Những điều cần biết về tăng sản thượng thận bẩm sinh
1 NGUYÊN LÝ CHUNG
Tăng sản thượng thận bẩm sinh bao gồm một nhóm các rối loạn di truyền tính lặn qua nhiễm sắc thể, thường được đặc trưng bằng tình trạng bất thường chức năng của các enzym tham gia vào quá trình sinh tổng hợp các steroid thượng thận, dẫn tới làm giảm quá trình sinh tổng hợp cortisol và gây tăng bù trừ nồng độ hormon hướng vỏ thượng thận (ACTH) huyết thanh [1-3].
Các kiểu hình lâm sàng và sinh hóa của hội chứng này phụ thuộc vào mức độ nặng và thể thiếu hụt enzym gây các biến đổi khác nhau về tổng hợp glucocorticoid, mineralocorticoid và steroid sinh dục [1]. Biểu hiện điển hình bao gồm thể nặng với mất muối nặng ở trẻ sơ sinh do thiếu hụt aldosteron xảy ra đồng thời và một thể với sinh tổng hợp aldosteron có vẻ bình thường dẫn tới cơ quan sinh dục không điển hình. Các thể nhẹ hơn và không điển hình có thể được biểu hiện với tình trạng tăng quá mức androgen ở người lớn [4].
Tăng sản thượng thận bẩm sinh điển hình là nguyên nhân gây ra hầu hết các trường hợp lưỡng tính giả/mơ hồ giới tính (pseudohermaphroditism) ở nữ giới và khoảng 50% tổng số các trường hợp cơ quan sinh dục ngoài không rõ ràng.
2 Dịch tễ học
Khoảng 95% các trường hợp là hậu quả của tình trạng thiếu hụt 21-hydroxylase và 5 - 8% là do thiếu hụt 118-hydroxylase [5,6]. Các thể hiếm gặp của tăng sản thượng thận bẩm sinh bao gồm thiếu hụt 17α-hydroxylase/17,20-lyase, thiếu hụt 3ẞ-hydroxysteroid dehydrogenase typ 2 (3B-HSD2), thiếu hụt P450 oxidoreductase (POR), tăng sản thượng thận dạng lipoid hoặc thiếu hụt enzym cắt chuỗi bên cholesterol (cholesterol side-chain cleavage [SCC]) [1].
Tham khảo Bảng 13.1 để có các thông tin và tỷ lệ bệnh.
Hầu hết các trường hợp tăng sản thượng thận bẩm sinh điển hình được phát hiện ở tuổi thơ ấu, tuy nhiên, những hiểu biết rõ về di truyền học và sinh lý bệnh của tăng sản thượng thận bẩm sinh sẽ phát hiện được nhiều hơn các thể nhẹ và các thể không điển hình trong độ tuổi trưởng thành của bệnh nhân [1].
Tần suất mắc tổng thể của tăng sản thượng thận bẩm sinh không điển hình hiện chưa được biết rõ, tuy nhiên vào khoảng 0,1 - 0,2% quần thể dân chúng chung.
