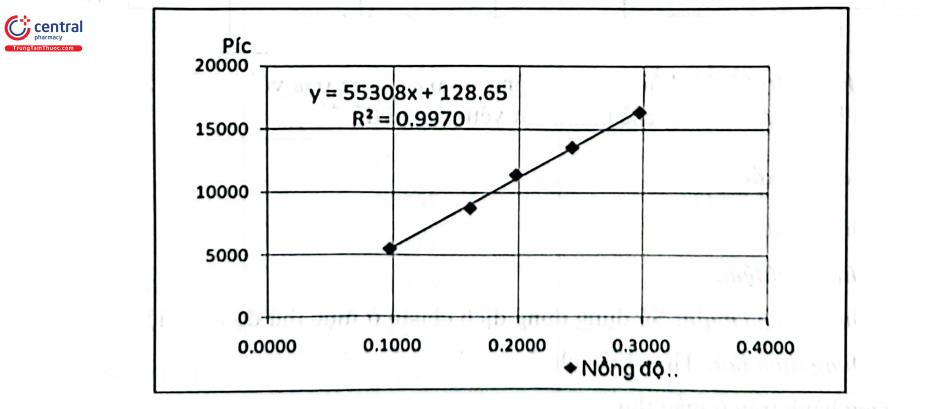
Khảo sát sự tương quan giữa y (diện tích píc API) và x (nồng độ API) bằng phương pháp bình phương tối thiểu, kết quả cho thấy: hệ số R2 (→r=0,9985); chứng tỏ có sự tương quan tuyến tính giữa nồng độ API và diện tích píc API tương ứng.
STT | % so với nồng độ định lượng | Lượng cân chuẩn API (mg) | Bình định mức | Nồng độ API (mg/ml) | Diện tích píc (mAU.s) |
|---|---|---|---|---|---|
1 | 50 | 24, 30 | 250 | 0,0967 | 5478,1 |
2 | 80 | 40,45 | 250 | 0.1609 | 8752,5 |
3 | 100 | 49,68 | 250 | 0,1976 | 11402,2 |
4 | 120 | 60,95 | 250 | 0,2425 | 13604,0 |
5 | 150 | 74,50 | 250 | 0,2964 | 16393,4 |
Hệ số tương quan: Hệ số góc: Hệ số chắn (intercept): %Y | 0,9985 (> 0,998) 55308 128,65 1,13 (<2%) | ||||
Độ đúng
* Thực nghiệm:
- Dung dịch chuẩn: Sử dụng dung dịch chuẩn ở mục thử độ chọn lọc.
- Chuẩn bị các mẫu thử:
Cân chính xác khoảng 50 mg; 62,5 mg; 75 mg chuẩn API vào các bình định mức 25 ml có chứa sẵn khoảng 62,3 mg placebo, thêm 2,5 ml nước, lắc kỹ cho bột viên phân tán đều, thêm 20 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
Mỗi mức nồng độ tiến hành 3 mẫu thử.
* Kết quả:
| Mẫu | Lượng cân mẫu placebo | Lượng chuẩn API thêm vào (mg) | Lượng API tinh khiết (mg) | Diện tích píc API (mAU.s) | Lượng API tìm lại (mg) | % Thu hồi | Kết quả thống kê |
|---|---|---|---|---|---|---|---|
| 80% | 63,42 | 50,31 | 50,05 | 8937,6 | 49,88 | 99,67 | TB: 99,30% RSD: 0,73% |
| 80% | 62,31 | 53,69 | 53,40 | 9546,6 | 53,28 | 99,77 | |
| 80% | 63,72 | 52,04 | 51,76 | 9132,6 | 50,97 | 98,46 | |
| 100% | 63,82 | 68,92 | 68,55 | 12311,3 | 68,71 | 100,23 | TB: 99,28% RSD: 0,97% |
| 100% | 62,81 | 64,99 | 64,65 | 11500,0 | 64,18 | 99,28 | |
| 100% | 62,41 | 63,58 | 63,24 | 11140,4 | 62,18 | 98,31 | |
| 120% | 63,52 | 78,10 | 77,69 | 13729,4 | 76,62 | 98,63 | TB: 98,96% RSD: 0,28% |
| 120% | 64,22 | 78,26 | 77,84 | 13820,9 | 77,13 | 99,09 | |
| 120% | 63,62 | 79,12 | 78,70 | 13981,1 | 78,03 | 99,15 |
* Nhận xét: % thu hồi trong khoảng 98,0% - 102,0% với RSD < 2% => Phương pháp định lượng API trong chế phẩm đạt yêu cầu về độ đúng.
Độ chính xác
Độ lặp lại
* Thực nghiệm:
- Dung dịch chuẩn: Sử dụng dung dịch chuẩn ở mục thử độ đặc hiệu.
- Dung dịch thử: Thực hiện chuẩn bị như qui trình.
Tiến hành trên 6 mẫu thử.
* Kết quả: Bảng PL 1.4. Kết quả thẩm định độ lặp lại của phương pháp
| Trung bình diện tích píc API của dung dịch chuẩn: 11402,2 Lượng cân mẫu chuẩn: 51,18 mg Khối lượng trung bình viên: 0,5018 g | |||
| STT | Lượng cân mẫu thử (g) | Diện tích píc | Kết quả định lượng (%) |
| 1 | 0,5105 | 11330,5 | 99,45 |
| 2 | 0,4922 | 10843,9 | 98,72 |
| 3 | 0,5292 | 11769,4 | 99,66 |
| 4 | 0,5050 | 11145,4 | 98,89 |
| 5 | 0,5062 | 11154,2 | 98,74 |
| 6 | 0,5084 | 11189,1 | 98,62 |
| Trung bình | 99,01 | ||
| RSD (%) | 0,44 | ||
* Nhận xét: RSD <2%=> Phương pháp định lượng API trong chế phẩm đạt yêu cầu về độ lặp lại.
Độ chính xác trung gian
* Thực nghiệm:
Xác định độ chính xác trung gian bằng cách xác định hàm lượng API trong chế phẩm bởi các kiểm nghiệm viên khác nhau, tại các thời điểm khác nhau.
