Sinh học phân tử ung thư phổi không tế bào nhỏ đột biến KRAS, EGFR, ALK và ứng dụng lâm sàng

Tác giả: Tiến sĩ, Bác sĩ NGUYỄN QUANG ĐỢI - Khoa Hô Hấp, Bệnh viện đa khoa tỉnh Hải Dương
Tải bản PDF TẠI ĐÂY
1 Giới thiệu
Cơ sở phân tử của ung thư phổi phức tạp và không đồng nhất. Những tiến bộ trong hiểu biết của chúng ta về sự thay đổi phân tử ở nhiều cấp độ (di truyền, ngoại di truyền, biểu hiện protein) và ý nghĩa chức năng của chúng có vai trò tác động đến chẩn đoán, tiên lượng và điều trị ung thư phổi. Ung thư phổi phát triển thông qua một quá trình nhiều giai đoạn liên quan đến sự phát triển của nhiều thay đổi di truyền và ngoại di truyền, đặc biệt là kích hoạt các con đường thúc đẩy tăng trưởng và ức chế các con đường ức chế khối u. Hiểu rõ hơn về nhiều con đường sinh hóa liên quan đến sinh bệnh học phân tử của ung thư phổi là rất quan trọng đối với sự phát triển của các chiến lược điều trị mà có thể gây ra các rối loạn (aberrations) phân tử đích và các con đường kích hoạt xuôi dòng (downstream) của chúng(1).
Những thay đổi phân tử đặc hiệu thúc đẩy sự phát triển của khối u và bộc lộ các đích cho điều trị đã được xác định rõ nhất trong ung thư biểu mô tuyến (ADC: Adenocarcinomas) nhưng ngày càng có mối quan tâm đến đặc điểm phân tử của ung thư biểu mô tế bào vảy (SCC: Squamous cell carcinoma) mở ra các đích điều trị tiềm năng mới. Trong ung thư phổi cũng như trong các khối u ác tính khác, sự hình thành ung thư (tumourigenesis) liên quan đến việc kích hoạt các protein thúc đẩy tăng trưởng (ví dụ, v-Ki-ras2 Kirsten rat sarcoma virus homolog (KRAS), thụ thể yếu tố tăng trưởng biểu bì (EGFR), BRAF, MEK-1, HER2, MET, ALK và được tái sắp xếp trong quá trình chuyển nạp (RET) cũng như bất hoạt các gen ức chế khối u (ví dụ: P53, phosphatase with tensin homology (PTEN), LKB-1)(2).
Kích hoạt tăng trưởng sinh ung thư có thể xảy ra bằng cách khuếch đại gen hoặc các thay đổi di truyền khác bao gồm đột biến điểm và sắp xếp lại cấu trúc, dẫn đến tín hiệu không được kiểm soát thông qua con đường gây ung thư. Sự phụ thuộc sinh ung thư (oncogene addiction) xảy ra khi sự sống sót của tế bào phụ thuộc vào việc tiếp tục kích hoạt tín hiệu sai lầm khiến chúng trở thành ứng cử viên lý tưởng cho các liệu pháp nhắm đích (targeted therapies) (3).
Đột biến điều khiển (driver mutations) gây ung thư đã được xác định ở hơn 50% ung thư biểu mô tuyến phế quản và hầu như luôn loại trừ các đột biến điều khiển khác. Các con đường truyền tín hiệu được điều hòa bởi gen gây ung thư và gen ức chế khối u thường được kết nối với nhau bằng cách đan chéo (cross-talk) giữa các con đường liên quan sinh ung thư. Thêm vào sự phức tạp là sự xuất hiện các đột biến mới của các khối u theo thời gian trong quá trình tiến triển tự nhiên của bệnh và đáp ứng với các biện pháp điều trị (4).