2.1 Bệnh căn
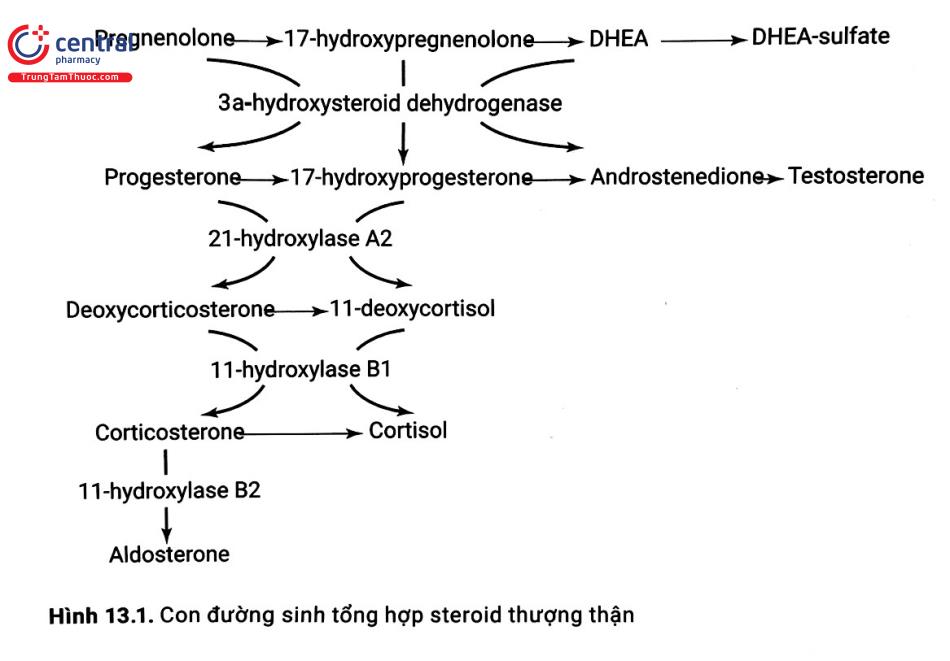
Sản xuất cortisol bị thiếu hụt là bất thường chính trong tăng sản thượng thận bẩm sinh và cần nắm vững quá trình sinh tổng hợp steroid thượng thận để hiểu rõ sinh bệnh học và cơ sở điều trị (xem Hình 13.1).
Các thể bệnh tăng sản thượng thận bẩm sinh được quyết định bởi điểm khác biệt mà quá trình sinh steroid thượng thận bị gián đoạn. Xem Bảng 13.1 để biết phân loại tăng sản thượng thận bẩm sinh, các thông tin về đột biến đặc hiệu, khiếm khuyết hay thiếu hụt enzym liên quan và các biểu hiện kiểu hình [1,5].
Các đột biến kết hợp với tình trạng giảm nặng trong hoạt tính enzym gây nên các thiếu hụt aldosteron và cortisol, trái lại các đột biến kết hợp với tình trạng giảm ít hơn sẽ gây các bất thường hormon ít nghiêm trọng hơn và diễn tiến bệnh không điển hình.
Một số bệnh nhân với tăng sản thượng thận bẩm sinh không điển hình có một alen bị đột biến “nặng” và một alen bị đột biến “nhẹ”. Vì vậy, mức độ nặng của kiểu hình ở thể dị hợp tử phối hợp được xác định bởi đột biến ít nghiêm trọng hơn [1].
Giảm nồng độ cortisol kích thích tuyến yên tiết ACTH gây tăng sản xuất các chất tiền thân cortisol như Progesterone và 17-hydroxyprogesterone (17-OHP), các chất này có thể được chuyển hóa thành những chất tiền thân của androgen (xem Hình 13.1).
2.2 Phân loại
Ba phần tư các bệnh nhân bị tăng sản thượng thận bẩm sinh thể điển hình là tình trạng “tăng thải muối [salt wasters/” do thiếu hoạt tính enzym 21-hydroxylase và sản xuất không đủ aldosteron để giữ lại natri.
Một phần tư các bệnh nhân còn lại là các đối tượng có hoạt tính enzym thấp song vẫn có thể phát hiện được và duy trì được khả năng sản xuất aldosteron, mặc dù bị nam hóa song không bị mất natri qua mức và được biết như tình trạng “nam hóa đơn thuần (simple virilizers]”.
Các đối tượng giữ lại được 20 - 60% hoạt tính enzym thường được biểu hiện ở tuổi ấu thơ bằng các thể kín đáo hơn và không điển hình, không thường gặp các cơn suy thượng thận cấp [3].
Bệnh nhân bị thiếu hụt 21- hydroxylase điển hình là các đối tượng có nguy cơ cao xuất hiện suy thượng thận nếu không được cho dùng glucocorticoid ngoại sinh.