* Kết quả: KNV2:
- Số đĩa lý thuyết của cột là 3957; hệ số kéo đuôi của píc chính là 1,9.
Bảng PL 1.5. Kết quả đánh giá độ thích hợp hệ thống
| STT | Thời gian lưu (phút) | Diện tích píc |
| 1 | 14,251 | 11148.2 |
| 2 | 14,239 | 11153,5 |
| 3 | 14,288 | 11156,4 |
| 4 | 14,265 | 11201,1 |
| 5 | 14,317 | 11176,7 |
| 6 | 14,320 | 11207,8 |
| TB | 14,280 | 11174,0 |
| RSD (%) | 0,22 | 0,23 |
Bảng PL 1.6. Kết quả thẩm định độ chính xác trung gian của phương pháp
| STT | KNV1 Ngày: 1 Lượng cân chuẩn: 51,18 mg Khối lượng trung bình viên: 0,5018 g Diện tích píc API TB mẫu chuẩn: 11402,2 | KNV2 Ngày: 2 Lượng cân chuẩn: 50,02 mg Khối lượng trung bình viên: 0,5025 g Diện tích pic API TB mẫu chuẩn: 11174,0 | ||||
| Lượng cân mẫu thử (g) | Diện tích píc API | % Hàm lượng | Lượng cân mẫu thử (g) | Diện tích píc API | % Hàm lượng | |
| 1 | 0,5105 | 11330,5 | 99,45 | 0,5042 | 11118,2 | 98,68 |
| 2 | 0,4922 | 10843,9 | 98,72 | 0,4925 | 10863,1 | 98,71 |
| 3 | 0,5292 | 11769,4 | 99,66 | 0,5003 | 11045,8 | 98,80 |
| 4 | 0,5050 | 11145,4 | 98,89 | 0,5228 | 11584,0 | 99,16 |
| 5 | 0,5062 | 11154,2 | 98,74 | 0,5095 | 11390,6 | 100,05 |
| 6 | 0,5084 | 11189,1 | 98,62 | 0,4916 | 10755,2 | 97,90 |
| Trung bình: 99,01% RSD: 0,44% | Trung bình: 98,88% RSD: 0,71% | |||||
| Kết quả định lượng trung bình (n=12): 98,95% RSD (n=12): 0,57% | ||||||
Nhận xét: RSD (n=12) ≤ 3,0% => Phương pháp đạt yêu cầu về độ chính xác trung gian.
Khoảng xác định
Kết quả thẩm định tính tuyến tính, độ đúng, độ chính xác cho thấy trong khoảng nồng độ API bằng 80% đến 120% so với nồng độ định lượng đáp ứng yêu cầu qui định do đó khoảng xác định của phương pháp là trong giới hạn từ 80% đến 120% so với nồng độ định lượng.
5.1.3.2 Kết luận
Qui trình phân tích theo Tiêu chuẩn cơ sở đáp ứng các yêu cầu về độ chọn lọc, độ tuyến tính và khoảng xác định, độ đúng, độ lặp lại và độ chính xác trung gian, thích hợp để định tính, định lượng API trong viên nén API 250 mg.
5.2 Phụ lục 2. Thẩm định phương pháp xác định tạp chất liên quan của thuốc thành phẩm theo tiêu chuẩn cơ sở (viên nén bao phim API 250 mg)
5.2.1 Quy trình phân tích
* Điều kiện sắc ký:
- Cột: Hypersil BDS C18 (250 x 4,6 mm; 5 pm)
- Tốc độ dòng: 1,0 ml/phút.
- Detector UV: 230 nm.
- Thể tích tiêm: 20 ul.
- Nhiệt độ cột: 30°C
* Chuẩn bị mẫu:
- Dung môi pha mẫu: Acetonitril - nước (1:1)
- Dung dịch chuẩn: Cân chính xác khoảng 20,0 mg chuẩn API vào bình định mức 100 ml, hòa tan và pha loãng đến vừa đủ thể tích bằng dung môi pha mẫu, lắc đều. Hút 2,0 ml dung dịch này pha loãng vừa đủ 50,0 ml bằng dung môi pha mẫu, lắc đều. Hút 1,0 ml dung dịch này pha loãng vừa đủ 20,0 ml dung môi pha mẫu, lắc đều. Lọc qua màng lọc 0,45 um.
- Dung dịch thử: Lấy 20 viên, xác định khối lượng trung bình viên, nghiền thành bột mịn. Cân chính xác một lượng bột viên tương ứng với 250 mg API vào bình định mức 100 ml, thêm 10 ml nước, lắc để bột viên phân tán đều, thêm 80 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
- Sự phù hợp của hệ thống sắc ký: Tiêm dung dịch chuẩn vào hệ thống sắc ký. Trên sắc đồ thu được hệ số kéo đuôi của píc API không được quá 2,0; số địa lý thuyết không được nhỏ hơn 2000, độ lệch chuẩn tương đối của diện tích píc giữa 6 lần tiêm lặp lại không được lớn hơn 5,0%.
- Tiến hành sắc ký lần lượt với dung dịch chuẩn và dung dịch thử. Thời gian sắc ký đối với dung dịch thử gấp 3 lần thời gian lưu của píc API.
Yêu cầu: Trên sắc ký đồ của dung dịch thử:
+ Diện tích của bất kì píc nào trừ píc chính không được lớn hơn diện tích của píc chính trên sắc ký đồ dung dịch chuẩn (0,2%).
+ Tổng diện tích của tất cả các píc trừ píc chính không được lớn hơn hai lần diện tích của píc chính trên sắc ký đồ của dung dịch chuẩn (0,4%).
5.2.2 Đề cương thẩm định
5.2.2.1 Độ chọn lọc
* Thực nghiệm:
- Tiến hành sắc ký các loại mẫu sau đây theo qui trình phân tích:
+ Mẫu trắng: Dung môi pha mẫu.
+ Dung dịch mẫu placebo: Chuẩn bị theo qui trình, thay mẫu thử bằng mẫu placebo.
+ Dung dịch chuẩn: Chuẩn bị theo qui trình.
+ Dung dịch thử: Chuẩn bị theo qui trình.