Có một sự đa dạng di truyền lớn trong ung thư phổi và chúng chứa đựng (harbour) số lượng lớn nhất về các sai sót di truyền của tất cả các khối u. Sự hiểu biết về sinh học phân tử của ung thư phổi đã được cách mạng hóa bằng phát triển và ứng dụng các công nghệ giải trình tự gen, từ đó cung cấp một phương tiện toàn diện để xác định sự thay đổi sinh dưỡng (somatic) trong toàn bộ bộ gen và bộ gen ung thư. Ung thư phổi có bộ gen rất phức tạp, một nghiên cứu giải trình tự bộ gen quy mô lớn gần đây về 31 trường hợp ung thư phổi không tế bào nhỏ (NSCLC) xác định được 727 gen đột biến không được báo cáo trước đây trong tài liệu hoặc trong cơ sở dữ liệu COSMIC (5).
Các nghiên cứu về bộ gen đã xác nhận những thay đổi rõ ràng trước đây trong ung thư phổi như KRAS, EGFR và BRAF và cũng đã xác định được các đột biến tần số thấp nhưng tái phát, là những phát hiện mới trong ung thư, bao gồm cả những thay đổi có thể nhắm đích như JAK2, ERBB4, RET, thụ thể yếu tố tăng trưởng nguyên bào sợi 1 (FGFR1) và thụ thể miền discoidin 2 (là một phần của vùng ngoại bào gắn kết với Collagen) (DDR2) (6,7).
Mặc dù các nghiên cứu này cung cấp một bức tranh toàn cảnh về sự thay đổi di truyền trong ung thư phổi, nhưng thách thức lớn nhất vẫn là xác định các đột biến điều khiển có liên quan đến sinh học phân tử từ phần lớn các đột biến thành viên. Mức độ tương