==> Bạn đọc có thể xem thêm: Nguyên nhân, triệu chứng và điều trị u tủy thượng thận
| Bảng 13.1. Phân loại tăng sản thượng thận bẩm sinh | ||||||
| Thiếu hụt 21-hydroxylase | Thiếu hụt 11ẞ-Hydroxylase | Thiếu hụt 17aHydroxylase/17, 20- lyase | Thiếu hụt 3B-Hydroxysteroid dehydrogenase typ 2 | Thiếu hụt P450 oxidoreductase | Thiếu hụt tăng sản thượng thận dạng hoặc SCC enzym | |
| Tần suất bị | Điển hình: Classic: 1/14.000 Không điển hình: 1/200-1.000 | Điển hình: 1/100.000 Không điển hình: Chưa rõ | 1/50.000 | Hiếm gặp | Hiếm gặp | Hiếm gặp |
| Gen bi tác động | CYP21A2 | CYP11B1 | CYP17A1 | HSD3B2 | P450 oxidoreducta (POR) | Steroidogenic acute regulatory protein (STAR) |
| Các biến đổi hormon | 117-0HP, 21- deoxycortisol, androstenedion và renin ICortisol và aldosteron | ↑Doc, 11- deoxycortisol, androstenedion, 17- OHP ↓Cortisol, aldosteron, corticosteron, renin | ↑ DOC, corticosteron và progesteron ↓ Cortisol, aldosteron, 17-OHP, 170-hydroxy pregnenolon, DHEA, androstenedion | ↑117- hydroxypregnenolon, DHEA, renin ↓Cortisol,aldosteron, progesterone, 17-OHP, androstenedion, DOC, 11-deoxycortisol | ↑Pregnenolone, progesterone, 17-0HP, DOC, corticosterone ↓DHEA and androstenedione | ↑Renin ↓tất cả các steroid |
| Biểu hiện lâm sàng | Điển hình: mất muối ở trẻ sơ sinh, nam hóa. Không điển hình: tăng quá mức androgen nhẹ. | Điển hình: nam hóa, tăng huyết áp, hạ kali máu. Không điển hình: hiếm gặp, tăng androgen quá mức. | Kiểu hình nữ vị thành niên song không có các đặc trưng sinh dục thứ phát và giảm chức năng sinh dục với tăng kích tố hưởng sinh dục, tăng huyết áp và giả Kali máu. | Điển hình: mất muối ở trẻ sơ sinh, cơ quan sinh dục ngoài của nam và nữ kém phát triển, nam hóa nhẹ cơ quan sinh dục ngoài nữ. Không điển hình: cực kỳ hiếm gặp. | Cơ quan sinh dục không điển hình nam hóa ở nữ không tiến triển, nam hoas ở mẹ nam hóa. các biểu hiện của khung xương | Điển hình: mất muối ở trẻ sơ sinh, cơ quan sinh dục ngoài của nữ. Không điển hình: suy thượng thận, cơ quan sinh dục ngoài và chức năng sinh dục thay đổi. |
3 CHẨN ĐOÁN
3.1 Biểu hiện lâm sàng

Tình trạng thiếu hụt 21-hydroxylase điển hình xuất hiện từ khi sinh hoặc trong vòng vài tuần sau sinh với tụt huyết áp do suy thượng thận.
Thiếu hụt 21-hydroxylase gây nam hóa đơn thuần thường được chẩn đoán ngay sau sinh ở các trẻ nữ do có các bất thường cơ quan sinh dục ngoài (âm vật to ra, hợp nhất các nếp môi sinh dục, hình thành xoang niệu sinh dục), song đối với trẻ nam chẩn đoán có thể bị chậm lại trong nhiều năm tới khi xuất hiện các dấu hiệu của tăng androgen quá mức.
Các triệu chứng của thiếu hụt 21-hydroxylase không điển hình có thể kín đáo hoặc thậm chí không có triệu chứng. Bệnh có thể được biểu hiện ở bất kỳ lứa tuổi nào sau sinh với các triệu chứng cường androgen như trứng cá nang, rậm lông, vô sinh và kinh nguyệt không đều. Các triệu chứng này có thể thay đổi lên xuống theo thời gian. Nam giới bị tăng sản thượng thận bẩm sinh không điển hình có thể bị vô sinh và trứng cá [14].