+ Dung dịch thử phân hủy ở điều kiện nhiệt độ:
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API vào bình định mức 100 ml. Đặt bình định mức này trong tủ sấy ở nhiệt độ 80 °C trong 4 giờ. Để nguội, thêm 10 ml nước, lắc để bột viên phân tán đều, thêm 80 ml acetonitril, siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
+ Dung dịch thử phân hủy ở điều kiện acid:
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API, vào bình định mức 100 ml. Thêm 5 ml dung dịch acid hydrocloric 1 M, đặt bình định mức này trong tủ sấy ở nhiệt độ 80°C trong 2 giờ. Tiếp tục tiến hành như đối với Dung dịch thử phân hủy ở điều kiện nhiệt độ.
+ Dung dịch thử phân hủy ở điều kiện base:
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API, vào bình định mức 100 ml. Thêm 5 ml dung dịch natri hydroxyd 1 M, đặt bình định mức này trong tủ sấy ở nhiệt độ 80°C trong 2 giờ. Tiếp tục tiến hành như đối với Dung dịch thử phân hủy ở điều kiện nhiệt độ.
+ Dung dịch thử phân hủy ở điều kiện khử.
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API, vào bình định mức 100 ml. Thêm 5 ml dung dịch H2O2 10%, đặt bình định mức này trong tủ sấy ở nhiệt độ 80°C trong 2 giờ. Tiếp tục tiến hành như đối với Dung dịch thử phân hủy ở điều kiện nhiệt độ.
+ Dung dịch thử phân hủy ở điều kiện oxi hóa:
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API, vào bình định mức 100 ml. Thêm 5 ml dung dịch KMnO4 5%, đặt bình định mức này trong tủ sấy ở nhiệt độ 80°C trong 2 giờ. Tiếp tục tiến hành như đối với Dung dịch thử phân hủy ở điều kiện nhiệt độ.
+ Dung dịch thử phân hủy ở điều kiện tia UV:
Cân chính xác một lượng bột viên tương ứng với khoảng 250 mg API, vào bình định mức 100 ml. Lắc nhẹ để lớp bột thành một lớp mỏng ở đáy bình. Để dưới đèn UV trong 4 giờ. Tiếp tục tiến hành như đối với chuẩn bị Dung dịch thử.
- Ghi lại các sắc ký đồ. Xác định thời gian lưu và diện tích píc của hoạt chất cần phân tích và các píc tạp khác; độ tinh khiết của píc hoạt chất cần phân tích trên sắc ký đồ các mẫu thử và mẫu đối chiếu.
* Yêu cầu:
- Sắc ký đồ dung dịch thử cho píc có thời gian lưu khác nhau không có ý nghĩa thống kê với thời gian lưu của píc chất chuẩn trên sắc ký đồ dung dịch mẫu chuẩn.
- Sắc ký đồ của mẫu trắng, placebo không xuất hiện píc ở trong khoảng thời gian lưu tương ứng với thời gian lưu của chất chuẩn. Nếu có đáp ứng píc (diện tích píc) phải < 1,0% so với đáp ứng píc của mẫu chuẩn.
- Píc của API thu được trên sắc ký đồ dung dịch thử, dung dịch thử khi phân hủy phải tinh khiết (purity index ≥ threshold)
5.2.2.2 Độ thích hợp của hệ thống
* Thực nghiệm:
- Dung dịch chuẩn; sử dụng dung dịch từ phần thẩm định độ chọn lọc, đi thi hành Tiến hành sắc ký dung dịch chuẩn, ghi lại sắc ký đồ, xác định giá trị thời gian lưu, diện tích píc trung bình, RSD, số đĩa lý thuyết và hệ số đối xứng.
* Yêu cầu:
- Số đĩa lý thuyết, N ≥ 2000 và hệ số đối xứng, T ≤ 2,0,
- Giá trị RSD của thời gian lưu ≤ 1,0% và của diện tích píc ≤ 5,0%,
5.2.2.3 Giới hạn phát hiện - Giới hạn định lượng
* Thực nghiệm:
Thêm chuẩn với lượng giảm dần vào mỗi lượng mẫu placebo tương đương với lượng bột viên chứa 250 mg API, Xử lý mẫu theo qui trình.
Tiến hành sắc ký dung dịch thu được, ghi lại các sắc ký đồ và xác định tỷ số S/N và RSD của diện tích píc.
* Yêu cầu:
- LOD: Tỷ số S/N ≥ 3 và RSDspic ≤ 10,0%.
- LOQ: LOD × 3,3.
5.2.3 Báo cáo kết quả thẩm định
5.2.3.1 Nguyên, vật liệu - thiết bị và dụng cụ
BY BUT HSI
* Chất chuẩn:
- API: Chuẩn Viện Kiểm nghiệm thuốc trung ương; Hàm lượng: 99,94%; Độ ẩm: 0,47%; SKS: WS.0218abc.02
* Mẫu thử, mẫu placebo:
- Mẫu thử: Viên nén API 250 mg.
- Mẫu placebo: Là mẫu chứa các tá dược bào chế viên được cân và trộn đều theo đúng tỷ lệ trong công thức. Khối lượng tương ứng một viên là 251,4 mg.
* Hóa chất thuốc thử: Acetonitril, acid acetic băng loại dùng cho HPLC của Merck, Đức
* Thiết bị phân tích: Máy HPLC Shimadzu SPD - M30A, máy HPLC Agilent 1260 hiệu chuẩn đạt.
5.2.3.2 Kết quả thẩm định
Độ chọn lọc
* Thực nghiệm:
- Mẫu trắng: Dung môi pha mẫu
Chuẩn bị các mẫu như chỉ dẫn ở phần đề cương. (Bảng PL2.1).