đối của các đột biến tái phát tần số cao làm nổi bật tính không đồng nhất và tính phức tạp của sinh học phân tử trong ung thư phổi, với các con đường phổ biến bị ảnh hưởng bởi một loạt các thay đổi di truyền khác nhau, đặt ra thách thức cho việc sử dụng các thuốc điều trị cá thể hóa (8-10).
2 Dịch tễ học ung thư phổi
2.1 Tình hình ung thư phổi trên thế giới
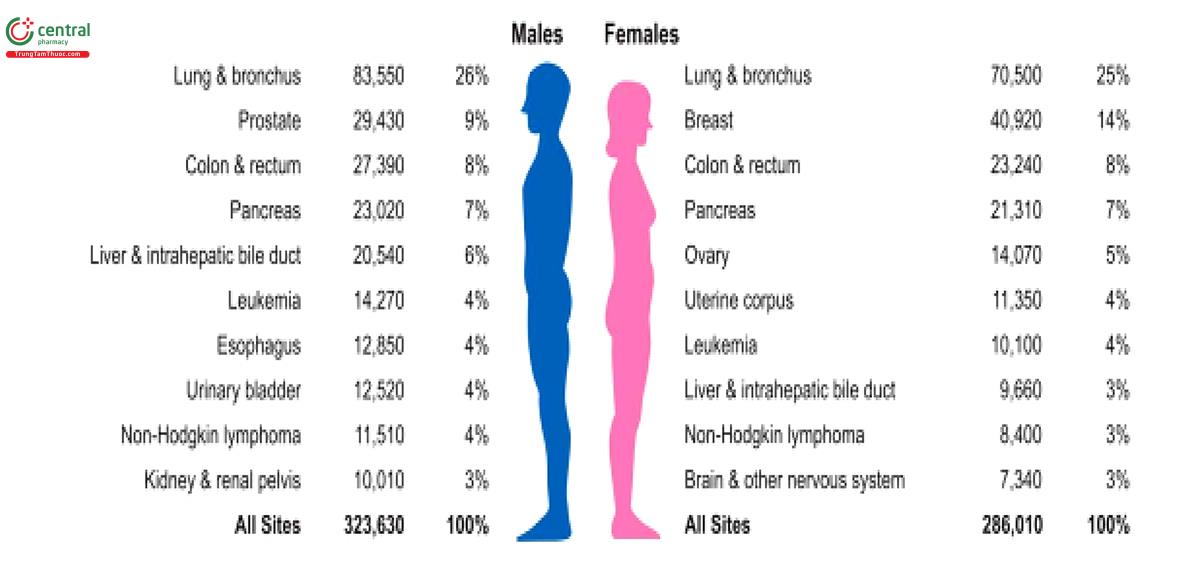
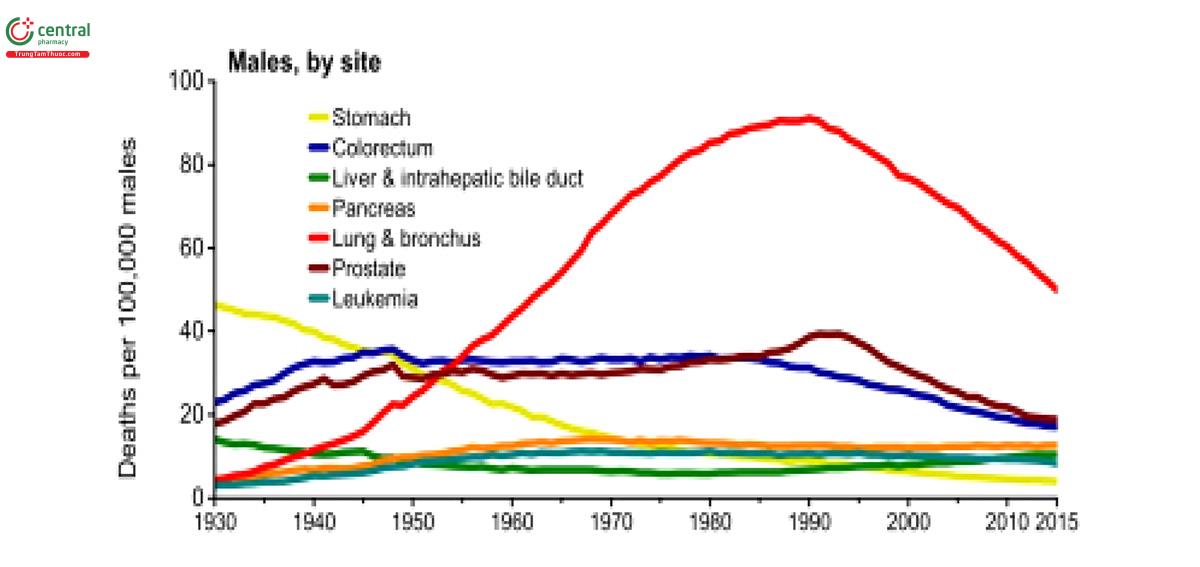
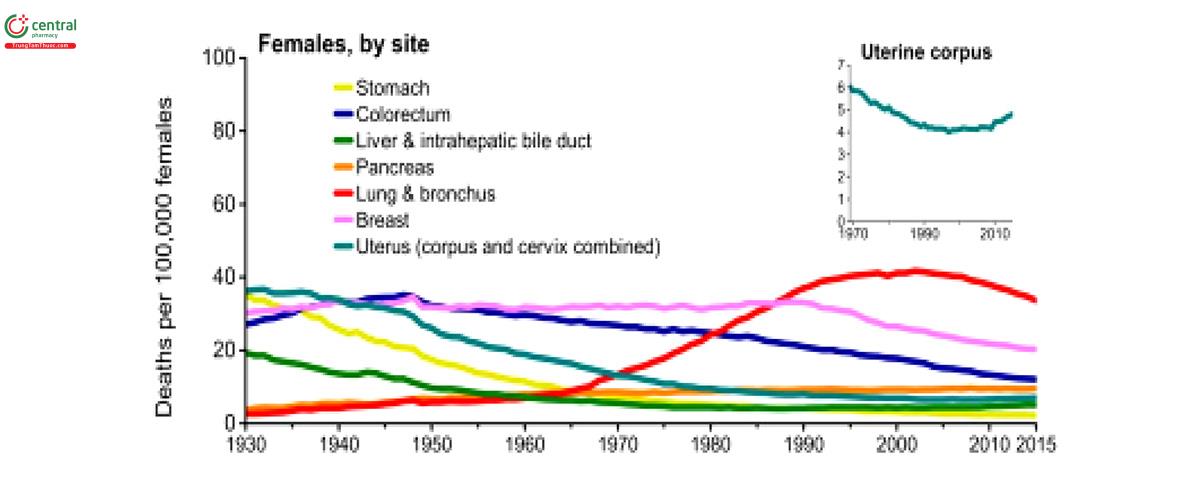
Năm 2012, ước tính trên toàn thế giới có khoảng 1,82 triệu trường hợp ung thư phổi (UTP) mới mắc và khoảng 1,59 triệu ca tử vong. Tại Mỹ, UTP là loại ung thư có tỷ lệ tử vong cao nhất và tỷ lệ mới mắc đứng thứ hai ở cả hai giới (11). Theo thống kê của Tổ chức Y tế thế giới (WHO) năm 2015, trên thế giới hàng năm có khoảng 1,37 triệu người tử vong do UTP chiếm 18% trong các trường hợp tử vong do các loại ung thư (12). Năm 2015, ở Mỹ có khoảng 221.200 trường hợp UTP mới được phát hiện và khoảng 158.040 ca tử vong do UTP, chiếm 28% tổng số tử vong do ung thư (13). Cũng theo số liệu thống kê tại Mỹ, ước tính trong năm 2016, số ca mới mắc của UTP khoảng 224.390 trường hợp, chiếm vị trí thứ 2 ở cả hai giới, với 117.920 ca mới mắc chiếm (14,0%) ở nam và 106.470 ca mới mắc ở nữ chiếm (13,0%) trong các loại ung thư. Tỷ lệ tử vong do UTP vươn lên vị trí đầu tiên, ở