Ở trẻ em cả hai giới, tăng sản thượng thận bẩm sinh không điển hình có thể được chẩn đoán trong quá trình tìm nguyên nhân mọc lông mu sớm hoặc tình trạng tăng trưởng tuyến tính nhanh với tuổi xương già hơn so với tuổi bệnh nhân.
Chẩn đoán phân biệt đối với tăng sản thượng thận bẩm sinh không điển hình ở người lớn không nhiều, bao gồm hội chứng buồng trứng đa nang, các khối u buồng trứng hoặc thượng thận gây nam hóa và sử dụng steroid đồng hóa ngoại sinh.
Trong tăng sản thượng thận bẩm sinh không điển hình ở nữ, khi khám thực thể các đặc điểm của tình trạng cường androgen chiếm uu thế. Rậm lông là triệu chứng thường gặp nhất, tiếp sau là thiểu kinh và trứng cá [1].
Nam giới có thể được biểu hiện bằng tinh hoàn nhỏ khi so sánh với dương vật, tinh trùng ít, vô sinh và thân hình lùn. Trong tăng sản thượng thận bẩm sinh điển hình, tỷ lệ các khối u còn lại của thượng thận ở tỉnh hoàn là 21 - 28% và hầu như chỉ được tìm thấy ở thể đột biến nặng [1,7].
Chiều cao cuối cùng của người trưởng thành có thể bị giảm bởi tình trạng cốt hóa sớm đầu xương do tăng androgen quá mức.
Thiếu hụt P450 oxidoreductase (POR) có thể gây ra các biểu hiện xương như các bất thường sọ mặt [1].
3.2 Chẩn đoán cận lâm sàng
Sàng lọc khả năng bị tăng sản thượng thận bẩm sinh ở trẻ sơ sinh bằng cách định lượng 17-OHP giúp làm giảm tới mức thấp nhất sự chậm trễ chẩn đoán và làm giảm tỷ lệ tàn phế do bị cơn suy thượng thận cấp. Một vài trường hợp tăng sản thượng thận bẩm sinh không điển hình đã được phát hiện bằng biện pháp sàng lọc trẻ sơ sinh, song hầu hết các trường hợp này bị bỏ lọt do 17-OHP bình thường hoặc ở mức ranh giới.
Nếu test sàng lọc cho trẻ sơ sinh cho kết quả dương tính hoặc nghi ngờ có tình trạng tăng sản thượng thận bẩm sinh, một gói xét nghiệm các steroid bằng kỹ thuật đo khối phổ (mass spectrometry) được chỉ định. Tuy nhiên, chẩn đoán thường yêu cầu phải tiến hành nghiệm pháp kích thích bằng cosyntropin như sẽ được mô tả ở dưới.
Đo khối phổ đối với các chất chuyển hóa steroid trong nước tiểu cung cấp một thăm dò thay thế cho nghiệm pháp huyết thanh và có thể đặc biệt hữu ích để phân biệt tình trạng thiếu hụt POR [1,8].
Định kiểu gen đối với trạng thái người mang hữu ích cho tư vấn di truyền cho bệnh nhân.
Tình trạng thiếu hụt 21-hydroxylase điển hình được đặc trưng bằng gia tăng nồng độ 17- OHP có ý nghĩa ở tuổi thơ ấu với tình trạng nam hóa ở các mức độ khác nhau, tình trạng này chỉ có thể trở nên rõ ràng tới giai đoạn sau này trong tuổi phát triển của trẻ. Nên lấy mẫu máu vào buổi sáng sớm để định lượng nồng độ 17- OHP trong giai đoạn tạo nang ở phụ nữ. Ít khả năng cho chẩn đoán tăng sản thượng thận bẩm sinh nếu kết quả nồng độ 17 - OHP < 200 ng/dL và một giá trị > 400 ng/dL đã được báo cáo là đạt độ đặc hiệu 100% và độ nhạy 90% cho chẩn đoán này [9].