Bảng PL 2.1. Cách chuẩn bị mẫu thẩm định độ chọn lọc
| Mẫu | Lượng cân (mg) | Thể tích pha (mL) | Lấy (mL) | Thể tích pha (mL) | Lấy (mL) | Thể tích pha (mL) |
| Chuẩn | 20,01 | 100 | 2 | 50 | 1 | 20 |
| Placebo | 252,1 | 100 | 2 | 25 | ||
| Thử | 502,0 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy bằng nhiệt độ | 505,5 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy bằng acid | 508,1 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy bằng base | 506,9 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy ở điều kiện thử | 502,6 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy ở điều kiện oxi hóa | 500,7 | 100 | 2 | 25 | ||
| Mẫu thử phân hủy ở điều kiện tia UV | 499,5 | 100 | 2 | 25 |
* Kết quả:
Bảng PL 2.2. Kết quả thẩm định độ chọn lọc
| Mẫu-píc | Thời gian lưu (phút) | Thời gian lưu tương đối | Diện tích píc | % Tạp | Độ tinh khiết píc ứng với API |
| Placebo | - | - | - | - | - |
| Chuẩn | 14,250 | 1 | 27,1 | - | - |
| Thử | |||||
| Tạp 1 | 7,313 | 0,51 | 7,3 | 0,054 | - |
| API | 14,315 | 1 | 1180,7 | - | 999,970 |
| Tổng tạp | 0,054 | ||||
| Thử xử lý trong dung dịch acid | |||||
| Tạp 1 | 3,447 | 0,24 | 12,2 | 0,090 | - |
| Tạp 2 | 7,581 | 0,53 | 9,5 | 0,070 | - |
| API | 14,315 | 1 | 1161,9 | - | 999,269 |
| Tổng tạp | 0,160 | ||||
| Thử xử lý trong dung dịch base | |||||
| Tạp 1 | 7,501 | 0,52 | 23,3 | 0,172 | - |
| API | 14,315 | 1 | 1152,4 | - | 999,688 |
| Tổng tạp | 0,172 | ||||
| Thử xử lý với nhiệt | |||||
| Tạp 1 | 3,897 | 0,27 | 23,2 | 0,171 | - |
| Tạp 2 | 7,581 | 0,53 | 9,9 | 0,073 | - |
| API | 14,297 | 1 | 1128,4 | - | 999,953 |
| Tổng tạp | 0,244 | ||||
| Thử xử lý trong dung dịch KMnO4 | |||||
| Tạp 1 | 3,893 | 0,27 | 23,2 | 0,171 | - |
| Tạp 2 | 7,575 | 0,53 | 40,2 | 0,297 | - |
| Tạp 3 | 8,246 | 0,58 | 13,5 | 0,100 | - |
| API | 14,2886 | 1 | 1061,1 | - | 999,552 |
| Tổng tạp | 0,568 | ||||
| Thử xử lý trong dung dịch H2O2 | |||||
| Tạp 1 | 3,892 | 0,27 | 20,6 | 0,152 | - |
| Tạp 2 | 7,543 | 0,53 | 32,8 | 0,242 | - |
| API | 14,295 | 1 | 1075,4 | - | 999,630 |
| Tổng tạp | 0,394 | ||||
| Thử xử lý bằng tia UV | |||||
| Tạp 1 | 3,889 | 0,27 | 16,6 | 0,123 | - |
| Tạp 2 | 7,585 | 0,53 | 10,5 | 0,077 | - |
| API | 14,315 | 1 | 1125 | - | 999,715 |
| Tổng tạp | 0,200 | ||||
-: Không phát hiện
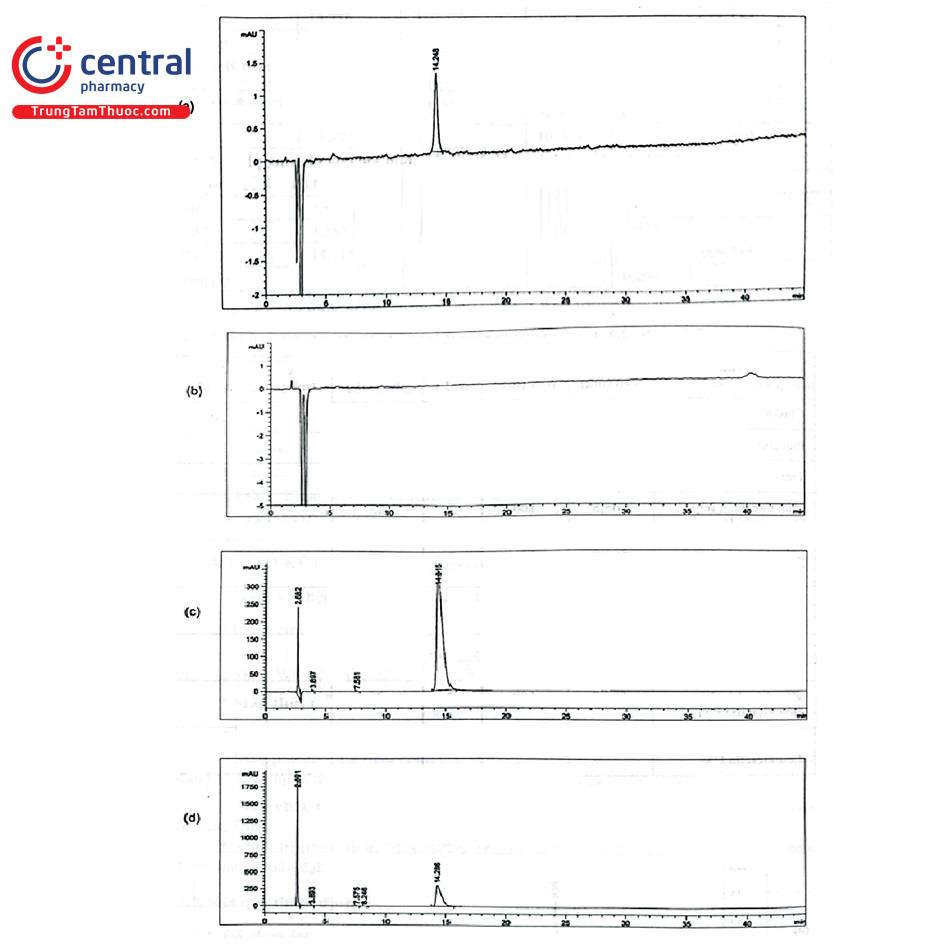
- Mẫu trắng và mẫu placebo không có píc nào có thời gian lưu tương ứng với thời gian lưu của píc API trên sắc ký đồ mẫu chuẩn.
