Bệnh Thalassemia: Cơ chế di truyền, cách chẩn đoán, điều trị và phòng bệnh
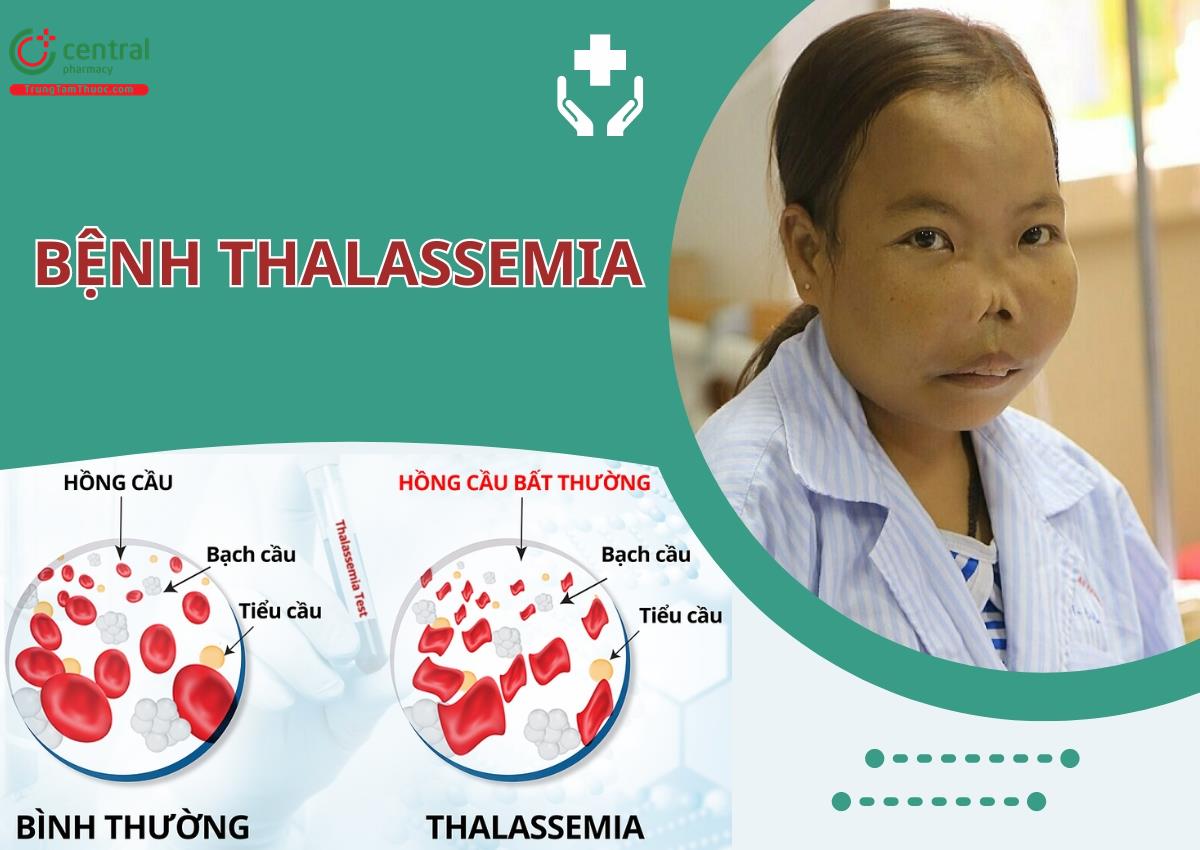
Trungtamthuoc.com - Thalassemia là một chứng rối loạn máu di truyền khiến cơ thể có ít hemoglobin hơn bình thường, gây thiếu máu và khiến cơ thể mệt mỏi. Bài viết dưới đây sẽ giúp bạn đọc hiểu cơ chế di truyền, cách chẩn đoán, điều trị và phòng bệnh Thalassemia.
1 TỔNG QUAN
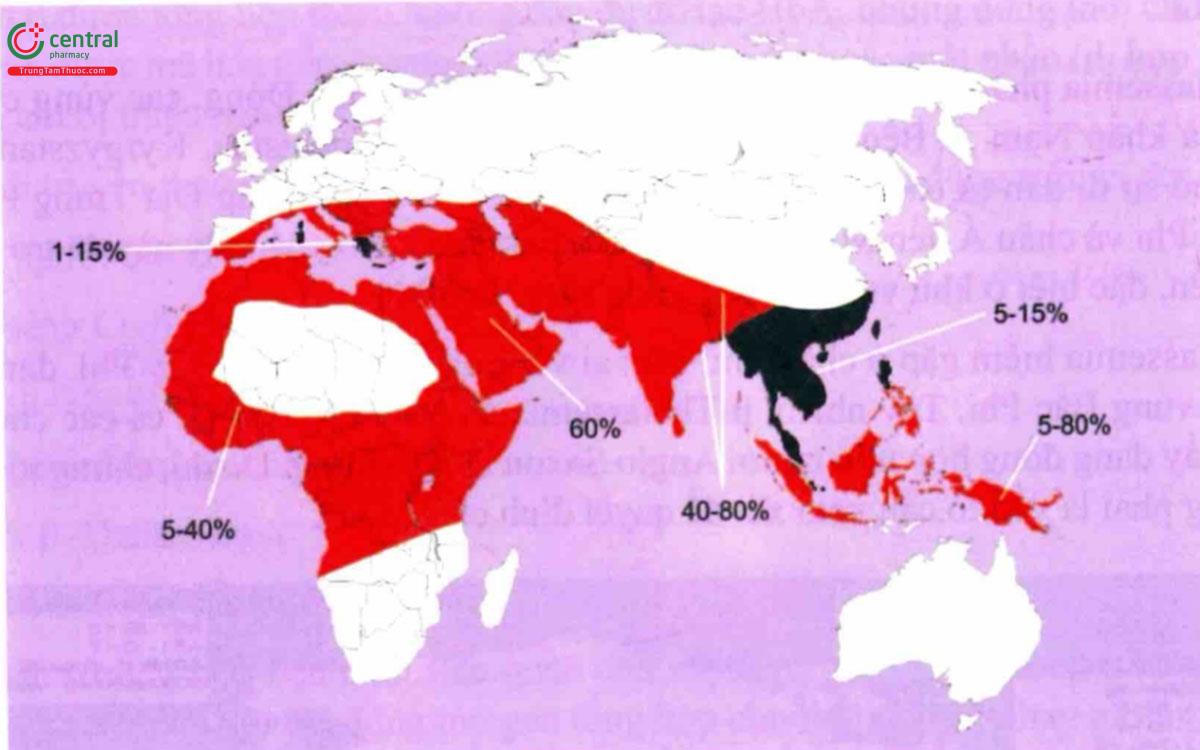
Bệnh Thalassemia là bệnh di truyền phổ biến nhất ở người. Bệnh xảy ra với tần suất cao khắp vùng Địa Trung Hải, Trung Đông, Ấn Độ, Myanmar, trải dọc từ Nam Trung Quốc qua Thái Lan đến Malaysia và các cộng đồng dân cư đào khu vực Thái Bình Dương. Bệnh cũng có thể gặp ở các nước có dân nhập cư từ các dân số có tần suất mắc bệnh cao.
Có hai loại Thalassemia chính, α và β (có liên quan đến gen α và β) và những dạng hiếm gặp hơn gây ra do bất thường gen globin khác. Tất cả những tình trạng này là do sự mất cân bằng trong việc sản xuất các chuỗi globin trong phân tử hemoglobin người lớn, làm dư thừa chuỗi α trong β-Thalassemia và dư chuỗi β trong α-Thalassemia.
Có hàng trăm loại đột biến khác nhau tại locus globin α và β đã được xác định là nguyên nhân của giàm hay mất sản xuất α và B. [1]
2 LỊCH SỬ PHÁT HIỆN VÀ ĐỊNH NGHĨA
Năm 1910, J.B. Herrick báo cáo một trường hợp thiếu máu nặng ở một sinh viên da đen người Jamaica với hồng cầu có dạng lạ: khum như hình trăng lưỡi liềm, kéo dài ra.
Năm 1917, Emmel báo cáo gặp hình dạng bất thường của hồng cầu ở người có vẻ ngoài bình thường, trường hợp này được biết sau này thuộc dạng dị hợp tử.
Năm 1927, Hahn và Gillespie ghi nhận có thể gây biến dạng in vitro hồng cầu ở một số bệnh nhân thành hồng cầu hình liềm bằng cách làm thay đổi pH môi trường, thay đổi nồng độ oxy,... Hai ông cũng sau đó nhận thấy có thể gây liềm hóa in vivo hồng cầu các bệnh nhân trên bằng cách gây giảm nồng độ oxy – máu. Hahn gọi tên những người lành có hồng cầu hình dạng bình thường nhưng có thể bị đổi dạng thành hình liềm nếu bị giảm oxy – máu là “sickle cell trait". Khi tiến hành khảo sát gia đình những bệnh nhân thì thấy đặc điểm này được di truyền theo tính trội.
Năm 1925, Cooley mô tà bệnh thiếu máu mạn tính, thường gặp ở các dân cư vùng Địa Trung Hài. Bệnh nhân có các đặc điểm biển dạng xương, thâm da, suy tim, lách to, gan to, hồng cầu hình dạng khác nhau, kích thước không đồng nhất, có hồng cầu nhân xuất hiện ở máu ngoại vi và bệnh nhân thường tử vong rất sớm.
Năm 1932, George H. Whipple và William L. Bradford xuất bản một bài báo về triệu chứng trong bệnh này. Whipple đưa ra thuật ngữ thiếu máu miền biển (thalassic anemia) và viết liền lại thành Thalassemia, vì tất cả những bệnh nhân đầu tiên đều là người Địa Trung Hài.
Năm 1940, M. Wintrobe mô tả một bệnh cảnh lâm sàng nhẹ hơn nhưng với đặc điểm tế bào tương tự như các trường hợp bệnh do Cooley mô tà, sau này được biết là dạng dị hợp tử. Đến thời điểm này, bệnh được mô tả bởi Cooley và Lee là dạng đồng hợp tử của 1 gen nằm trên NST thường, trong khi dạng dị hợp tử thường kèm với những biến đổi huyết học nhẹ hơn. Tình trạng đồng hợp tử nặng được gọi là Thalassemia major (thể nặng). Tình trạng dị hợp tử, Thalassemia trait, để chỉ Thalassemia minor (thể nhẹ) hay Thalassemia minima (rất nhẹ). Sau này, thuật ngữ Thalassemia intermediate (thể vừa) được dùng để mô tả những rối loạn nhẹ hơn thể nặng nhưng nặng hơn Thalassemia trait.
Năm 1949, Linus Pauling và Itano điện di và phân tách ra được các hemoglobin (Hb) khác nhau, đặt nền móng cho khái niệm về cơ chế sinh lý của bệnh ở mức độ sinh học phân từ.
Năm 1956, Ingram chứng minh có sự thay đổi một hoặc một đoạn acid amin trên chuỗi phân tử Hb và đối chiếu với các công trình nghiên cứu của Watson và Crick về gen, đưa ra khái niệm về bệnh lý gen đầu tiên là bệnh Hemoglobin (Hemo-globinopathy).
3 ĐẶC ĐIỂM DI TRUYỀN VÀ DANH PHÁP
Các gen điều khiển tổng hợp Hb nằm ở các nhiễm sắc thể 11 và 16. Ở người, Hb có 6 tiểu đơn vị do 6 nhóm gen tạo ra: nhóm gen trên nhiễm sắc thể 16 được gọi là nhóm gen “giống α" (α-like), còn nhóm gen trên nhiễm sắc thể 11 được gọi là “giống β” (β-like) hoặc “không-α" (non-α).