nam là 85.920 người chiếm (27,0%), ở nữ 7.160 người chiếm (26,0%) trong tất cả các trường hợp tử vong do UTP (14). Năm 2018, ước tính các trường hợp mới mắc ung thư phổi ở Mỹ là 121.680 đối với nam và 112.350 đối với nữ, trên tổng số là 234.030 ung thư phổi, tương đương 641 ca ung thư phổi được chẩn đoán mỗi ngày. Ung thư biểu mô phổi là loại ung thư phổ biến đứng thứ 2 ở cả 2 giới, sau ung thư tuyến tiền liệt ở nam và ung thư vú ở với nữ. Năm 2018, ung thư phổi chiếm 14% ung thư mới ở nam giới và 13% ung thư mới ở phụ nữ ở Mỹ (13).
Tại Trung Quốc năm 2014, có khoảng 3.804.000 người được chẩn đoán mới ung thư phổi (2.114.000 nam và 1.690.000 nữ), tương đương với hơn 10.422 trường hợp ung thư phổi được chẩn đoán mỗi ngày. Ước tính có 2.296.000 trường hợp tử vong do ung thư phổi, tương ứng 6.290 trường hợp tử vong mỗi ngày (15).
Tại Nhật Bản năm 2014 có 368.103 trường hợp tử vong do ung thư, trong đó 218.397 nam và 149.7706 nữ. Tỷ lệ tử vong chuẩn hóa theo độ tuổi (dân số thế giới) đối với nam và nữ lần lượt là 114,8 và 63/100.000. Trong số nam giới, năm 2014, vị trí ung thư hàng đầu là phổi (24,0%), tiếp theo là dạ dày (14,4%), đại trực tràng (12,0%), gan (8,8%) và tụy (7,5%). Năm 2014, vị trí ung thư hàng đầu ở nữ giới là đại trực tràng (14,9%), tiếp theo là phổi (14,0%), dạ dày (11,0%), tuyến tụy (10,0%) và vú (8,8%) (16).