- Mẫu thử cho pic chính có thời gian lưu tương ứng với thời gian lưu của API trên sắc ký đồ mẫu chuẩn (khoảng 14,3 phút). Một tạp có thời gian lưu 7,313 phút có hàm lượng 0,054%. Pic API tinh khiết với hệ số tinh khiết 999,170.
- Mẫu thử được xử lý trong các điều kiện khắc nghiệt, ngoài píc chính ứng với API và píc tạp có thời gian lưu khoảng 7,3 phút còn xuất hiện các píc tạp khác. Các píc tạp đều tách riêng biệt. Píc API đều có hệ số tinh khiết > 999.
* Kết luận: Phương pháp thử giới hạn tạp chất liên quan trong viên nén API 250 mg là chọn lọc.
Độ thích hợp của hệ thống
* Thực nghiệm:
- Dung dịch chuẩn: Sử dụng dung dịch chuẩn ở mục thử Độ chọn lọc.
- Tiêm lặp lại 6 lần dung dịch chuẩn vào hệ thống sắc ký, Ghi lại các SKĐ, xác định các thông số thời gian lưu, diện tích píc, số đĩa lý thuyết, hệ số đối xứng.
* Kết quả:
- Số đĩa lý thuyết của cột là 10754; hệ số kéo đuôi của píc chính là 1,2.
- Kết quả khảo sát độ lặp lại của dung dịch chuẩn:
Bảng PL 2.3. Kết quả khảo sát độ thích hợp hệ thống
| STT | Thời gian lưu (phút) | Diện tích píc (mAU.s) |
| 1 | 14,336 | 27,4 |
| 2 | 14,321 | 27,2 |
| 3 | 14,341 | 27,1 |
| 4 | 14,344 | 27,2 |
| 5 | 14,382 | 26,5 |
| 6 | 14,383 | 26,7 |
| Trung bình | 14,351 | 27,0 |
| RSD (%) | 0,16 | 1,27 |
* Nhận xét: RSD thời gian lưu < 1,0%; RSDdiện tích píc < 5,0%; N>2000; T<2,0=> hệ thống phù hợp thử giới hạn tạp chất liên quan trọng viên nén API 250 mg.
LOD-LOQ
* Thực nghiệm:
- Dung dịch chuẩn: Cân 20,01 mg chuẩn API vào bình định mức 100 ml, hòa tan và làm vừa đủ thể tích bằng dung môi pha mẫu, lắc đều.
- Mẫu thử 1: Cân 252,5 mg mẫu placebo, chuyển vào bình định mức 100 ml, thêm 0,50 ml dung dịch chuẩn, lắc đều. Thêm 10 ml nước, lắc để bột viên phân tán đều, thêm 80 ml acetonitril, lắc siêu âm 30 phút, để nguội đến nhiệt độ phòng, thêm acetonitril vừa đủ, lắc đều. Ly tâm. Hút chính xác 2 ml dịch trong phía trên vào bình định mức 25 ml, thêm dung môi pha mẫu vừa đủ đến vạch, lắc đều. Lọc qua màng lọc 0,45 um.
- Mẫu thử 2: Cân 252,0 mg mẫu placebo, chuyển vào bình định mức 100 ml, thêm 1 ml dung dịch chuẩn, lắc đều, tiếp tục tiến hành như đối với mẫu thử 1.
- Mẫu thử 3: Cân 251,8 mg mẫu placebo, chuyển vào bình định mức 100 ml, thêm ml dung dịch chuẩn, lắc đều, tiếp tục tiến hành như đối với mẫu thử 1.
Tiến hành sắc ký 6 lần mỗi dung dịch thử, ghi lại các sắc ký đồ, xác định tỷ số S/N và RSD của diện tích píc.
* Kết quả:
- Dung dịch thử 1 có nồng độ ~ 0,08 ug/ml cho đáp ứng píc gấp khoảng 3 lần độ nhiễu đường nền.
- Kết quả độ lặp lại của diện tích píc của dung dịch có nồng độ ở mức giới hạn định lượng được ghi trong bảng sau.
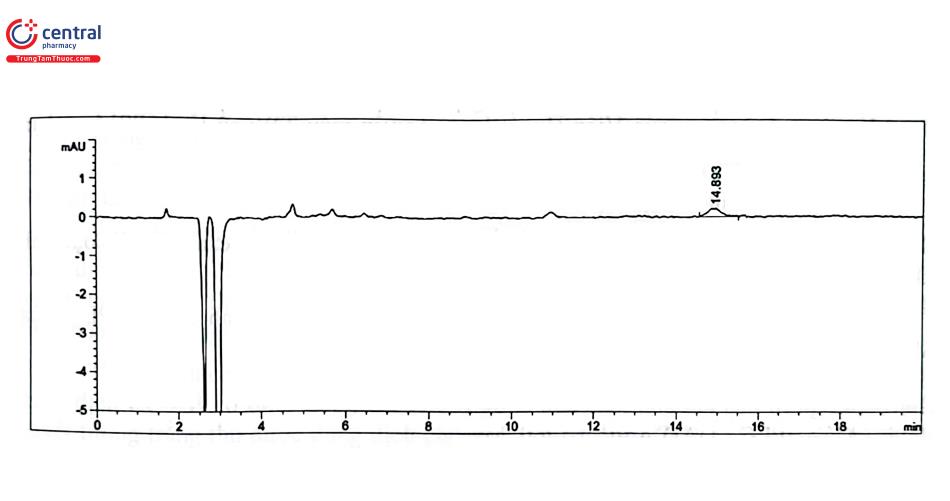
Bảng PL 2.4. Kết quả thẩm định độ lặp lại tại LOD
| STT | Thời gian lưu (phút) | Diện tích píc | S/N |
| 1 | 14,393 | 5,2 | 3,0 |
| 2 | 14,354 | 5,8 | 3,3 |
| 3 | 14,323 | 5,8 | 4,4 |
| 4 | 14,342 | 5,0 | 2,9 |
| 5 | 14,338 | 4,8 | 3,0 |
| 6 | 14,371 | 4,7 | 3,6 |
| Trung bình | 14,354 | 5,2 | 3,4 |
| RSD (%) | 0,18 | 9,27 (< 10,0%) |
=> LOD≈ 0,08 μg/ml
LOQ = 0,08×3,3≈ 0,264 μg/ml.