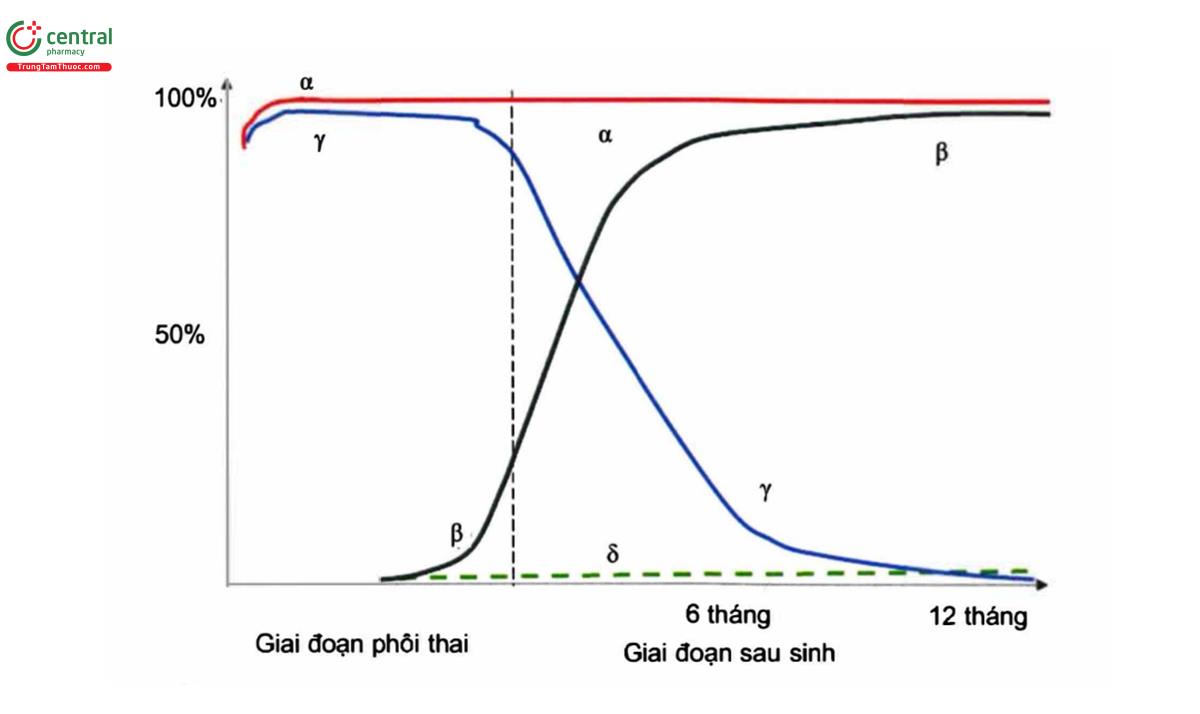
Việc sản xuất các chuỗi Hb thay đổi tuần tự từ giai đoạn phôi, thai, sơ sinh, nhũ nhi đến người trường thành. Thực tế, trẻ con qua khỏi 6 tháng tuổi đã phải có các thành phần Hb tương tự như người lớn bình thường.
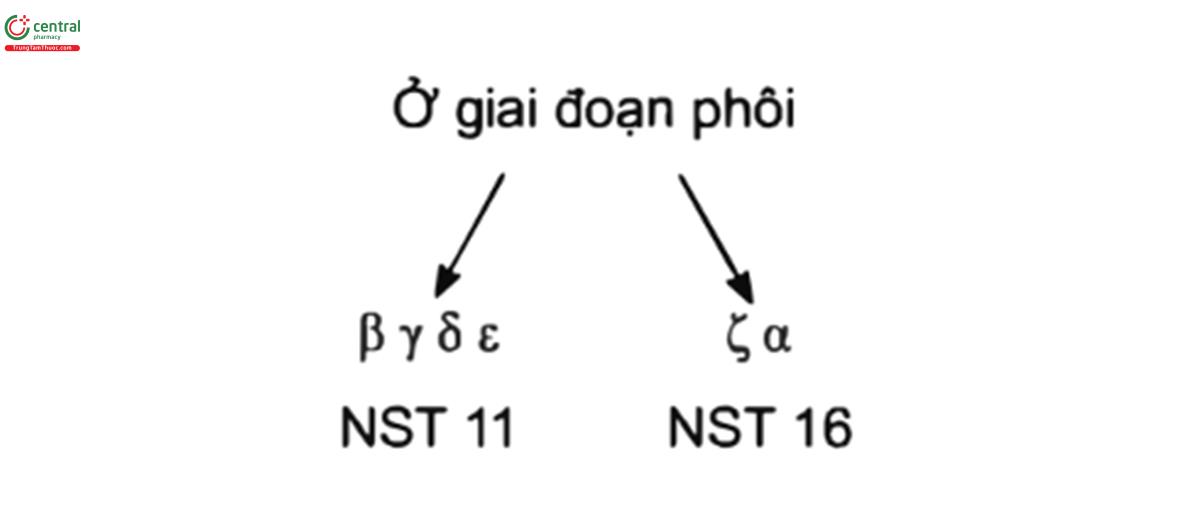
Theo sơ đồ tổng hợp các chuỗi Hb, bình thường các Hb sau đây sẽ tuần tự xuất hiện và biến mất đi theo thời gian phát triển của phôi thai và giai đoạn sau sinh:
* Giai đoạn phôi
Hb Gower 1:E2e2
Hb Gower 2: α2ε2
Hb Portland: E2γ2
* Giai đoạn thai:
HbF: α2γ2
* Giai đoạn sau sinh:
HbA: α2β2
HbA2: α2δ2
Do tính cân đối trong quá trình sản xuất các chuỗi, ở người bình thường tỉ lệ α/non-α = 1. Các rối loạn di truyền Hb xảy ra có thể được chia làm ba nhóm lớn:
Bệnh Hb (hemoglobinopathy): có đột biến gây thay đổi cấu trúc polypeptid của globin.
Thalassemia: giảm hoặc không tổng hợp được một hoặc vài chuỗi globin làm mất cân bằng tỉ lệ tương đối giữa các chuỗi globin.
Tồn lưu di truyền Hb thai (HPFH: Hereditary Persistance of Fetal Hb), một nhóm bệnh lành tính; cơ chế chuyển đảo tổng hợp từ y sang β-globin sau khi chào đời không được thực hiện, chuỗi β được tổng hợp thiếu, không đầy đủ để tạo HbA, nhưng đồng thời chuỗi y không biến mất vẫn được mã hóa tiếp ở lượng nhiều (đồng hợp tử) hoặc một phần (dị hợp tử), do vậy bệnh nhân chỉ bị thiếu máu nhẹ và HbF vẫn tồn tại.
Bệnh di truyền theo định luật Mendel và hầu hết các gen đều đồng trội, ví dụ:
- AA: người lành bình thường
- FF: bệnh Cooley, Thalassemia major
- AF: dị hợp tử, có bệnh nhưng nhẹ
- AS: bệnh hồng cầu hình liềm nhẹ
- SF: S-B-Thalassemia
- SS: bệnh hồng cầu hình liềm nặng.
90% các trường hợp bệnh chỉ liên quan đến một gen, gây thay thế một acid amin trên chuỗi globin, hay thay đổi mã đóng mở gen tổng hợp chuỗi globin này hay globin khác. 10% các trường hợp còn lại là các sai lệch, đào đoạn, đứt đoạn, ghép đoạn gen hay kéo dài gen mã hóa một globin bất thường,...
Hemoglobin giữ vai trò tối quan trọng trong cơ thể là giao – nhận – vận chuyển khí; nhận oxy ở phổi, vận chuyển trong hồng cầu và nhường oxy cho mô, làm động tác ngược lại đối với CO,. Lượng oxy khuếch tán trong huyết tương cũng làm vai trò tương tự, nhưng ở một lượng rất thấp. Hb có khả năng vận chuyển oxy cao gấp 80 lần so với huyết tương, do vậy biến động trong thành phần và cấu trúc phân tử của Hb có thể làm thay đổi nghiêm trọng sinh lý hồng cầu, gây ra ba hậu quả cơ bản:
- Tán huyết: thiếu máu nặng
- Ái lực Hb với oxy tăng cao: bệnh nhân bị đa hồng cầu thứ phát, da niêm có màu đỏ rực
- Ái lực Hb với oxy giàm thấp: bệnh nhân bị tím tái.
4 DANH PHÁP
Các chuỗi globin được gọi theo chữ Hy Lạp: α, β, γ, δ,... Các đột biến trên chuỗi được ghi chú lên trên như số mũ, ví dụ: βA là chuỗi β-globin bình thường, βs là chuỗi β bị đột biến gây bệnh hồng cầu liềm (HbS). Để minh họa cụ thể hơn loại đột biến, người ta ghi chú vị trí acid amin bị đột biến và tên acid amin thay vào bất thường, ví dụ: HbS có thể được ghi là β6Glu ->Val nghĩa là trên chuỗi β-globin của bệnh hồng cầu liềm, acid amin thứ 6 là glutamin bị thay thế bởi valin; tương tự như vậy HbE có thể ghi là β26Glu ->Lys cùng ý nghĩa như trên.
Theo quy ước đó, ta có thể hiểu như sau:
βA/βA: người bình thường.
βA/βS: dị hợp tử hồng cầu liềm.
βS/βS: đồng hợp từ hồng cầu liềm.
βA/βE: dị hợp tử HbE.
5 DỊCH TỄ
β-Thalassemia phân bố rộng ở dân số Địa Trung Hải, Trung Đông, các vùng của Ấn Độ, Pakistan và khắp Nam Á. Bệnh phổ biến ở Tajikistan, Turkmenistan, Kyrgyzstan và Trung Quốc. Vì có sự di dân từ các khu vực có tần suất bệnh cao như vùng Địa Trung Hải (Ý, Hy Lạp), châu Phi và châu Á đến Mỹ, gen a và β-Thalassemia cùng bệnh lý này đã trở nên tương đối phổ biến, đặc biệt ở khu vực Bắc Mỹ cũng như Nam Mỹ.
β-Thalassemia hiếm gặp ở châu Phi, trừ vài vùng riêng biệt trong Tây Phi, đáng chú ý là Liberia và vùng Bắc Phi. Tuy nhiên, B-Thalassemia xảy ra lác đác ở tất cả các chủng tộc và quan sát thấy dạng đồng hợp từ ở người Anglo Saxon thuần chủng. Do đó, chủng tộc của bệnh nhân không phải là yếu tố cần xem xét để quyết định chẩn đoán.
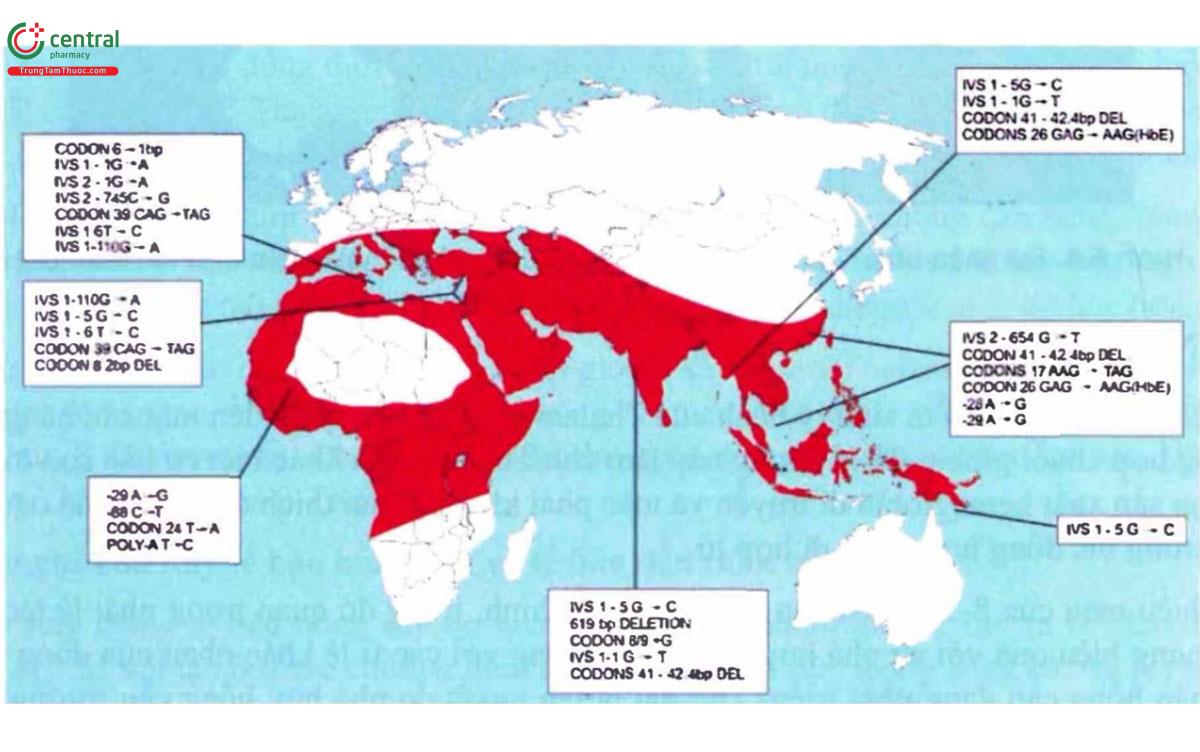
Bệnh α-Thalassemia xảy ra khắp châu Phi, Địa Trung Hải, Trung Đông và Nam Á. αº Thalassemia được tìm thấy hầu như phổ biến ở cộng đồng người Địa Trung Hải và phương Đông, nhưng cực kỳ hiếm ở cộng đồng người Trung Đông và châu Phi. Tuy nhiên, dạng α+ Thalassemia xảy ra với tần suất cao khắp Tây Phi, Địa Trung Hải, Trung Đông và Nam Á. Ở Mỹ, khoảng 30% người Mỹ gốc Phi mang gen α+ Thalassemia. Có đến 80% cộng đồng người ở một số vùng của Papua New Guinea mang gen của at Thalassemia. Mức phổ biến của α+ Thalassemia ở những cộng đồng riêng biệt thì chưa được xác định, nhưng bệnh được báo cáo khá nhiều ở một số cộng đồng người Trung Đông và Nam Á. Những đột biến điểm kết thúc chuỗi α như hemoglobin Constant Spring, dường như đặc biệt hay gặp ở Nam Á. Khoảng 4% cộng đồng người ở Thái Lan mang gen.