Tiêu chuẩn vàng đối với chẩn đoán hormon là nghiệm pháp kích thích bằng kích tố thượng thận (cosyntropin) và nghiệm pháp phải cho kết quả với giá trị trong khoảng từ 200 ng/dL - 400 ng/dL.
- Test bằng ACTH tổng hợp 250 pg đường tĩnh mạch và tiến hành định lượng nồng độ 17-OHP huyết tương ở thời điểm trước và 60 phút sau tiêm thuốc, chú ý tham khảo giá trị xét nghiệm theo từng phòng xét nghiệm. Bệnh nhân bị tăng sản thượng thận bẩm sinh điển hình có giá trị>300 ng/dL, bệnh nhân bị tăng sản thượng thận bẩm sinh không điển hình sẽ thường có giá trị 17 -0HP> 1000 ng/dL. Nếu giá trị 17-OHP dưới 50 ng/dL có thể loại trừ chẩn đoán tăng sản thượng thận bẩm sinh [1].
- Nồng độ cortisol huyết tương được định lượng ở mức nền và tại thời điểm 30 và 60 phút sau nghiệm pháp kích thích.
- Nếu nồng độ cortisol sau nghiệm pháp ACTH <20 ug/dL, lưu ý là bệnh nhân cần được bổ sung thêm glucocorticoid trong thời gian bị stress thực thể. Xem thêm Chương 12 để biết chi tiết về chẩn đoán suy thượng thận.
- Chỉ nên tiến hành xác định kiểu gen nếu kết quả nghiệm pháp kích thích bằng cosyntropin không thật rõ ràng và nhằm mục đích tư vấn di truyền cho bệnh nhân [1].
- Các thành viên thế hệ F1 trong gia đình của bệnh nhân bị bệnh nên được sàng lọc về khả năng bị tăng sản thượng thận bẩm sinh do tiềm ẩn khả năng không có các dấu hiệu lâm sàng và có thể gây nguy hiểm nếu bệnh nhân không được điều trị.
4 ĐIỀU TRỊ
Đích điều trị đối với tăng sản thượng thận bẩm sinh là điều chỉnh tình trạng tiết cortisol bị thiếu hụt và ức chế sản xuất ACTH quá mức. Điều trị bao gồm việc làm cân bằng giữa điều trị thay thế đầy đủ glucocorticoid để ức chế tình trạng tăng sản xuất androgen và ngăn ngừa cơn suy thượng thận cấp ở các trường hợp điển hình, tránh các biến chứng liên quan với tăng glucocorticoid quá mức. Những bệnh nhân này nên mang theo người giấy cảnh báo y tế và được hướng dẫn sử dụng liều corticoid thích hợp khi bị stress.
Điều trị đối với tăng sản thượng thận bẩm sinh “gây tăng mất muối” điển hình là điều trị thay thế glucocorticoid và mineralocorticoid và bô sung natri clorua [1].
Điều trị bệnh nhân người lớn bị thiếu hụt 21- hydroxylase thể không điển hình chỉ nên tiến hành đối với bệnh nhân bị vô sinh hoặc cường androgen khi bệnh nhân mong muốn được điều trị tình trạng này. Hiện chưa có báo cáo trong y văn về tử vong do suy thượng thận như một hậu quả của tăng sản thượng thận bẩm sinh thể không điển hình, do đó không cần sử dụng liều steroid khi bị stress trừ khi đáp ứng cortisol dưới mức bình thường khi làm test kích thích bằng kích tố thượng thận (cosyntropin). Xem Chương 12 để biết chi tiết về chẩn đoán và xử trí suy thượng thận.
Hydrocortison là điều trị được ưu tiên lựa chọn ở trẻ em do thuốc cải thiện kết cục lâm sàng song vẫn đảm bảo trẻ đạt được chiều cao cuối cùng của người trưởng thành (chiều cao được đo 1 năm sau khi trẻ kết thúc giai đoạn dậy thì).
- Liều thường dùng là 10 - 15 mg/m2/ngày hydrocortison chia nhỏ thành 2 - 3 lần/ngày.