Kết luận: Qui trình phân tích theo Tiêu chuẩn cơ sở đáp ứng các yêu cầu về độ chọn lọc, LOD và LOQ để xác định tạp chất trong viên nén API 250 mg.
5.3 Phụ lục 3. Thẩm định phương pháp phân tích dịch sinh học (Xây dựng và lựa chọn mô hình đường chuẩn trong thẩm định qui trình định lượng gliclazid trong huyết tương)
5.3.1 Nguyên, vật liệu - thiết bị và dụng cụ
* Chất chuẩn:
- Chuẩn (CC): Gliclazid của Viện Kiểm nghiệm thuốc trung ương; SKS: WS.02151yy.01; hàm lượng: 99,53% (Nguyên trạng).
- Chuẩn nội (IS): Glipizid của Viện Kiểm nghiệm thuốc trung ương; SKS: 01072xx; hàm lượng: 99,17% (Nguyên trạng).
Huyết tương trắng: của Viện Huyết học - truyền máu TW; số lô: 16P014xx; hạn dùng: 05/08/20xy; bảo quản: - 35°C ± 5°C
* Hóa chất, thuốc thử:
- Methanol (MeOH), acetonitril (MeCN), diethyl ether, n - hexan đều là loại tinh khiết HPLC của Merck, Đức.
- Kali dihydrophosphat và acid phosphoric đều là loại tinh khiết HPLC của Merck, Đức.
* Thiết bị phân tích, dụng cụ:
- Máy HPLC - DAD Agilent 1260, Mỹ
- Cân phân tích Mettler Toledo XPE105 - Thụy Sỹ
- Máy đo pH Mettler
- Máy lắc xoáy Labinco
- Máy ly tâm lạnh Sigma
- Bộ cô mẫu bằng khí nitơ của Thermo
- Tủ lạnh sâu
- Tủ lạnh bảo quản chuẩn
- Micropipette: 10 - 100 μL; 100-1000 μL ; 500 - 5000 μL
- Ống nghiệm thủy tinh borosilicat 15 mL có nắp xoáy
- Bình định mức
- Pipet chính xác bằng thủy tinh.
5.3.2 Quy trình phân tích
Chuẩn bị các dung dịch chuẩn
Chuẩn tự tạo để xây dựng đường chuẩn
- Dung dịch chuẩn gốc để pha đường chuẩn (CC - A): Cân chính xác khoảng 50 chất chuẩn gliclazid, hòa tan trong vừa đủ 50,0 mL bằng MeOH thu được dung dịch chuẩn gốc có nồng độ CC chính xác khoảng 1000 kg/mL. Bảo quản ở 2 - 8°C và sử dụng trong 30 ngày.
- Dung dịch chuẩn nội gốc: Cân chính xác khoảng 50 mg chất chuẩn glipizid, hòa tan trong vừa đủ 50,0 mL bằng MeOH thu được dung dịch chuẩn nội gốc có nồng độ IS chính xác khoảng 1000 ug/mL. Bảo quản ở 2 - 8°C và sử dụng trong 30 ngày.
- Dung dịch chuẩn nội làm việc: Pha loãng 1,00 mL dung dịch chuẩn nội gốc trong vừa đủ 25,0 mL bằng hỗn hợp MeOH : nước (1:1) thu được dung dịch chuẩn nội làm việc có nồng độ chính xác khoảng 40 ug/mL.
- Dung dịch chuẩn CC - B : Lấy 1,00 mL dung dịch chuẩn CC - A pha trong vừa đủ 25,0 mL MeOH thu được dung dịch CC - B có nồng độ CC chính xác khoảng 40 μg/mL.
- Dung dịch chuẩn làm việc trong huyết tương WS - CC1: Lấy chính xác 2 mL dung dịch CC - B cô dưới dòng khí nitơ ở 40°C đến cắn, hòa tan cắn trong 20,00 mL huyết tương trắng thu được dung dịch chuẩn làm việc WS - CC1 có nồng độ CC chính xác khoảng 4000 ng/ml
- Dung dịch chuẩn làm việc trong huyết tương WS - CC2: Lấy chính xác 500 LL dung dịch CC - B cô dưới dòng khí nitơ ở 40°C đến cắn, hòa tan cắn trong 20,00 mL huyết tương trắng thu được dung dịch chuẩn làm viêc WS -CC2, có nồng độ CC chính xác khoảng 1000 ng/ml
Từ dung dịch chuẩn làm việc WS - CC1 và WS - CC2, phối hợp với huyết tương trắng theo bảng PL 3.1 để được các mẫu đường chuẩn trong khoảng nồng độ 30 – 4000 ng/mL.
Bảng PL 3.1. Chuẩn bị đường chuẩn
| Mẫu | Blank | Zero | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 |
| Nồng độ (ng/mL) | Blank | Zero | 30 | 100 | 500 | 1000 | 2000 | 2500 | 3000 | 4000 |
| V HT trắng (uL) | 1000 | 1000 | 970 | 900 | 500 | 1000 | 500 | 375 | 250 | 0 |
| V WS - CC1 (uL) | - | - | - | - | - | - | 500 | 625 | 750 | 1000 |
| V WS - CC2 (uL) | - | - | 30 | 100 | 500 | 1000 | - | - | - | - |
Xử lý mẫu
Thêm chính xác 50 ul dung dịch chuẩn nội làm việc vào 1 mL huyết tương đã chuẩn bị. Lắc xoáy 5 giây. Thêm 5 ml diethyl ether : n - hexane (80 : 20). Lắc ngang 5 phút, ly tâm 5000 vòng/ phút × 5 phút ở 4°C. Hút 3,5 ml lớp dung môi phía trên, cô khô dưới dòng khí N2 ở 40°C. Cho vào cắn 500 ul pha động (MeCN - dung dịch đệm phosphate 20 mM pH 4,2 tỷ lệ 55:45), lắc xoáy 30 giây, ly tâm 5000 vòng/ phút × 5 phút ở 4°C. Lấy lớp dịch phía trên để sắc ký.