6 BỆNH HỌC
Hầu hết các đặc điểm sinh lý bệnh của Thalassemia có liên quan đến mất cân bằng trong sự tổng hợp chuỗi globin. Hiện tượng này làm cho Thalassemia khác biệt cơ bản so với tất cả rối loạn sản xuất hemoglobin di truyền và mắc phải khác và giải thích được mức độ nặng của bệnh trong thể đồng hợp tử và dị hợp tử.
Thiếu máu của B-Thalassemia có ba cơ chế chính, trong đó quan trọng nhất là tạo hồng cầu không hiệu quả với sự phá hủy trong tủy xương với các tỉ lệ khác nhau của dòng tế bào tiền thân hồng cầu đang phát triển. Thứ hai là tán huyết do phá hủy hồng cầu trưởng thành có chứa thể vùi của chuỗi a và cuối cùng là hồng cầu nhỏ và nhược sắc do giảm tổng hợp hemoglobin chung.
Vì sự khiếm khuyết ban đầu trong β-Thalassemia liên quan đến sự giảm sản xuất chuỗi β, việc tổng hợp hemoglobin F và A2 không bị ảnh hưởng. Sự sản xuất hemoglobin thai nhi trong tử cung diễn ra bình thường. Biểu hiện lâm sàng của Thalassemia chỉ xuất hiện khi trẻ sơ sinh có sự thay đổi sản xuất từ chuỗi y sang B. Các trường hợp β-Thalassemia dị hợp tử có sự tăng nồng độ hemoglobin A2. Mức gia tăng có vẻ không chỉ phản ánh sự giảm tương đối hemoglobin A do tổng hợp thiếu hụt chuỗi β mà còn là sự tăng tuyệt đối đầu ra của chuỗi ô ở cả hai dạng cis và trans với gen globin β đột biến.
Vì chuỗi α được chia cho các hemoglobin F, A và A2, nên không có sự gia tăng hemoglobin F trong α-Thalassemia. Chuỗi y và β dư thừa được tạo ra là kết quả của thiếu hụt sản xuất chuỗi a tạo thành thể vùi lắng đọng trên màng hồng cầu trưởng thành. Do đó, sự tạo hồng cầu không hiệu quả ở α -Thalassemia ít hơn trong β-Thalassemia, nguyên nhân chính của tình trạng thiếu máu là tình trạng tán huyết và nồng độ hemoglobin thấp trong hồng cầu.
6.1 Sự tổng hợp chuỗi globin không cân bằng
Việc đo sự tổng hợp chuỗi globin trong máu hay tủy xương in vitro ở các bệnh nhân thuộc các nhóm bệnh Thalassemia khác nhau kết hợp với các nghiên cứu khảo sát gia đình cho phép khám phá hoạt động của các gen Thalassemia đã cung cấp một bức tranh rõ ràng về hoạt động của các yếu tố quyết định bệnh.
Ở β-Thalassemia đồng hợp tử, sự tổng hợp globin β có thể bị giảm nhiều hoặc hoàn toàn không có. Hậu quả là chuỗi globin α bị sản xuất dư thừa. Chuỗi globin α tự do có thể tồn tại, kết tủa và lắng đọng trong tế bào tiền thân hồng cầu. Các thể vùi được tạo ra có thể quan sát được dưới cả kính hiện vi quang học và điện tử. Trong tủy xương, sự lắng đọng chuỗi α có thể nhìn thấy ở các tế bào tiền thân hồng cầu và trong suốt con đường biệt hóa trưởng thành của hồng cầu. Những thể vùi này chịu trách nhiệm trong việc tế bào tiền thân hồng cầu trong tủy xương bị phá hủy, đồng thời chịu trách nhiệm cho tình trạng tạo hồng cầu không hiệu quả trong tất cả các dạng β-Thalassemia. Một tỉ lệ lớn erythroblast đang phát triển bị hủy trong tùy xương ở các trường hợp bệnh nặng. Một số hồng cầu đã bị phá hủy trước khi trưởng thành.
β-Thalassemia dị hợp tử cũng có sự tổng hợp chuỗi globin không cân bằng, nhưng độ thừa chuỗi α thì ít hơn nhiều và có thể được giải quyết bởi enzyme của tế bào tiền thân hồng cầu. Tuy nhiên, việc tạo hồng cầu không hiệu quả vẫn xảy ra nhưng ở mức độ nhẹ hơn.
Dù vẫn có sự mất cân bằng về tỉ lệ chuỗi globin xảy ra ở α-Thalassemia thể nặng, chuỗi y và β thừa sẽ vẫn tạo ra thể vùi nhưng lại không kết tủa trong tế bào tiền thân hồng cầu giống như trong β-Thalassemia. Do đó, sinh bệnh học của thiếu máu khác nhau cơ bản giữa hai nhóm α-Thalassemia và β-Thalassemia.
6.2 Cơ chế của hủy tế bào hồng cầu và tế bào tiền thân hồng cầu
Sự hủy màng tế bào hồng cầu do quá trình kết tủa chuỗi globin xảy ra bởi hai đường chính: (1) thể vùi tạo bởi các chuỗi α thừa làm phá hủy cấu trúc màng hồng cầu và (2) sự phá hủy tương tự qua trung gian sản phẩm thoái gián của chuỗi α thừa. Các sản phẩm thoái giáng của chuỗi globin α tự do, heme, hemin (oxidized heme) và Fe tự do cũng đóng vai trò trong quá trình phá hủy màng hồng cầu. Chuỗi globin thừa gắn kết với các protein màng khác nhau làm thay đổi cấu trúc và chức năng của chúng. Sắt thừa, bằng cách tạo ra các gốc oxy hóa tự do, sẽ phá hủy thành phần màng hồng cầu (gồm lipid và protein) và các tiều cơ quan nội bào. Heme và sản phẩm của nó có thể xúc tác sự hình thành các dạng oxygen phản ứng có thể phá hủy màng hồng cầu. Các hồng cầu mất khả năng thay đổi hình dạng và đàn hồi, trở lên cứng và mất nước (underhydrated), rò ri Kali, tăng nồng độ calcium và nồng độ ATP thấp, không bền. Sự phá hủy hồng cầu cũng có thể qua sự hiện diện các thể vùi cứng khi hồng cầu di chuyển ngang qua lách.
Kết quả của việc sản xuất chuỗi non-α thừa trong α-Thalassemia khá khác biệt. Vì chuỗi α được chia sẻ bởi cả hai hemoglobin thai và người lớn, việc sản xuất chuỗi α thiếu hụt biểu hiện ở cả thai nhi và người lớn. Trong thai kỳ, việc này dẫn đến việc sản xuất thừa chuỗi y trong khi ở người lớn, việc này dẫn đến việc sản xuất thừa chuỗi β. Chuỗi y thừa tạo nên homotetramer y, hay hemoglobin Bart's; chuỗi β dư thừa tạo nên homotetramer β, hay hemoglobin H. Thực tế chuỗi y và β tạo nên homotetramer là nguyên nhân của sự khác nhau cơ bản trong sinh bệnh học của a và β-Thalassemia. Vi tetramer y, và β, có thể hòa tan, nên chúng không tạo kết tủa trong tế bào tiền thân hồng cầu trong tủy xương, do đó, α-Thalassemia không đặc trưng bởi sự tạo hồng cầu không hiệu quả. Tuy nhiên, tetramer β, kết tủa khi hồng cầu già sự hình thành các thể vùi. Chính vì vậy, thiếu máu của α-Thalassemia nặng hơn ở người lớn do hồng cầu bị phá hủy trong các vi mạch của lách bởi sự hiện diện thể vùi. Tương tự như trong β-Thalassemia, chuỗi α thừa làm phá hủy, oxy hóa và không ổn định cơ học nhiều loại protein màng, bệnh nhân α-Thalassemia có màng hồng cầu rất bền và không có bằng chứng oxy hóa hay rối loạn chức năng protein màng hồng cầu. Hơn nữa, sự lắng đọng các chuỗi β gây nên tình trạng hydrat hóa gia tăng ở α-Thalassemia.
Yếu tố khác làm nặng thêm sự thiếu oxy của tình trạng thiếu máu của α-Thalassemia, chính là cả hai hemoglobin Bart's và hemoglobin H không thể hiện sự tương tác heme-heme và hầu như đường cong phân ly oxygen hình hyperbol với ái lực với oxy rất cao. Do đó, chúng không thể phóng thích oxygen tại mô sinh lý. Vì vậy, trong thực tế, chúng chuyên chở oxy vô dụng. Hậu quả, trẻ em có hemoglobin Bart's cao thường thiếu oxy trong thai kỳ nghiêm trọng. Đây là điều cơ bàn trong bệnh cành lâm sàng & Thalassemia đồng hợp từ, dẫn đến thai phù, chết lưu muộn trong thai kỳ hay trong lúc sinh. Sự thiếu oxy được phản ánh bằng tình trạng phủ nghiêm trọng ở trẻ, có thể là hậu quả của sự gia tăng tính thẩm mao mạch và gia tăng số lượng hồng cầu nghiêm trọng. Sự cung cấp oxy cho thai thiếu hụt có khả năng dẫn đến loạn dưỡng nhau thai và có thể đi kèm bất thường phát triển thường xảy ra ở bệnh nhân α-Thalassemia thể nặng trong tử cung.
6.2.1 Hậu quả của cơ chế bù trừ tình trạng thiếu máu trong Thalassemia
Thiếu máu nặng trong β-Thalassemia đồng hợp tử và ái lực với oxy tương đối cao của hemoglobin F tạo nên tình trạng thiếu oxy mô trầm trọng. Tương tự, hemoglobins Bart's và H trong bệnh lý α-Thalassemia thể nặng cũng có ái lực rất cao với oxy, cũng tạo ra tình trạng thiếu hụt oxy mô trầm trọng. Để thích nghi với tình trạng thiếu oxy mô này, Erythropoietin được gia tăng sản xuất. Điều này đưa đến sự tăng tạo hồng cầu. Kết quả là sự biến dạng của xương sọ và mặt và sự xốp các xương dài. Ngoài việc gây ra các biến dạng xương, tủy xương còn có thể gây gãy xương bệnh lý và nhiễm trùng xoang và tai giữa do việc dẫn lưu không hiệu quả.
6.2.2 Lách to
Sự tiếp xúc liên tục của lách với hồng cầu có chứa thể vùi của chuỗi globin kết tủa là nguyên nhân gây lách to. Lách to tiến triển xảy ra ở cả hai dạng α và β-Thalassemia và làm trầm trọng thêm thiếu máu. Lách to hoạt động như một hố chứa hồng cầu, cô lập một tỉ lệ đáng kể khối hồng cầu. Hơn nữa, lách to có thể gây ra sự gia tăng thể tích huyết tương. Sự phối hợp pha trộn hồng cầu trong lách và sự gia tăng thể tích huyết tương có thể làm nặng thêm cả hai nhóm bệnh lý α và β-Thalassemia.
6.2.3 Chuyển hóa sắt bất thường
Thiếu máu trong β-Thalassemia đồng hợp tử thường làm gia tăng hấp thu sắt qua đường ruột. Sự hấp thu sắt bị giàm do truyền máu. Sự hấp thu tăng gây nên tình trạng ứ sắt, trước tiên ở tế bào Kupffer của gan, đại thực bào ở lách và sau đó là các tế bào võng nội mô ở gan. Hầu hết bệnh nhân đồng hợp tử β-Thalassemia phải truyền máu định kỳ; do đó, làm nặng thêm tình trạng ứ sắt. Sắt lắng đọng trong các tuyến nội tiết, đặc biệt là tuyến phó giáp, tuyến yên, tuyến tụy, gan và quan trọng nhất là ở cơ tim.