- Sau khi kết thúc được tình trạng tăng trưởng tuyến tính, glucocorticoid loại tác dụng kéo dài được ưu tiên sử dụng cho bệnh nhân.
Khiếm khuyết sinh tổng hợp aldosteron rõ rệt trên lâm sàng chỉ được gặp ở thể “bị tăng mất muối”. Tuy nhiên, tình trạng thiếu hụt aldosteron dưới lâm sàng được thấy ở tất cả các thể và có thể được đánh giá bằng tỷ số aldosteron/hoạt tính renin huyết tương.
- Tất cả các bệnh nhân có tăng hoạt tính renin huyết tương hoặc giảm tỷ số aldosteron/ hoạt tính renin huyết tương có thể được hưởng lợi nhờ điều trị bằng mineralocorticoid và bổ sung thỏa đáng natri trong chế độ ăn [1].
- Duy trì cân bằng natri làm giảm nồng độ Vasopressin và ACTH và cho phép sử dụng các liều glucocorticoid thấp hơn.
- Tính nhạy cảm với mineralocorticoid có thể thay đổi qua thời gian và nhu cầu về điều trị thay thế mineralocorticoid nên được tái đánh giá định kỳ.
Điều trị thay thế các steroid sinh dục được bắt đầu vào thời gian dậy thì ở các đối tượng bị thiếu hụt 17- OH, tăng sản thượng thận bẩm sinh thể lipoid, thiếu hụt SCC, thiếu hụt POR và thiếu hụt 3B -HSD2 [1].
Phẫu thuật tạo hình cơ quan sinh dục hiện vẫn tiếp tục gây ra nhiều tranh cãi. Gia đình bệnh nhân nên được hướng dẫn về các ưu và nhược điểm của phậu thuật như một lựa chọn cho bệnh nhân có các rối loạn phát triển giới tính.
5 CÁC BIẾN CHỨNG
Không may là, kết cục điều trị tăng sản thượng thận bẩm sinh không đạt được tối ưu hoặc do ức chế không hoàn toàn tình trạng cường androgen hoặc ngược lại, cường cortisol do điều trị quá mức. Cả hai tình trạng trên có thể gây giảm chiều cao cuối cùng ở tuổi trưởng thành của bệnh nhân từ 1,7 - 2,0 SD dưới giá trị trung bình của quần thể người bình thường [2].
Chiều cao cuối cùng ở tuổi trưởng thành có thể được cải thiện ở trẻ em
với tuổi xương già hơn so với tuổi thực của bệnh nhân, được dự kiến ít nhất là 1 độ lệch chuẩn ở dưới chiều cao đích trung vị của cha mẹ, nhờ việc sử dụng GH và chất chủ vận hormon gây giải phóng LH [10].
Xem Chương 12 để biết về xử trí cơn suy thượng thận ở bệnh nhân bị tăng sản thượng thận bẩm sinh thể điển hình gây mất muối.
6 KIỂM TRA VÀ THEO DÕI
Theo dõi các tác dụng của điều trị thay thế steroid rất quan trọng. Nồng độ 17- OHP kháng lại với tình trạng ức chế bởi glucocorticoid và vì vậy khi nồng độ 17-OHP bình thường biểu hiện tình trạng điều trị quá mức. Điều trị quá mức có thể gây hội chứng Cushing do thuốc và dẫn tới làm giảm tỷ trọng khoáng của xương.
Nên duy trì nồng độ 17-OHP ở mức cao của giới hạn bình thường trên đến mức bất thường nhẹ trong khi duy trì nồng độ androstenedion ở giới hạn bình thường [1].
Có thể theo dõi điều trị thay thế mineralocorticoid bằng cách theo dõi huyết áp và nồng độ kali máu.
Nồng độ renin tăng cao hoặc tụt huyết áp có thể gợi ý là điều trị thay thế bằng mineralocorticoid không thoả đáng.
Chỉ nên tiến hành thăm dò hình ảnh học thượng thận cho các đối tượng có diễn biến sinh hóa hoặc lâm sàng không điển hình. Không có đủ thông tin để khuyến cáo tiến hành sàng lọc thường quy tìm kiếm khối u thượng thận.