Điều kiện phân tích:
- Cột: Atlantis dC18 (250 x 4,6 mm; 5 um)
- Tốc độ dòng: 1,0 ml/phút.
- Detector UV: 230 nm.
- Thể tích tiêm: 100 ul.
-Pha động: Acetonitril - nước được điều chỉnh đến pH = 2,5 ± 0,2 bằng acid phosphoric (55:45).
5.3.3 Báo cáo kết quả
5.3.3.1 Chuẩn bị mẫu xây dựng đường chuẩn
- Lượng cân chuẩn CC: 50,49 mg
- Lượng cân chuẩn IS: 50,28 mg
- Tiến hành chuẩn bị mẫu chuẩn để xây dựng đường chuẩn như chỉ 2. Chuẩn bị 05 đường chuẩn riêng biệt. Xử lý mẫu theo qui trình.
5.3.3.2 Kết quả sắc ký mẫu xây dựng đường chuẩn
Sắc ký các mẫu theo phương pháp qui định. Ghi lại sắc ký đồ và đáp ứng píc.
Kết quả:
Bảng PL 3.2. Diện tích píc các chất phân tích của các đường chuẩn thực nghiệm
| Nồng độ (ng/mL) | CC1 | CC2 | CC3 | CC4 | CC5 | |||||
| CC | IS | CC | IS | CC | IS | CC | IS | CC | IS | |
| 30,2 | 3495 | 194719 | 3153 | 183871 | 3700 | 182908 | 3050 | 172109 | 3042 | 172152 |
| 100,5 | 9922 | 196788 | 8980 | 187888 | 9471 | 185137 | 9095 | 174774 | 8886 | 173223 |
| 502,5 | 44493 | 197353 | 39337 | 171389 | 39790 | 171122 | 41048 | 171027 | 40438 | 168341 |
| 1005 | 88364 | 194284 | 84889 | 184980 | 85303 | 182885 | 79507 | 172724 | 79539 | 172733 |
| 2010 | 172711 | 194834 | 170200 | 186851 | 170909 | 185475 | 161175 | 176133 | 156061 | 174285 |
| 2513 | 232421 | 208025 | 215826 | 187374 | 221116 | 189331 | 200927 | 172509 | 200624 | 172528 |
| 3015 | 278740 | 212701 | 256308 | 185331 | 260231 | 185148 | 240267 | 166813 | 247023 | 174599 |
| 4020 | 339700 | 189422 | 340470 | 182974 | 315409 | 167034 | 320949 | 172762 | 331929 | 177773 |
5.3.3.3 Xử lý số liệu
Tính F
Từ dữ liệu thực nghiệm, dùng F - test để xác định mức độ đồng nhất của tập hợp kết quả thu được dựa trên kết quả của hai mức nồng độ LLOQ và ULOQ để kiểm tra.
Bảng PL 3.3. Kết quả đánh giá giá trị F
| Đường chuẩn | LLOQ | ULOQ | ||||
| CC | IS | CC/IS | CC | IS | CC/IS | |
| 1 | 3495 | 194719 | 0,0179 | 339700 | 189422 | 1,793 |
| 2 | 3153 | 183871 | 0,0171 | 340470 | 182974 | 1,861 |
| 3 | 3700 | 182908 | 0,0202 | 315409 | 167034 | 1,888 |
| 4 | 3050 | 172109 | 0,0177 | 320949 | 172762 | 1,858 |
| 5 | 3042 | 172152 | 0,0177 | 331929 | 177773 | 1,867 |
| VAR | 0,00000144 | 0,00127 | ||||
| F thực | 879,9 | |||||
| F bảng | 15,6 | |||||
Nhận xét: F thực >F bảng: Cần sử dụng hệ số trọng số.
Khảo sát và lựa chọn mô hình trọng số
- Khảo sát trên các mô hình trọng số: 1, 1/x, 1/x2, 1/x1/2 để lựa chọn mô hình phù hợp nhất.
Bảng PL 3.4. Kết quả mô hình trọng số: 1
| Dung dịch | Độ đúng (%) của đường chuẩn | Tổng | ||||
| CC1 | CC2 | CC3 | CC4 | CC5 | ||
| S1 | 101,6 | 140,4 | 151,0 | 114,0 | 132,2 | |
| S2 | 103,8 | 108,4 | 111,3 | 107,7 | 111,9 | |
| S3 | 99,7 | 100,2 | 99,6 | 102,0 | 103,5 | |
| S4 | 101,6 | 99,6 | 99,6 | 98,2 | 99,0 | |
| S5 | 99,5 | 98,6 | 98,3 | 97,8 | 96,2 | |
| S6 | 100,4 | 99,7 | 99,6 | 99,6 | 99,9 | |
| S7 | 98,2 | 99,7 | 99,9 | 102,6 | 101,3 | |
| S8 | 100,9 | 100,6 | 100,6 | 99,3 | 100,3 | |
| Slope | 0,000441 | 0,000461 | 0,000467 | 0,000465 | 0,000463 | |
| Intercept | 0,004418 | - 0,00238 | - 0,00107 | 0,001717 | - 0,00083 | |
| r | 0,9998 | 0,9999 | 0,9999 | 0,9997 | 0,9998 | |
| RE | 10,8 | 51,8 | 66,0 | 31,6 | 54,0 | 214,2 |
Bảng PL 3.5. Kết quả mô hình trọng số: 1/x
| Dung dịch | Độ đúng (%) của đường chuẩn | Tổng | ||||
| CC1 | CC2 | CC3 | CC4 | CC5 | ||
| S1 | 97,3 | 104,4 | 105,4 | 97,9 | 98,4 | |
| S2 | 102,5 | 98,0 | 98,1 | 103,1 | 102,2 | |
| S3 | 99,5 | 98,6 | 97,6 | 101,3 | 102,1 | |
| S4 | 101,6 | 99,2 | 99,1 | 98,0 | 98,6 | |
| S5 | 99,5 | 98,7 | 98,4 | 97,8 | 96,3 | |