Sự lắng đọng sắt ở cơ tim dẫn đến tử vong do liên quan dẫn truyền hoặc do suy tim khó chữa. Hậu quả khác của quá tài sắt gồm tiểu đường, suy tuyến phó giáp, suy giáp và bất thường chức năng vùng hạ đồi – tuyến yên dẫn đến chậm phát triển và suy sinh dục. Đo trữ lượng sắt ở gan giúp phản ảnh chính xác lượng sắt trong cơ thể, góp phần xác định bệnh nhân ó nguy cơ mắc các biến chứng nặng do quá tải sắt. Kết quả nghiên cứu trên các bệnh nhân có tình trạng ứ sắt di truyền cho thấy những trường hợp có LIC # 80 µmol/g gan tươi (~15 mg/g gan khô), làm tăng nguy cơ bệnh gan và các bệnh nội tiết khác. Bệnh nhân với tình trạng ứ sắt nặng hơn sẽ gia tăng nguy cơ bệnh tim và tử vong sớm. Rối loạn chuyển hóa sắt ít gặp hơn ở bệnh nhân người lớn bị α-Thalassemia. Nguyên nhân chưa rõ, nhưng có thể do mức độ thiếu máu nhẹ hơn, truyền máu ít hơn và sự gia tăng tạo hồng cầu ít hơn.
6.2.4 Nhiễm trùng
Tất cả các dạng Thalassemia thể nặng thường gia tang nguy cơ nhiễm trùng. Lý do không rõ ràng và vẫn đang được xác định. Nồng độ sắt huyết thanh cao tương đối có lẽ thích hợp với sự phát triển vi trùng. Cũng có một cơ chế khác là việc phong tỏa hệ thống monocyte-macrophage như là hậu quả của tình trạng gia tăng phá hủy hồng cầu. Không có khiếm khuyết nào trong chức năng bạch cầu hay miễn dịch được báo cáo. Bệnh nhân Thalassemia phụ thuộc truyền máu có nguy cơ cao mắc các bệnh lây nhiễm do truyền máu như viêm gan B, viêm gan C, HIV/AIDS và sốt rét.
6.2.5 Bất thường đông máu
Hiểu biết ngày càng nhiều về tình trạng tăng đông tiềm ẩn trong một số trường hợp Thalassemia đã được xem xét. Nhiều bằng chứng cho thấy các ở các bệnh nhân này, đặc biệt sau cắt lách và có số lượng tiểu cầu tăng cao, có thể có bệnh động mạch phổi tiến triển. Đây có thể là hậu quả của sự kết cụm tiểu cầu trong tuần hoàn phổi.
Càng ngày càng có nhiều bằng chứng cho thấy trong thiếu máu hồng cầu hình liềm, sự phóng thích hemoglobin và arginase gây ra tăng áp phổi tiến triển với nitric oxide bị hủy và rồi loạn chức năng nội mạc. Có nhiều yếu tố khác góp phần gây nên biến chứng này gồm tình trạng tăng đông và phá hủy cấu trúc phổi do lắng đọng sắt dư thừa.
6.3 Đặc điểm lâm sàng
6.3.1 β-Thalassemia
Dạng β-Thalassemia có kiểu hình lâm sàng nặng nhất là Thalassemia thể nặng. Một bệnh ảnh lâm sàng nhẹ hơn, đặc trưng bởi khởi phát trễ và không đòi hỏi truyền máu hay số lần truyền máu ít hơn so với thể nặng, là β-Thalassemia trung gian. Bên cạnh đó, β-Thalassemia thể nhẹ là thuật ngữ mô tà tình trạng người mang dị hợp tử β-Thalassemia.
β-Thalassemia thể nặng
Thalassemia thể nặng gây ra bệnh cảnh lâm sàng được mô tả lần đầu bởi Cooley và Lee vào năm 1925. Trẻ bị bệnh không có bất cứ biểu hiện lâm sàng nào lúc sinh. Thiếu máu thường phát triển trong những tháng đầu của cuộc đời và ngày càng trở nên nặng hơn. Phần lớn trẻ β-Thalassemia phụ thuộc truyền máu đến khám vì triệu chứng trong năm đầu đời. Thiếu máu nặng và kéo dài khiến trẻ chậm phát triển về thể chất.
Diễn tiến bệnh ở trẻ em tùy thuộc gần như hoàn toàn vào việc trẻ có được duy trì một chế độ truyền máu và thài sắt hợp lý hay không. Nếu được truyền máu hợp lý, trẻ có thể tăng trưởng và phát triển bình thường và không có bất kỳ dấu chứng bất thường nào. Các triệu chứng sẽ rõ ràng hơn khi có tình trạng quá tài sắt vào cuối thập niên đầu đời do tạo hồng cầu không hiệu quả và truyền máu lặp đi lặp lại nhiều lần. Trẻ được truyền máu không hợp lý sẽ phát triển triệu chứng điển hình của thiếu máu trong bệnh cành Cooley. Quá trình tăng trưởng bị chậm lại, kết hợp với sự phát triển quá mức của xương sọ và xương hàm trên khiến gương mặt dần dần có vẻ ngoài như “mongoloid". Những thay đổi này thường kèm với hình ảnh X-quang đặc trưng của sọ, xương dài và bàn tay. Mô xốp xương sọ dãn rộng, với hình ảnh “hair on end” và hình ảnh “sun ray” cho thấy sự rỗ của xương dài và xương đốt ngón. Biến dạng xương, gan lách to và xạm da là các triệu chứng thường gặp bên cạnh tình trạng thiếu máu. Một số triệu chứng khác của tình trạng tăng chuyển hóa có thể gặp như sốt, đổ mồ hôi và tăng acid uric máu.
Những trẻ này đặc biệt dễ bị nhiễm trùng và đây cũng là nguyên nhân gây tử vong thường gặp nhất. Gãy xương tự phát thường xảy ra do sự dãn rộng của khoang tủy và sự mỏng đi của các xương dài, xương sọ. Biến dạng xương hàm trên thường dẫn đến các vấn đề về răng do mọc sai vị trí. Lách to dẫn đến tình trạng giảm tiểu cầu và giảm bạch cầu thứ phát, có thể làm gia tăng nguy cơ và mức độ của xuất huyết và nhiễm trùng. Cắt lách được chỉ định khi bệnh nhân được chẩn đoán cường lách, nhằm mục tiêu làm giảm số lần truyền máu và giảm nguy cơ xuất huyết do giảm tiểu cầu; tuy nhiên, nhiễm trùng sau cắt lách đặc biệt thường gặp. Khuynh hướng chảy máu có thể có cả khi không có giảm tiểu cầu, nguyên nhân do suy giảm chức năng gan trong các trường hợp ứ sắt nặng.
Trẻ tăng trưởng và phát triển bình thường trong khoảng 10 năm đầu đời nhờ truyền máu đều đặn sẽ bắt đầu có biểu hiện do ứ sắt khi bước vào tuổi dậy thì, đặc biệt, nếu không có liệu pháp thải sắt phù hợp. Biểu hiện đầu tiên của quá tải sắt thường là dậy thì muộn và không có kinh ở trẻ gái. Qua thời gian, nhiều rối loạn nội tiết khác có thể xuất hiện, đặc biệt là đái tháo đường, suy sinh dục và thiếu hormone tăng trưởng. Suy giáp cũng có thể xảy ra nhưng ít phổ biến hơn. Đến cuối thập kỉ thứ hai, biến chứng suy tim xuất hiện. Đa số bệnh nhân thường chết trong thập kỷ thứ hai hay ba do suy tim, loạn nhịp hoặc rối loạn dẫn truyền. Các biến chứng này có thể xuất hiện sớm nếu bệnh nhân bị nhiễm trùng lặp đi lặp lại.
Kể cả khi trẻ được truyền máu và thài sắt hợp lý cũng có thể có một số biến chứng, đặc biệt là các bệnh lây nhiễm do truyền máu như viêm gan B, C, HIV hay sốt rét. Tần suất lây nhiễm này đang giảm nhờ các chương trình tầm soát rộng rãi máu người cho.
β-Thalassemia thể trung gian
Kiểu hình bệnh nhân Thalassemia trung gian nặng hơn Thalassemia thể ẩn không triệu chứng nhưng nhẹ hơn Thalassemia thể nặng phụ thuộc vào truyền máu. Bệnh nhân thường đến khám muộn hơn do thiếu máu trễ hơn so với bệnh nhân đồng hợp từ β-Thalassemia phụ thuộc truyền máu. Có thể duy trì Hb # 6 g/dL mà không cần truyền máu. Tuy nhiên, trẻ vẫn sẽ có biểu hiện chậm tăng trưởng và chậm phát triển do thiếu máu. Bệnh nhân trở nên nặng với các triệu chứng như biến dạng xương rõ, viêm khớp, đau xương; lách to tiến triển; chậm tăng trưởng và loét mạn tính vùng trên mắt cá chân. Mặt khác, bệnh nhân vẫn có thể hoàn toàn không có triệu chứng cho đến khi lớn và không phụ thuộc truyền máu với Hb 10 - 12 g/dL. Một số ca trở nên nặng hơn do cường lách tiến triển.
Nhìn chung, đặc điểm lâm sàng của B-Thalassemia thể trung gian tương tự như B-Thalassemia thể nặng. Ở những trường hợp nặng, bệnh nhân cần phải được truyền máu định kỳ như β-Thalassemia thể nặng. Một số biến chứng quan trọng như cường lách tiến triển có thể gặp. Ứ sắt do tăng hấp thu có thể có ở những bệnh nhân không phụ thuộc truyền máu. Ứ sắt gây biến chứng đái tháo đường và rối loạn nội tiết đặc biệt hay gặp ở thập kỷ thứ tư của cuộc đời. sỏi mật, biến dạng xương, loét chân, huyết khối,... có thể xuất hiện sau cắt lách.
β-Thalassemia thể nhẹ
Tình trạng dị hợp tử của B-Thalassemia thường được xác định trong các nghiên cứu phà hệ gia đình bệnh nhân B-Thalassemia thể nặng, nghiên cứu dân số hoặc do tình cờ phát hiện trong xét nghiệm thường quy khi khám sức khoẻ tổng quát. Bệnh nhân có thể có triệu chứng thiếu máu, có biểu hiện mệt mỏi, lách có thể to nhẹ hoặc không to.
6.3.2 α-Thalassemia
Hội chứng phù thai Hb Bart's (Hb Bart's hydrops fetalis syndrome)
Đây là nguyên nhân thường gây thai chết lưu ở khu vực Đông Nam Á. Trẻ có thể chết lưu giữa tuần 34 - 40 của thai kỳ hoặc sinh ra vẫn sống nhưng chết trong vài giờ đầu sau sinh. Xanh xao, phù và gan lách to là các biểu hiện thường gặp. Bệnh cành lâm sàng tương tự phù nhau thai do bất đồng nhóm máu Rh. Từ thiết thường cho thấy có tạo máu ngoài tùy và phù nhau thai. Một số bất thường di truyền có thể có kèm theo.
Một vài trường hợp đã được cứu nhờ chẩn đoán trước sinh và truyền thay máu đã được báo cáo. Những trẻ này tăng trưởng và phát triển bình thường dù phải phụ thuộc truyền máu.
Tình trạng này thường đi kèm tỉ lệ nhiễm độc thai cho mẹ và khó sinh vì nhau lớn. Nguyên nhân của tăng kích thước nhau thai vẫn chưa biết, dù thiếu oxy trong tử cung nặng bị nghi ngờ nhất vì biểu hiện tương tự xảy ra ở trẻ phù do bất thuận hợp Rh với mẹ.
Bệnh hemoglobin H
Bệnh hemoglobin H được mô tà độc lập tại Mỹ và Hy Lạp năm 1956. Triệu chứng lâm sàng rất đa dạng. Một số bệnh nhân bị ảnh hưởng nghiêm trọng như β-Thalassemia thể nặng, nhưng đa số có diễn tiến nhẹ hơn nhiều. Thiếu máu kéo dài với lách to nhiều mức độ có thể xảy ra. Biến dạng xương thường ít gặp.
Nhiều nỗ lực đã được thực hiện để tìm mối liên hệ giữa kiểu gen và kiểu hình bệnh hemoglobin H. Nhìn chung, bệnh nhân α-Thalassemia không đột biến có gen a, kết hợp với yếu tố quyết định αº Thalassemia (αTα/--, hay αConstantspringα/- -) có nồng độ hemoglobin H cao hơn, mức độ thiếu máu nhiều hơn và diễn tiến biểu hiện lâm sàng nặng hơn bệnh nhân có kiểu gen - -/-α.