Ở bệnh nhân có thai bị tăng sản thượng thận bẩm sinh thể điển hình phải được các thầy thuốc chuyên khoa nội tiết có kinh nghiệm trong lĩnh vực này thực hiện. Trong khi tình trạng hiếm muộn ở các phụ nữ bị tăng sản thượng thận bẩm sinh thể không điển hình thường nhẹ, một số bằng chứng cho thấy điều trị bằng glucocorticoid giúp giảm tỷ lệ sảy thai [11]. Điều trị tăng sản thượng thận bẩm sinh trước sinh hiện được coi như còn trong giai đoạn thực nghiệm [1].
Thảo luận về tư vấn trước sinh, chẩn đoán và điều trị đối với tăng sản thượng thận bẩm sinh ngoài phạm vi của chương này, độc giả nên tham khảo các bài báo tổng quan trọng tài liệu tham khảo số [12,13].
7 TÀI LIỆU THAM KHẢO
1. El-Maouche D, Arlt W, Merke DP. Congenital adrenal hyperplsia. Lancet 2017;390:2194-2210.
2. Speiser PW, Azziz R, Baskin LS, et al. Congenital adrenal hyperplasia due to steroid 21-hydroxylase defi- ciency: an endocrine society clinical practice guideline. J Clin Endocrinol Metab 2010;95:4133-4160.
3. Speiser PW, White PC. Congenital adrenal hyperplasia. N Engl J Med 2003;349:776–788.
4. Carmina E, Dewailly D, Escobar- Morreale HF, et al. Non-classic congenital adrenal hyperplasia due to 21-hydroxylase deficiency revisited: an update with a special focus on adolescent and adult women. Hum Reprod Update 2017;23(5):580–599.
5. Hannah-Shmouni F, Chen W, Merke DP. Genetics of congenital adrenal hyperplasia. Endocrinol Metab Clin North
Am 2017;46(2):435–458.
6. Bulsari K, Falhammar H. Clinical perspectives in congenital adrenal hyperplasia due to 11ẞ-hydroxylase deficiency. Endocrine 2017;55(1): 19-36.
7. Dumic M, Duspara V, Grubic Z, Oguic SK, Skrabic V, Kusec V. Testicular adrenal rest tumors in congenital adrenal hyperpla- sia-cross-sectional study of 51 Croatian male patients. Eur J Pediatr 2017;176:1393-1404.
8. Hines JM, Bancos I, Bancos C, et al. High-resolution, accurate-mass (HRAM) mass spectometry urine steroid profiling in the diagnosis of adrenal disorders. Clin Chem 2017;63:1824-1835.
9. Azziz R, Hincapie LA, Knox- henhauer ES, Dewailly D, Fox L, Boots LR. Screening for 21-hy- droxylase-deficient nonclassic adrenal hyperplasia among hyper- androgenic women: a prospective study. Fertil Steril 1999;72(5): 915-925.
10. Turcu AF, Auchus RJ. The next 150 years of congenital adrenal hyperplasia. J Steroid Biochem Mol Biol 2015;153:63-71.
11. Bidet M, Bellane Chatelet C, Galand Porter MB, et al. Fertility in women with nonclassical con- genital adrenal hyperplasia due to 21-hydroxylase deficiency. J Clin Endocrinol Metab 2010;95: 1182-1190.
12. Yau M, Khattab A, New MI. Prena- tal diagnosis of congenital adrenal hyperplasia. Endocrinol Metab Clin North Am 2016;45:267–281.
13. Nimkarn S, New MI. Congenital adrenal hyperplasia due to 21-hydroxylase deficiency: a paradigm for prenatal diagnosis and treatment. Ann N Y Acad Sci 2010;1192:5-11.
14. Thomas J.Braranski, MD, PhD; Janet B.McGill, MD, MA, FACE; Julie M.Silverstein, MD và các tác giả khác tham gia biên soạn, Khoa nội tiết chuyển hóa và nghiên cứu