| S6 | 100,4 | 99,9 | 99,9 | 99,7 | 100,1 | |
| S7 | 98,2 | 100,1 | 100,3 | 102,8 | 101,6 | |
| S8 | 100,9 | 101,0 | 101,2 | 99,5 | 100,6 | |
| Slope | 0,000441 | 0,000458 | 0,000463 | 0,000463 | 0,000461 | |
| Intercept | 0,00500 | 0,002722 | 0,00549 | 0,004024 | 0,003978 | |
| r | 0,9999 | 1,0000 | 0,9999 | 0,9998 | 0,9998 | |
| RE | 10,9 | 10,9 | 13,7 | 14,4 | 16,8 | 66,7 |
Bảng PL 3.6. Kết quả mô hình trọng số: 1/x2
| Dung dịch | Độ đúng (%) của đường chuẩn | Tổng | ||||
| CC1 | CC2 | CC3 | CC4 | CC5 | ||
| S1 | 99,2 | 100,8 | 100,8 | 99,0 | 99,3 | |
| S2 | 102,8 | 97,5 | 97,4 | 103,3 | 102,3 | |
| S3 | 99,3 | 99,2 | 98,2 | 101,2 | 102,0 | |
| S4 | 101,2 | 99,9 | 99,9 | 97,8 | 98,5 | |
| S5 | 99,1 | 99,5 | 99,3 | 97,6 | 96,1 | |
| S6 | 100,0 | 100,7 | 100,8 | 99,4 | 99,9 | |
| S7 | 97,9 | 100,8 | 101,3 | 102,5 | 101,4 | |
| S8 | 100,5 | 101,8 | 102,1 | 99,3 | 100,5 | |
| Slope | 0,000443 | 0,000454 | 0,000458 | 0,000465 | 0,000461 | |
| Intercept | 0,00469 | 0,003329 | 0,00627 | 0,003823 | 0,00384 | |
| r | 0,9999 | 0,9999 | 0,9999 | 0,9997 | 0,9998 | |
| RE | 9,1 | 8,1 | 10,2 | 13,9 | 12,4 | 53,7 |
Bảng PL 3.7. Kết quả mô hình trọng số: 1/x1/2
| Dung dịch | Độ đúng (%) của đường chuẩn | Tổng | ||||
| CC1 | CC2 | CC3 | CC4 | CC5 | ||
| S1 | 96,2 | 113,2 | 116,9 | 100,8 | 103,7 | |
| S2 | 102,2 | 100,5 | 101,4 | 103,9 | 103,7 | |
| S3 | 99,5 | 98,9 | 98,0 | 101,4 | 102,2 | |
| S4 | 101,5 | 99,2 | 99,1 | 98,0 | 98,6 | |
| S5 | 99,5 | 98,6 | 98,3 | 97,7 | 96,2 | |
| S6 | 100,4 | 99,8 | 99,7 | 99,6 | 100,0 | |
| S7 | 98,2 | 99,9 | 100,1 | 102,7 | 101,5 | |
| S8 | 100,9 | 100,8 | 100,9 | 99,4 | 100,5 | |
| Slope | 0,000441 | 0,000459 | 0,000465 | 0,000464 | 0,000461 | |
| Intercept | 0,005141 | 0,001465 | 0,003831 | 0,003602 | 0,003218 | |
| r | 0,9999 | 1,0000 | 0,9999 | 0,9998 | 0,9998 | |
| RE | 11,7 | 18,1 | 24,2 | 14,1 | 16,8 | 84,9 |
Bảng PL 3.8. Kết quả tổng hợp xác định hệ số
| 1/x2 | 53,8 |
| 1 | 213,9 |
| 1/x1/2 | 84,7 |
| 1/x | 63,2 |
| Lựa chọn hệ số trọng số: 1/x2 | |
.jpg)
Kết quả xây dựng đường chuẩn của mô hình lựa chọn (1/x2)
Bảng PL 3.9. Kết quả khảo sát đường chuẩn theo mô hình trọng số 1/x2
| Đường chuẩn | Mẫu | S1 | S2 | S3 | S4 | S5 | S6 | S7 | S8 |
| Nồng độ (ng/mL) | 30,2 | 100,5 | 502,5 | 1005 | 2010 | 2513 | 3015 | 4020 | |
| CC1 | Độ đúng % | 99,2 | 102,8 | 99,3 | 101,2 | 99,1 | 100,0 | 97,9 | 100,5 |
| PTHQ, r | y = 0,000443*x + 0,004691; r = 0,9999 | ||||||||
| CC2 | Độ đúng % | 100,8 | 97,5 | 99,2 | 99,9 | 99,5 | 100,7 | 100,8 | 101,8 |
| PTHQ, r | y = 0,000454*x + 0,003330; r = 0,9999 | ||||||||
| CC3 | Độ đúng % | 100,0 | 97,4 | 98,2 | 99,9 | 99,3 | 100,9 | 101,2 | 102,1 |
| PTHQ, r | y = 0,000458*x + 0,006269; r = 0,9999 | ||||||||
| CC4 | Độ đúng % | 99,0 | 103,3 | 101,2 | 97,7 | 97,6 | 99,4 | 102,5 | 99,3 |
| PTHQ, r | y = 0,000465*x + 0,003823; r = 0,9997 | ||||||||
| CC5 | Độ đúng % | 99,3 | 102,4 | 102,0 | 98,5 | 96,1 | 99,9 | 101,4 | 100,5 |
| PTHQ, r | y = 0,000461*x + 0,003839; r = 0,9998 | ||||||||
y: tỷ số diện tích píc CC/IS; x: Nồng độ CC trong huyết tương
Kết luận: Đường chuẩn thực nghiệm theo mô hình trọng số 1/x2 đều thoả mãn các yêu cầu qui định
- Độ đúng so với giá trị thực của các nồng độ đều đạt từ 85 - 115%,trừ tại điểm LLOQ được chấp nhận 80% - 120%.
- Có hệ số tương quan r≥ 0,98
6 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Thẩm định phương pháp phân tích”, Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 182 - 230. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược Điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