Thể α-Thalassemia nhẹ hơn, gồm trait α'α/ - - hay αConstantspringα/- -
Vì hai gen α globin tồn tại trên mỗi nửa kiểu gen (haploid genome), một loạt nhiều tình trạng khác nhau với các kiểu hình trùng lắp được tạo nên bởi sự tương tác giữa chúng. Người mang α-Thalassemia mất đoạn và không mất đoạn, -α/αα và αTα/αα thường không có triệu chứng lâm sàng. Tương tự, tình trạng đồng hợp tử của α+ Thalassemia mất đoạn, -α/-α và dị hợp tử của αº Thalassemia, - -/αα, cũng không có triệu chứng, dù một số bệnh nhân có thể có thiếu máu nhẹ và thay đổi hồng cầu. Mặt khác, tình trạng đồng hợp tử của dạng α-Thalassemia không mất đoạn, αTα/αTα, thường có kiểu hình cực kỳ đa dạng. Một số bệnh nhân thường chỉ có thiếu máu nhược sắc nhẹ. Dạng đồng hợp tử của đột biến kết thúc chuỗi, đáng chú ý là he- moglobin Constant Spring, là một trường hợp đặc biệt vì nó tạo ra kiểu hình đặc trưng: thiếu máu tán huyết mức độ vừa kèm lách to nhẹ.
7 ĐẶC ĐIỂM CẬN LÂM SÀNG
7.1 β-Thalassemia
7.1.1 β-Thalassemia thể nặng
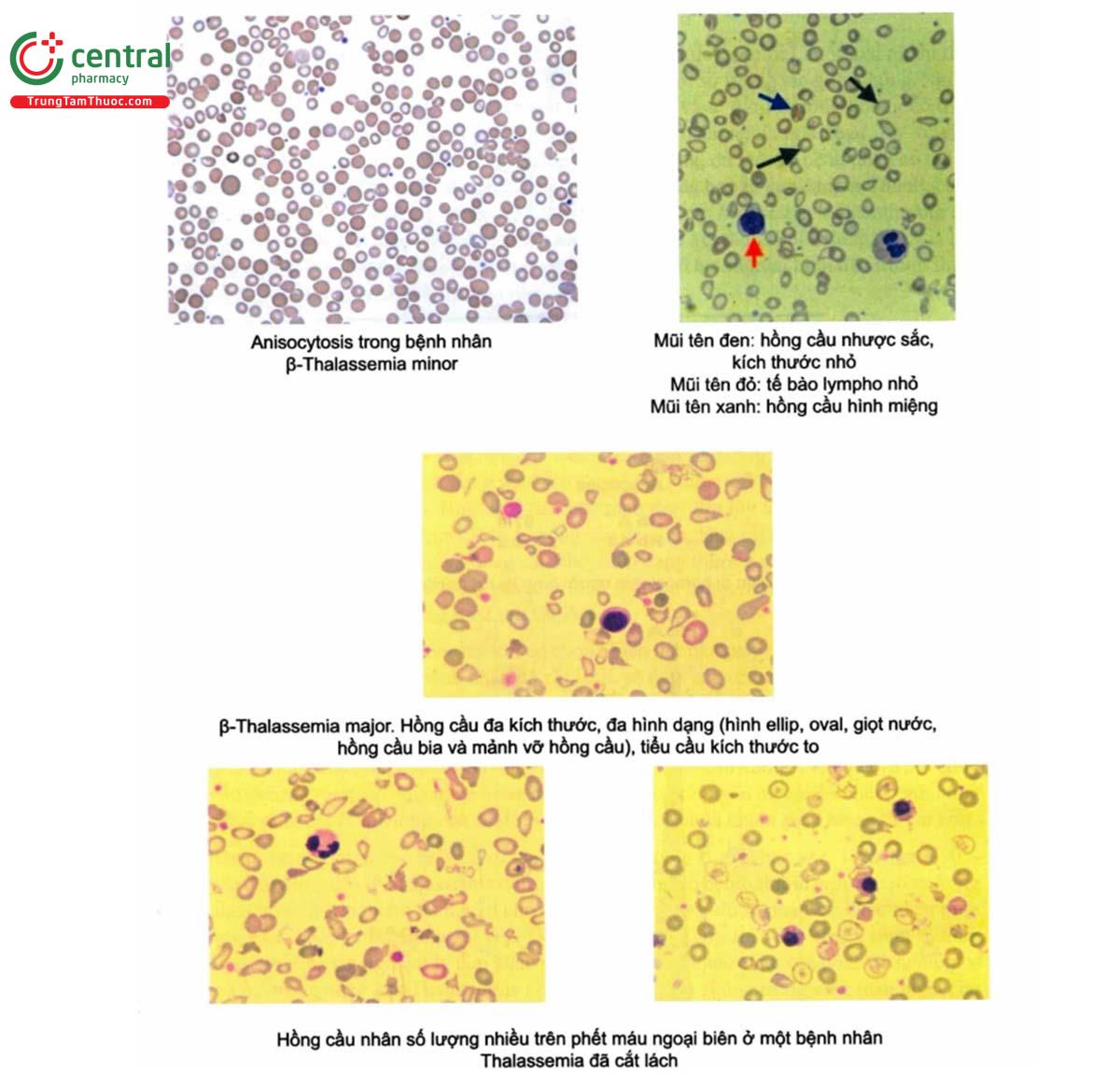
Mức hemoglobin của bệnh nhân khi đến khám trung bình thường từ 2 – 3 g/dL hoặc thậm chí thấp hơn. Hồng cầu đa hình dạng, đa kích thước rõ, nhược sắc, có hồng cầu bia và các chấm basophile nhiều mức độ. Hình thái tiêu bản máu thay đổi tùy thuộc lách tình trạng cắt lách hay chưa. Ở bệnh nhân chưa cắt lách, hồng cầu đa hình dạng rất hay gặp. Tuy nhiên, sau khi cắt lách, hồng cầu to, mỏng và hồng cầu nhỏ, biến dạng thường gặp. Đếm số lượng hồng cầu lưới tăng vừa và hồng cầu nhân gần như luôn luôn có trong máu. Những loại hồng cầu này thường có mức độ cao trong máu sau cắt lách.
Bạch cầu và tiểu cầu tăng nhẹ trừ khi có cường lách thứ phát xảy ra. Nhuộm tiêu bản máu với methyl violet, đặc biệt ở bệnh nhân đã cắt lách, sẽ thấy các chấm hay các thể vùi rải rác trong hồng cầu. Những thể vùi này luôn luôn có trong tế bào tiền thân hồng cầu trong tủy.
Tủy thường tăng sinh hồng cầu với bất thường hình thái erythroblasts và tăng ứ đọng sắt. Các nghiên cứu về chuyển hóa sắt cho thấy quá trình tạo hồng cầu không hiệu quả và đời sống hồng cầu thường rút ngắn. Quần thể hồng cầu với đời sống rất ngắn hiện diện chung với hồng cầu có đời sống dài hơn. Hồng cầu có đời sống dài hơn có chứa nhiều fetal hemoglobin hơn.
Điện di hemoglobin cho thấy có sự gia tăng hemoglobin F, giới hạn từ < 10% đến > 90% và đây là đặc trưng của β-Thalassemia đồng hợp tử. Không có hemoglobin A được tạo ra trong βº Thalassemia. Xét nghiệm tách acid cho thấy hemoglobin F được phân bố khác nhau giữa các hồng cầu. Mức hemoglobin A2 ở β-Thalassemia đồng hợp tử có thể thấp, bình thường hoặc tăng cao. Các xét nghiệm ly tâm khác nhau cho thấy sự khác nhau của phân bố hemoglo- bin F và A2 trong hồng cầu Thalassemia nhưng nồng độ của chúng trong máu toàn phần cho thấy tỉ lệ tổng hợp của chúng.
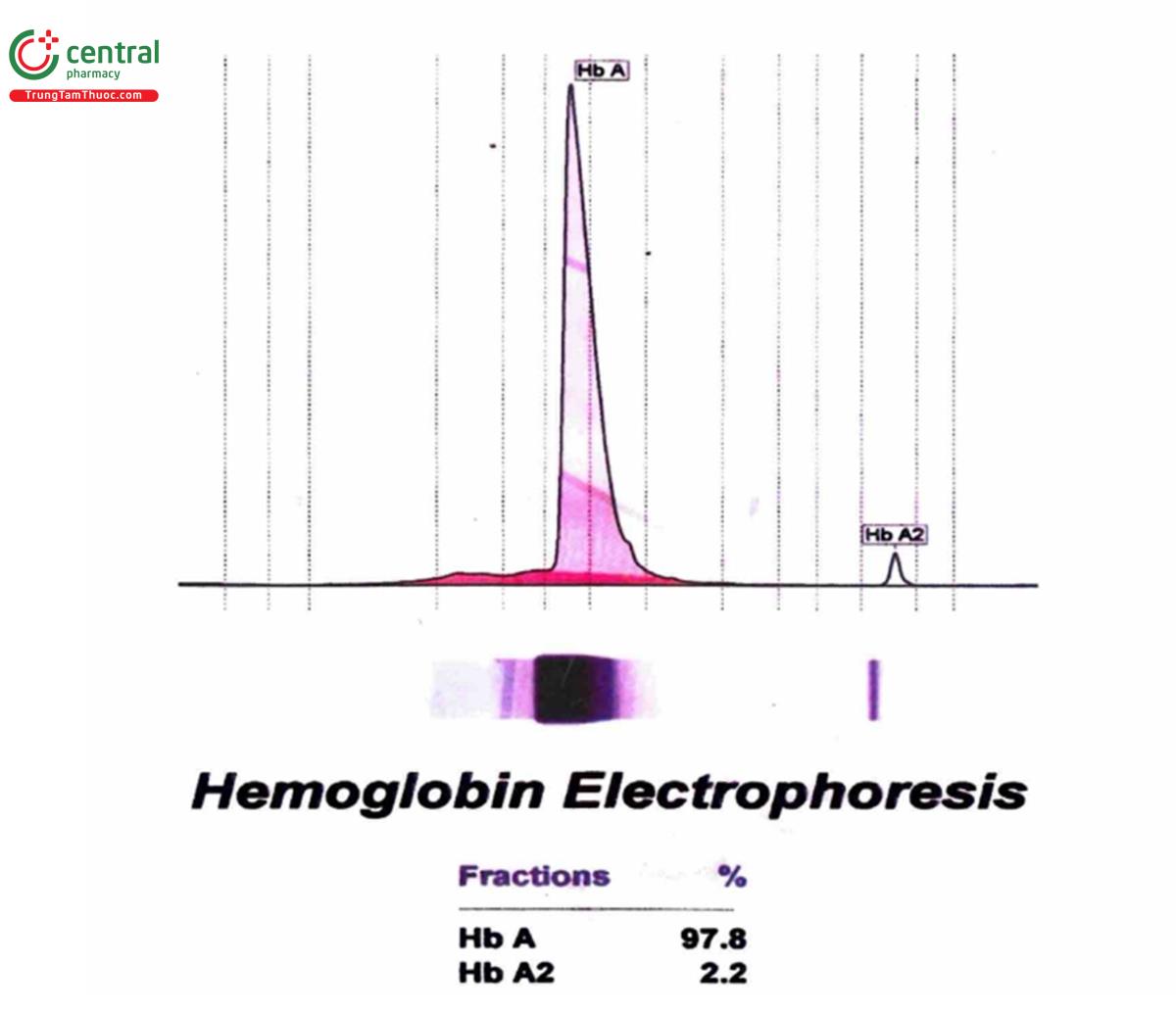
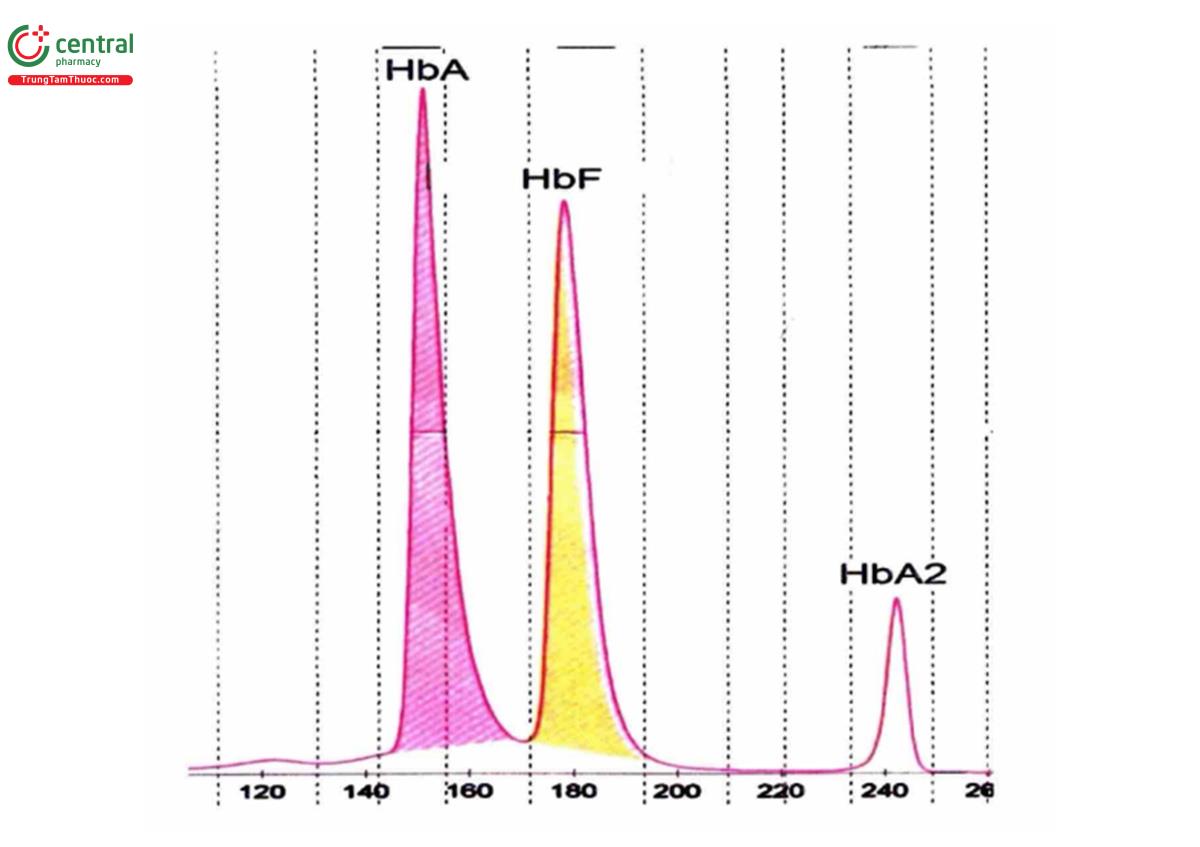
Việc thăm khám anh chị em ruột, cha mẹ và con có thể rất quan trọng trong chẩn đoán nhờ phát hiện bất thường của các thành viên khác trong gia đình. Xét nghiệm đếm tế bào máu trên các thành viên này là cần thiết. Ngoại trừ mức hemoglobin cao hơn, các thay đổi huyết học trong β-Thalassemia trung gian tương tự như trong β-Thalassemia thể nặng.
7.1.2 β-Thalassemia thể nhẹ
Nồng độ hemoglobin ở bệnh nhân β-Thalassemia thể nhẹ thường dao động từ 9 –11 g/ dL. Đa số hồng cầu thường nhỏ và nhược sắc, giá trị trung bình của MCH từ 20 – 22 pg và của MCV từ 50 – 70 fl. Số lượng hồng cầu bình thường hoặc tăng, hemoglobin và hematocrit thường giảm nhẹ. Trong dân số, các chỉ số về hồng cầu rất hữu dụng trong việc tầm soát người mang dị hợp tử Thalassemia.
Tủy xương trong β-Thalassemia dị hợp từ thường tăng sinh hồng cầu nhẹ, hiếm gặp thể vùi hồng cầu. Thỉnh thoảng có sự thay đổi hình dạng nguyên hồng cầu khổng lồ là do thiếu Acid Folic, đặc biệt trong thai kỳ. Lưu ý có thể có tạo hồng cầu không hiệu quả mức độ nhẹ, nhưng đời sống hồng cầu bình thường.
Trên xét nghiệm điệm di hemoglobin, nồng độ hemoglobin A2 tăng từ 3,5 – 7%. Mức hemoglobin F tăng, có thể gặp trong 50% trường hợp, thường dao động từ 1 – 3% và hiểm khi > 5%.
7.2 α-Thalassemia
7.2.1 Hội chứng phù nhau thai hemoglobin Bart's
Ở trẻ có hội chứng phù nhau thai, phết máu ngoại vi thường có hình ảnh thay đổi của Thalassemia nặng với nhiều hồng cầu nhân. Thành phần hemoglobin cấu tạo chính là hemoglobin Bart's, với khoảng 10 20% hemoglobin Portland. Thường không có hemoglobin A hoặc F, dù một số trường hợp hiếm có hiện diện lượng nhỏ hemoglobin A.
7.2.2 Bệnh hemoglobin H
Phết máu ngoại vi thể hiện hồng cầu đa hình dạng, đa kích thước, nhược sắc. Tỉ lệ hồng cầu lưới thường khoảng 5%. Ủ hồng cầu với brilliant cresyl blue cho thấy các thể vùi nằm rài rác trong hầu hết các tế bào. Các thể này được tạo thành do sự kết tụ của hemoglobin H do phản ứng oxy hóa khử của thuốc nhuộm.
Sau cắt lách, một số tế bào có thể có thể Heinz nhỏ, đơn độc. Các thể vùi này được hình thành là do sự kết tủa của phân tử hemoglobin H không bền và chỉ gặp sau cắt lách. Hemoglobin H chiếm từ 5 – 40% tổng số hemoglobin. Lượng rất ít hemoglobin Bart's có thể có và hemoglobin A2 thường giảm nhẹ.
7.2.3 αº Thalassemia và α Thalassemia thể ẩn
αº Thalassemia thể ẩn đặc trưng bởi sự hiện diện từ 5 – 15% hemoglobin Bart's lúc sinh. Hemoglobin này biến mất lúc trưởng thành và không không được thay thế bởi một lượng hemoglobin H tương đương. Thinh thoảng tế bào có thể vùi hemoglobin H có thể xuất hiện sau khi ủ với brilliant cresyl blue. Hiện tượng này thường được dùng như một test chẩn đoán Thalassemia thể ẩn. Tuy nhiên, test này khó chuẩn hóa và cần phải có nhiều kinh nghiệm để đánh giá. Ở bệnh nhân người lớn, hồng cầu dị hợp từ có thay đổi hình thái với MCH và MCV thấp. Điện di hemoglobin thì bình thường.
Bệnh nhân α+ Thalassemia thể ẩn (-α/αα) đặc trưng bởi không thay đổi hay thay đổi huyết học rất ít, 1 - 2% hemoglobin Bart's lúc sinh trong một số trường hợp. Giảm nhẹ tỉ lệ sản xuất giữa α/β, do đó, kiểu gen này thường gợi ý đến người lành mang gen bệnh (silent carrier). Các nghiên cứu so sánh mức hemoglobin Bart's lúc sinh với phân tích DNA chứng tỏ không có hemoglobin Bart's với số lượng có ý nghĩa có thể phát hiện được ở trẻ mới sinh bị α+ Thalassemia dị hợp tử.
8 CHẨN ĐOÁN PHÂN BIỆT
Các dấu hiệu lâm sàng và huyết học trong β-Thalassemia đồng hợp từ và bệnh hemoglo- bin H rất đặc trưng nên chẩn đoán thường không khó.
Ở trẻ nhỏ, chẩn đoán phân biệt Thalassemia với thiếu máu nguyên bào sắt di truyền (congenital sideroblastic anemia) có thể rất khó, nhưng hình ảnh tủy ở trường hợp sau khá đặc trưng.
Trong JMML có tăng hemoglobin F nên bệnh này có thể hơi giống β-Thalassemia. Tuy nhiên, không có tăng hemoglobin A2 trên điện di hemoglobin, có sự giảm carbonic anhydrase và các đặc điểm in vitro cho thấy sự đáp ứng của các tế bào tiền thân tủy in vitro với GM-CSF (granulocyte-monocyte colony-stimulating factor) sẽ giúp chẩn đoán phân biệt bệnh này với β-Thalassemia.
9 ĐIỀU TRỊ, TIẾN TRIỂN, TIÊN LƯỢNG
Cách thức điều trị duy nhất hiện nay đối với trẻ Thalassemia là truyền máu định kỳ, thài sắt để phòng ngừa quá tài sắt, cân nhắc cắt lách trong trường hợp biến chứng cường lách và thực hiện chăm sóc nhi khoa tổng quát tiêu chuẩn. Ghép tùy có vai trò quan trọng trong những trường hợp được chọn lọc.
9.1 Truyền máu
Trẻ bị β-Thalassemia được duy trì mức hemoglobin 9,5 - 14 g/dL sẽ tăng trưởng và phát triển bình thường và gần như sẽ không có các đặc điểm biến dạng xương của Thalassemia. Việc duy trì hemoglobin thấp hơn mức này mà không có bất cứ hậu quà có hại nào trên sự phát triển và mang lại thuận lợi là giàm tình trạng quá tài sắt cũng có thể được chấp nhận.
Việc truyền máu không nên bắt đầu quá sớm và nó chỉ nên được bắt đầu khi mức hemoglobin quá thấp ảnh hưởng cho sự phát triển bình thường của trẻ. Nếu truyền máu được bắt đầu quá sớm, Thalassemia thể trung gian có thể bị bỏ sót và trẻ có thể được truyền máu không cần thiết. Việc truyền máu nên được thực hiện thường xuyên, mỗi 4 tuần ở ngoại trú. Để tránh phản ứng truyền máu, nên truyền hồng cầu đông lạnh hay rửa, bộ lọc nên được dùng để loại bỏ bạch cầu và các thành phần protein huyết tương.
9.2 Thải sắt
Trẻ được điều trị truyền máu cuối cùng sẽ dẫn tới quá tài sắt và chết vì các biến chứng ứ sắt ở các cơ quan quan trọng. Do đó, những trẻ này phải được bắt đầu chương trình thài sắt trong 2 – 3 năm đầu đời. Dù nỗ lực tìm kiếm thuốc thải sắt dạng uống, deferoxamin (desferri- oxamine) được chứng minh là thuốc duy nhất có giá trị điều trị lâu dài cho Thalassemia. Thuốc được dùng tốt nhất là tiêm truyền dưới da thành bụng trước qua bơm tiêm suốt đêm từ 8 – 12 giờ. Liệu pháp thải sắt nên được khởi động khi nồng độ ferritin huyết thanh đạt 1.000 mcg/dL. Trong thực hành lâm sàng, mức ferritin này thường thấy sau 12 đến 15 lần truyền máu.
Để ngừa độc tính, trẻ không nên thải sắt khi gánh nặng sắt vẫn còn thấp. Liều khởi đầu thường là 20 mg/kg × 5 đêm/tuần, kèm 100 mg Vitamin C uống (người lớn và trẻ lớn là 200 mg) vào ngày truyền thuốc, sau khi tiêm truyền thuốc được bắt đầu. Ở các bệnh nhân ứ sắt nặng, đặc biệt nếu có biến chứng tim hay nội tiết, lượng sắt dự trữ trong cơ thể có thể giảm một cách hiệu quả bằng deferoxamin truyền tĩnh mạch với liều tới 50 mg/kg trọng lượng cơ thể.
Kinh nghiệm dùng deferoxamin ngày càng nhiều và hậu quả độc tính của thuốc đã được báo cáo. Không có biến chứng nghiêm trọng nào xảy ra ngoài nốt dưới da đau và đỏ da tại nơi tiêm truyền. Phản ứng dị ứng cực kỳ hiếm gặp. Các phản ứng này đều có thể kiểm soát. Độc tính thần kinh cảm giác có thể gặp ở 30% trường hợp. Trong một vài trường hợp có thể gây điếc vĩnh viễn. Độc tính ở mắt cũng được ghi nhận. Các triệu chứng bao gồm mất thị giác, mù màu và mất thị trường và trong một số trường hợp sẽ mất đi khi bệnh nhân ngưng thuốc.
Deferoxamin có thể gây biến đổi xương kèm đau xương và chậm tăng trưởng. Những thay đổi này có thể thấy được qua hình ảnh bất thường trên X-quang cột sống. Có thể phòng ngừa bằng cách theo dõi cẩn thận ở những bệnh nhân sử dụng desferrioxamin lâu dài. Các chuyên gia khuyến cáo bệnh nhân nên được khám tai và mắt mỗi 6 tháng.
Việc sử dụng deferoxamin tiêm dưới da ban đêm gặp nhiều khó khăn, do đó người ta đã nỗ lực tìm kiếm các thuốc thải sắt uống hiệu quả. Hai thuốc hiện đang có là deferipron và deferasirox. Deferipron được dùng với liều 75 mg/kg chia 3 lần trong ngày. Không có nhiều nghiên cứu thực nghiệm so sánh về hiệu quả của thuốc so với desferrioxamin trong thời gian dài, nhưng nhìn chung nó có vẻ kém hiệu quả hơn trong việc duy trì lượng sắt an toàn trong cơ thể. Tác dụng phụ quan trọng nhất là giàm bạch cầu hạt. Vì vậy, người ta khuyến cáo nên kiểm tra số lượng bạch cầu mỗi tuần. Deferipron cũng gây viêm khớp nhiều mức độ trong các chủng tộc khác nhau. Tuy nhiên, vì có ưu điểm là khả năng xuyên màng tế bào nên thuốc được cho là hiệu quả hơn trong việc loại bỏ sắt ở tim. Tuy nhiên, cho đến nay, tất cả các nghiên cứu cho rằng thuốc làm giàm tỉ lệ biến chứng tim ở bệnh nhân Thalassemia phụ thuộc truyền máu đều là hồi cứu và không có dữ liệu nào được kiểm chứng trong thời gian dài. Ngày nay các chuyên gia gợi ý nên dùng deferipron phối hợp với desferrioxamin, đặc biệt là vì hiệu quả thải sắt ở tim trong thời gian dài.
Deferasirox mang lại nhiều hứa hứa hẹn với hiệu quả nổi bật ở liều 5 – 10 mg/kg/ngày, hoặc liều cao hơn đối với bệnh nhân ứ sắt nặng. Kết quả ngang với desferrioxamin trong việc đạt được mức sắt ở gan hợp lý. Những nghiên cứu ban đầu cũng cho thấy thuốc hiệu quả trong việc loại bỏ sắt dư thừa ở tim. Các phản ứng bất lợi thường gặp nhất liên quan deferasirox gồm rối loạn dạ dày – ruột, sần da thoáng qua, tăng creatinine máu không tiến triển. Tuy nhiên, cũng còn quá sớm để chắc chắn về hiệu quả toàn bộ hay đánh giá độ an toàn lâu dài của thuốc.
Vì các dữ liệu đáng tin cậy về hiệu quả lâu dài của deferoxamin trên các bệnh nhân Thalassemia phụ thuộc truyền máu được điều trị hợp lý nên đến nay thuốc này vẫn là lựa chọn đầu tiên. Vai trò thật sự của phác đồ phối hợp deferoxamin và deferipron, hay dùng deferasirox đơn độc vẫn cần nhiều nghiên cứu tiền cứu để xác định.
Việc theo dõi cẩn thận tình trạng ứ sắt trong lúc dùng liệu pháp thải sắt là yếu tố rất quan trọng. Cách đánh giá đơn giản nhất là đánh giá thường quy nồng độ ferritin huyết thanh, mục tiêu nên duy trì ferritin < 1.500 mcG/L. Các kỹ thuật không xâm lấn khác để đánh giá sắt trong cơ thể đã được phát triển thêm. Ngày nay, có nhiều bằng chứng mạnh mẽ chứng minh rằng với việc đo và vẽ lên bản đồ LIC (liver iron concentration) bằng MRI là cách hiệu quả cực kỳ để đánh giá thường quy hiệu quả của liệu pháp thải sắt. Tương tự, có nhiều tiến bộ trong việc đánh giá sắt ở tim bằng phương pháp không xâm lấn T2* MRI.
Ngày càng nhiều bằng chứng cho thấy trẻ được duy trì mức hemoglobin cao thì không bị cường lách. Tuy nhiên, việc lách to kèm với nhu cầu truyền máu ngày càng nhiều, xảy ra phổ biển ở bệnh nhân có mức hemoglobin thấp hơn. Việc cắt lách nên được thực niện nếu nhu cầu truyền máu tăng đột ngột hay có triệu chứng đau do lách quá to. Vì nguy cơ cao nhiễm trùng pneumococcal, cắt lách không nên thực hiện ở trẻ < 5 tuổi. Những trẻ này nên được chủng ngừa pneumococcal trước khi cắt lách, sau đó bệnh nhân phải được uống penicillin phòng ngừa. Chủng ngừa Haemophilus influenzae type B và meningococcal cũng được khuyến cáo.
10 SO SÁNH BỆNH ALPHA THALASSEMIA VÀ BETA THALASSEMIA
Thalassemia còn gọi là tan máu bẩm sinh, là bệnh đột biến di truyền trên nhiễm sắc thể, giảm quá trình tổng hợp alpha hoặc beta globin, gây ra khuyết thiếu 1 hoặc nhiều chuỗi này. Khi đó, cơ thể có các biểu hiện như thiếu máu, thiếu oxy do lượng lớn các tế bào hồng cầu đã bị phá hủy. Bệnh gồm 2 loại chính là alpha Thalassemia và Beta Thalassemia, điểm khác biệt của 2 nhóm này là sự suy giảm của chuỗi alpha globin và beta globin. Từ đó với mỗi thể bệnh sẽ có những biểu hiện, cách điều trị và chẩn đoán khác nhau. Dưới đây là bảng so sánh chi tiết bệnh thể alpha thalassemia và beta thalassemia.
| Tiêu chí | Alpha Thalassemia | Beta Thalassemia |
| Giống nhau | Cả 2 tình trạng đều gây ra thiếu máu do suy giảm nồng độ hemoglobin | |
| Đặc điểm | Đột biến alpha Thalassemia được gây ra bởi việc mất hoặc đột biến ở một hoặc nhiều trong bốn gen alpha globin | Đột biến beta Thalassemia được gây ra bởi đột biến ở một hoặc cả hai gen beta globin |
| NST đột biến | NST số 16 | NST số 11 |
| Phân loại | 4 loại Thể ẩn: 1 gen bị ảnh hưởng Thể nhẹ: 2 gen bị ảnh hưởng Thể Hemoglobin H: 3 gen bị ảnh hưởng Thể phù thai (hydrops fetalis): 4 gen bị ảnh hưởng | 3 loại Thể nhẹ Thể trung gian Thể nặng |
| Chẩn đoán | Công thức máu Phết tế bào ngoại biên Đo lường việc lưu trữ và sử dụng sắt của cơ thể Điện di Hemoglobin Phân tích DNA | |
| Xét nghiệm phân tử | Phân tích hoặc giải trình tự gen | |
| Đặc điểm lâm sàng | Thể ẩn: không có dấu hiệu hay triệu chứng của Thalassemia, có nồng độ hemoglobin và các chỉ số hồng cầu bình thường nhưng có thể truyền gen bệnh cho con cái họ. Thể nhẹ: thiếu máu nhẹ, tế bào hồng cầu có kích thước nhỏ hơn và nhạt hơn bình thường Thể HbH: gây thiếu máu từ trung bình đến nặng gây ra mệt mỏi, lách to, biến dạng xương. Thể nặng: thai nhi thường bị sảy thai, chết non hoặc chết ngay sau khi sinh. | Thể nhẹ: không có vấn đề sức khỏe, chỉ phát hiện các tế bào hồng cầu nhỏ bất thường và thiếu máu nhẹ Thể trung gian: kiểu hình lâm sàng giữa dạng thể ẩn và thể nặng, thiếu máu từ nhẹ đến trung bình. Thể nặng: truyền máu thường xuyên, mắt xếch Mongoloid, ứ sắt gây tổn thương cơ quan mãn tính |
| Điều trị | Bổ sung axit folic thường được đưa ra, nhưng bổ sung sắt không được khuyến khích. Bệnh nhân bị thiếu máu nhẹ thường không cần bất kỳ phương pháp điều trị nào. Những người mắc bệnh alpha thalassemia nặng cần phải truyền máu suốt đời | Truyền máu thường xuyên Liệu pháp thải sắt Axit folic (nếu chế độ ăn uống không đủ axit folic) Cắt lách (đôi khi được sử dụng để giảm nhu cầu máu) Ghép tuỷ Liệu pháp gen cho mục đích sàng lọc và điều trị |
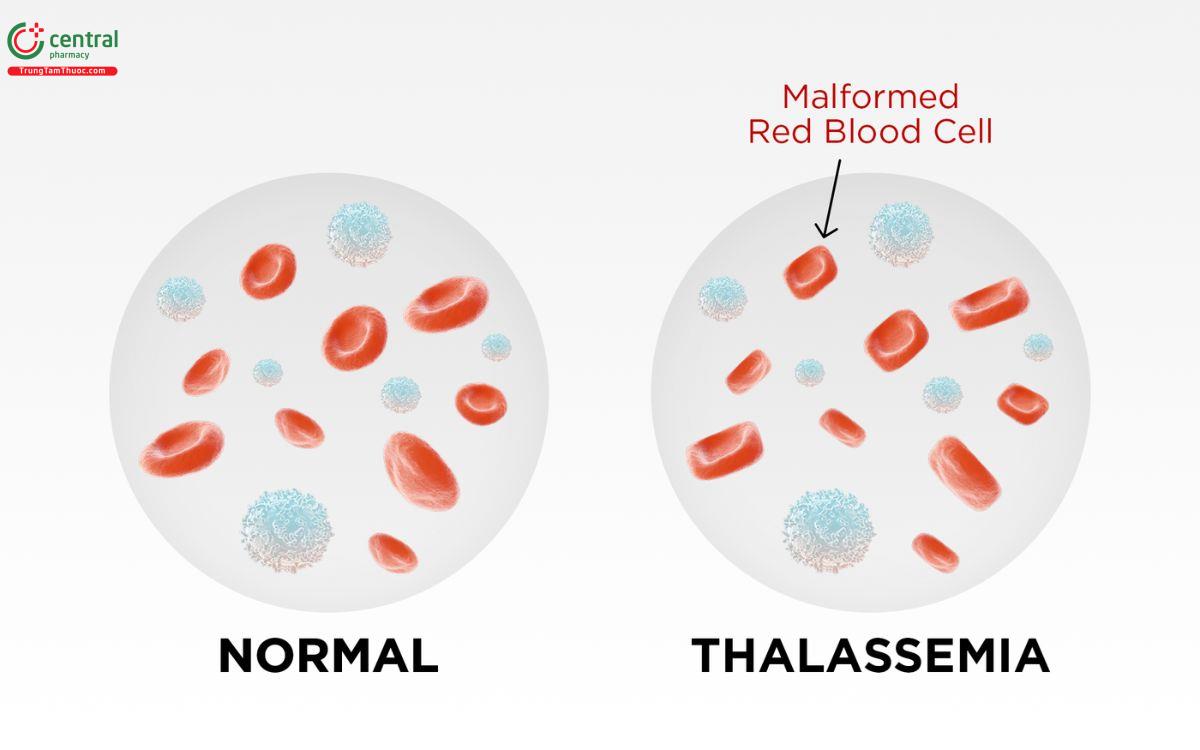
11 PHÂN BIỆT ĐẶC ĐIỂM BỆNH THALASSEMIA VÀ THIẾU MÁU THIẾU SẮT
Bệnh Thalassemia thường bị gắn nhãn sai là thiếu máu thiếu sắt do các giá trị trong phòng thí nghiệm rất giống nhau giữa 2 bệnh này. Ngay từ cái nhìn đầu tiên về công thức máu toàn phần (CBC), giá trị huyết sắc tố sẽ thấp. Điều này dẫn đến tình trạng thiếu máu và thực tế là thể tích hồng cầu (MCV) thấp. Bên cạnh đó, Thalassemia có nhu cầu tạo hồng cầu rất cao, nên thiếu sắt trong hồng cầu là hoàn toàn có thể tìm thấy.
Khuyến cáo gần nhất cho phép sàng lọc Thalassemia khi MCV<85 fL, do thiếu máu hồng cầu nhỏ gặp ở nhiều bệnh như IDA, Thalassemia, ACD. Một vài trường hợp thiếu máu thiếu sắt nhưng MCV không thấp, do chúng có thể kèm thiếu các chất khác như B9,B12. Vì vậy gây khó khăn trong quá trình chẩn đoán, nên có thể cứ bù sắt trước, thiếu máu hồng cầu to sẽ xuất hiện sau.
Dưới đây là sự phân biệt bệnh thiếu máu do thiếu sắt và bệnh Thalassemia trên công thức máu:
1. Sự khác biệt đầu tiên là số lượng hồng cầu ( RBC). Khi thiếu sắt, giá trị này thấp còn bệnh Thalassemia, hồng cầu ở mức bình thường đến tăng cao.
- Trong bệnh IDA thiếu nguyên liệu tạo máu, tuỷ không tăng sản xuất hồng cầu, nên MCV không giảm nhiều, chỉ số Mentzer cao
- Trong bệnh Thalassemia do sự tổng hợp globin bị thiếu nên tuỷ đáp ứng rất mạnh. Quan sát sẽ thấy nhiều hồng cầu lưới và hồng cầu nhân trên lam máu. Từ đó làm RBC tăng cao, chỉ số Mentzer thấp. Nếu chỉ số này dưới 13 có thể thực hiện điện di huyết sắc tố để chẩn đoán Thalassemia.
2. Chiều rộng phân bố hồng cầu (RDW) tăng lên trong IDA và gần mức bình thường hoặc tăng nhẹ trong Thalassemia.
- Do bệnh IDA gây ra tình trạng hỗn loạn trong quần thể hồng cầu và đời sống của chúng kéo dài 120 ngày, làm RDW tăng cao (tùy giai đoạn)
- Còn bệnh Thalassemia có quần thể hồng cầu với nhiều kích thước khác nhau, RDW sẽ thấp. Tuy nhiên tiêu chí này vẫn có những khuyết điểm do các chỉ số MCV, RDW biến đổi trong các đợt tan huyết.
3. Ngoài ra gặp tình trạng tăng tiểu cầu phản ứng ở IDA và giảm các dòng tế bào máu ở Thalassemia ở giai đoạn cường lách.
Quan sát 2 huyết đồ bên dưới để hiểu hơn sự khác nhau của 2 bệnh này.
12 Ghép tế bào gốc
Vào năm 1997, hơn 1.000 ca ghép tùy được thực hiện tại ba trung tâm của Italy. Dựa vào kinh nghiệm này và các dữ liệu sau đó, tiên lượng của cuộc ghép tùy thuộc vào liệu pháp thải sắt phù hợp. Do đó, bệnh nhân được chia ra làm ba nhóm: nhóm I bao gồm bệnh nhân thải sắt phù hợp và không có xơ gan hay gan to; nhóm II gồm bệnh nhân có một hay hai trong ba đặc điểm trên và nhóm III gồm bệnh nhân không có cả ba đặc điểm. Trong số bệnh nhân nhóm I đã được ghép sớm trong quá trình bệnh, DFS 5 năm (disease-free survival) đạt 90 – 93% và nguy cơ tử vong do ghép là 4%. Ở nhóm II, nhóm nguy cơ vừa, OS và DFS là 86% và 82%. Với nhóm III, nhóm nguy cơ cao, OS và DFS là 62% và 51%. Ngoài các biến chứng nhiễm trùng nặng sau ghép, phần lớn các trường hợp tử vong có liên quan đến GVHD (graft-versus- host disease) cấp hay mạn. Tỉ lệ chung của GVHD độ nhẹ đến nặng từ 27 – 30%. Việc điều chỉnh phác đồ điều kiện hóa đã làm giảm tỉ lệ độc tính của thuốc. Không ghi nhận có trường hợp nào mắc bệnh huyết học ác tính trong vòng 15 – 20 năm sau ghép.
13 Chăm sóc tổng quát
Việc điều trị Thalassemia đòi hỏi sự chăm sóc tổng quát đa phương diện. Nhiễm trùng cần được điều trị sớm. Nếu chế độ ăn thiếu folate thì cần được bổ sung. Việc bổ sung có thể không cần thiết ở những trẻ duy trì phác đồ có tốc độ truyền máu cao. Nên chú ý đặc biệt vùng tai mũi họng vì nhiễm trùng xoang mạn tính và các bệnh tai giữa do khiếm khuyết xương sọ dễ xảy ra.
Bên cạnh đó, chăm sóc răng định kỳ rất cần thiết ở trẻ truyền máu không đủ, có khiếm khuyết đa dạng xương hàm trên và răng kém phát triển. Trong các giai đoạn sau của bệnh, liệu pháp nội tiết thay thế cần thiết khi quá tải sắt trở thành vấn đề chính. Điều trị triệu chứng loãng xương và suy tim cũng cần thiết.
14 Điều trị các thể Thalassemia đặc biệt
Bệnh hemoglobin H thường không cần trị liệu đặc hiệu, dù cắt lách có thể có giá trị trong những trường hợp có kèm thiếu máu nặng và lách to. Vì cắt lách có thể làm gia tăng tỉ lệ huyết khối so với nhóm β-Thalassemia không cắt lách, nên chỉ thực hiện phẫu thuật này trong các trường hợp thiếu máu nhiều và kèm theo cường lách. Các thuốc oxy hóa không nên dùng ở bệnh nhân bị hemoglobin H.
Vấn đề trị liệu Thalassemia thể trung gian khá phức tạp. Việc quyết định truyền máu hay không ở trẻ có hemoglobin ổn định ở mức 6 – 7 g/dL thật sự khó khăn. Có lẽ điều tốt nhất là phải chấp nhận theo dõi sát các trẻ này trong những năm đầu đời. Nếu bệnh nhân tăng trường, phát triển bình thường và không có dấu hiệu biến dạng xương, thì không nên truyền máu. Tuy nhiên, nếu sớm có dấu hiệu chậm tăng trưởng hay giới hạn hoạt động vì thiếu máu, thì nên truyền máu định kỳ. Cường lách góp phần làm trầm trọng hơn tình trạng thiếu máu thì nên thực hiện cắt lách khi trẻ lớn và cường lách tiến triển. Thêm vào đó, các bệnh nhân này sẽ có tình trạng quá tải sắt do tăng hấp thu sắt từ dạ dày ruột nên cần đánh giá định kỳ nồng độ sắt huyết thanh và ferritin để có thể khởi động thải sắt khi có chỉ định.
15 PHÒNG NGỪA
Ở những vùng trên thế giới có tần suất mắc Thalassemia cao, bệnh tạo ra một gánh nặng kinh tế lớn lên xã hội. Ví dụ, nếu tất cả trẻ Thalassemia được sinh ra ở Cyprus được truyền máu và thài sắt đều đặn, ước tính trong 15 năm, tổng gói chi phí y tế của nước này chỉ đủ điều có thể đạt được theo hai cách: trị cho một mình bệnh này. Rõ ràng là phương pháp này không khả thi, vì vậy nỗ lực hướng đến phát triển chương trình phòng ngừa các thể Thalassemia khác nhau. Mục tiêu phòng ngừa
(1) Tư vấn di truyền, tầm soát dân số toàn bộ trong khi trẻ đang tuổi đi học và cành báo người mang gen về nguy cơ khi kết hôn với người mang gen khác. Khá ít dữ liệu về phương pháp này. Rõ ràng phương pháp này không thành công ở nhiều quốc gia, do đó, người ta hướng đến phát triển chương trình chẩn đoán tiền sản. Chẩn đoán tiền sàn để phòng ngừa Thalassemia đòi hỏi tầm soát người mẹ ở lần khám tiền sàn đầu tiên. Tầm soát người cha trong trường hợp người mẹ là người mang a-Thalassemia và đề nghị họ làm chẩn đoán tiền sản, kết thúc thai kỳ nếu cà cha và mẹ đều là người mang gen Thalassemia thể nặng. Hiện nay, chương trình này chủ yếu dành cho chẩn đoán tiền sàn dạng đồng hợp từ β' hoặc βº Thalassemia phụ thuộc truyền máu. Nỗ lực ban đầu lúc phát hiện tiền sản β-Thalassemia dùng mẫu máu thai và phân tích tổng hợp chuỗi globin diễn ra ở tuần 18 của thai kỳ. Dù khó khăn, phương pháp này được áp dụng thành công ở nhiều nước và dẫn đến giảm tỉ lệ trẻ sinh ra bị β-Thalassemia. Kỹ thuật đi kèm với bệnh suất mẹ thấp, từ suất thai khoảng 3 – 4% và tỉ lệ sai sót là 1 – 2%. Khuyết điểm chính của phương pháp này là được thực hiện tương đối trễ trong thai kỳ. Vì vậy, nên bắt đầu làm chẩn đoán tiền sản trong tam cá nguyệt thứ nhất.
(2) Kỹ thuật DNA có khả năng chẩn đoán các rối loạn hemoglobin quan trọng trong từ cung bằng cách phân tích DNA. Dù sự phân tích có thể làm trên DNA chiết xuất từ dịch ối (phương pháp này có mặt hạn chế, vì một lần nữa, nó phải thực hiện tương đối trễ trong thai kỳ và thường tế bào dịch ối phải được tăng trưởng trong nuôi cấy để đạt đủ lượng DNA). Tuy nhiên, DNA có thể ly trích được rất sớm vào khoảng tuần 9 của thai kỳ bằng mẫu gai nhau. Dù sự an toàn của kỹ thuật này vẫn được đánh giá đầy đủ và khiếm khuyết chỉ có thể xảy ra khi thủ thuật được làm rất sớm, sinh thiết gai nhau đã trở thành phương pháp chính chẩn đoán tiền sản cho bệnh nhân có nguy cơ Thalassemia dựa trên kinh nghiệm sau đó của kỹ thuật này.
Các tiến bộ vượt bậc trong kỹ thuật DNA đã cung cấp nhiều phương pháp khác nhau để xác định trực tiếp đột biến DNA của thai. Kể cả ở các gia đình có đột biến cực hiếm, kỹ thuật giải trình tự DNA nhanh chóng cho phép thực hiện chẩn đoán rất nhanh. Tỉ lệ sai sót khi dùng các phương pháp khác nhau này rất thay đổi, chủ yếu tùy vào kinh nghiệm của phòng xét nghiệm; tỉ lệ thấp dưới 1% được báo cáo từ hầu hết các trung tâm. Việc áp dụng kỹ thuật mới này đã làm giảm chủ yếu tỉ lệ sinh trẻ bị Thalassemia khắp vùng Địa Trung Hải, Trung Đông, một phần của tiểu lục địa Ấn Độ và Đông Nam Á.
16 THALASSEMIA LÀ VẤN ĐỀ SỨC KHỎE TOÀN CẦU
Những tiến bộ đáng kể trong chẩn đoán, phòng ngừa và điều trị Thalassemia đã cải thiện tiên lượng nhóm bệnh lý này ở nhiều quốc gia trên thế giới. Tuy nhiên, ở các nước đang phát triển có tần suất Thalassemia cao vẫn còn nhiều điều giới hạn trong chẩn chẩn đoán và điều trị. Các phương pháp nhằm kiểm soát và quản lý Thalassemia ở các nước nghèo đã và đang được xem xét, bao gồm phát triển mối liên hệ giữa các nước giàu và nghèo để huấn luyện cho nhân viên, trang bị các phương tiện và kỹ thuật chẩn đoán tầm soát sớm, điều trị thài sắt hiệu quả,... và một khi các điều này được phát triển, thì sẽ tiếp tục với các nước nghèo hơn. Nếu không tổ chức theo các đường hướng này thì Thalassemia sẽ tiếp tục gây ra cái chết trước khi trưởng thành cho hàng trăm trẻ trong số hàng ngàn trẻ mắc bệnh trên toàn thế giới.
17 CÂU HỎI TỰ LƯỢNG GIÁ
1. Trong bệnh thalassemia, nguyên nhân gây thiếu máu là gì?
A. Thiếu sắt
B. Thiếu vitamin B12
C. Chảy máu
D. Tán huyết
2. Loại bệnh nào không phải bệnh hemoglobin?
A. Alpha-Thalassemia
B. Hồng cầu hình cầu
С. НЫС
D. Beta-Thalssemia
3. Bệnh nhân nữ 50 tuổi, điện di Hb thấy: HbA: 70%, Hb E: 26%, Hb A 2: 3%, HbF: 1%, bệnh nhân có thể có bệnh gì?
A. Bình thường
B. Hb H
С. НЬЕ
D. Alpha-Thalassemia
4. Bệnh Thalassemia là do:
A. Không tổng hợp được heme
B. Không tổng hợp được sắt
C. Giảm tổng hợp một chuỗi globin
D. Thiếu men tổng hợp một chuỗi globin
5. Hình dạng hồng cầu trong bệnh Thalassemia:
A. Nhỏ
B. Bình thường
C. Nhỏ, nhược sắc
D. To, nhược sắc
18 ĐÁP ÁN
1D 2B 3C 4C 5C
19 TÀI LIỆU THAM KHẢO
1. David J. Weatherall, (2010), The Thalassemia: Disorder of Globin synthesis, William Hematology, 48, 725-758.
2. Fuchargeń G et al. (2004), A Simplified Screening Strategy for Thalassemia and Hemoglobin E in Rural Communities in South-East-Asia, Bull World Health Organ, 82(5): 364-372.
3. Winichagoon P, et al. (2008), Rapid diagnosis of Thalassemias and other hemoglobinopathies by capillary electrophoresis system, Transl Res, 152(4):178-184.
4. Edward J Benz, (2018), Clinical manifestations and diagnosis of the Thalassemias, http://www. uptodate.com
5. Aisha Ibrahim Al Naami, Dr. Ohood Othman Felemban, Disease Knowledge and Treatment Adherence Among Thalassemia Patients: A Literature Review, Journal of Nursing and Health Science. Truy cập ngày 3 tháng 5 năm 2024
6. Dharmesh Chandra Sharma, Anita Arya, Purnima Kishor, Poonam Woike, Jyoti Bindal, Overview on Thalassemias: a Review article, Medico Research Chronicles. Truy cập ngày 3 tháng 5 năm 2024