Tại Hàn Quốc, năm 2012, tỷ lệ thô mắc ung thư phổi là 43,9/100.000. Tỷ lệ tử vong chuẩn hóa theo tuổi của ung thư phổi là 19,8/100.000. Tỷ lệ sống sót tương đối sau 5 năm là 11,3% giai đoạn 1993 - 1995 và tăng lên 21,9% trong giai đoạn 2008 - 2012. Adenocarcinoma có xu hướng tăng đều đặn ở cả nam và nữ và đã thay thế ung thư biểu mô tế bào vảy, trở thành loại ung thư phổi phổ biến nhất ở Hàn Quốc. Ở những bệnh nhân mắc ung thư biểu mô tuyến, tần số đột biến EGFR là 43% (20-56%), trong khi đột biến gen EMK4-ALK <5% (17).

2.2 Tình hình ung thư phổi tại Việt Nam
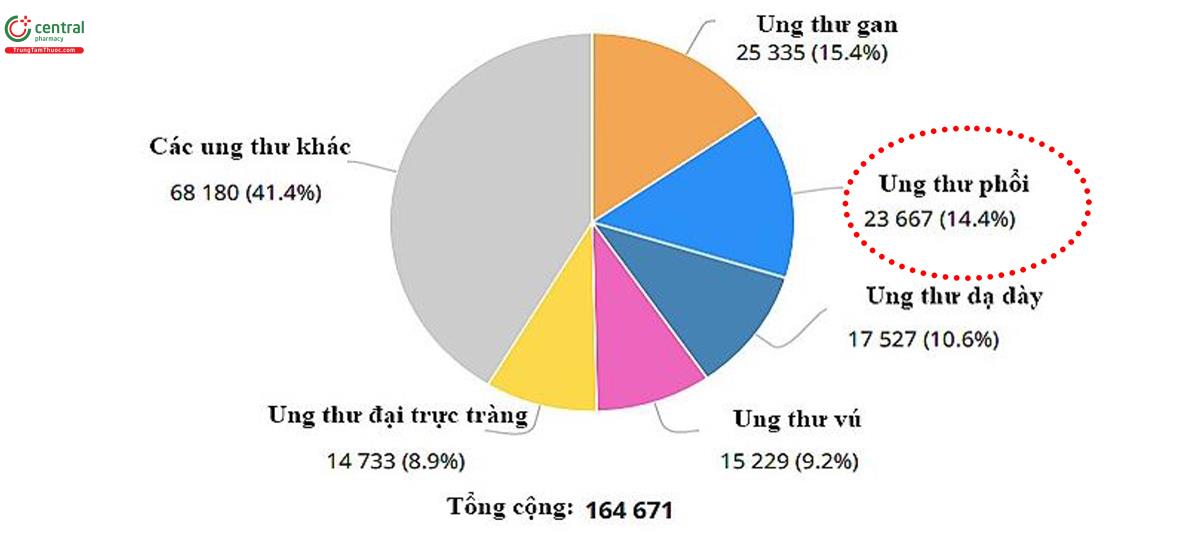
Tại Việt Nam, theo Nguyễn Bá Đức và cs (2006), UTP chiếm 20% trong tổng số các ung thư, là ung thư phổ biến nhất ở nam giới và đứng hàng thứ ba trong số các ung thư ở nữ giới sau ung thư vú và ung thư dạ dày (19). Theo thống kê của Bộ Y Tế (2009), UTP đứng thứ 2 về tỷ lệ tử vong của các loại ung thư hàng năm ở cả hai giới. Mỗi năm cả nước có trên 20.000 bệnh nhân UTP mới được phát hiện và có tới 17.000 trường hợp tử vong. Tại Bệnh viện Phổi Trung ương, tính đến năm 2012, số người mắc bệnh này đến khám và điều trị lên tới 16.677 người. Theo số liệu ghi nhận ung thư tại Hà Nội giai đoạn 2001 - 2005, ước tính hàng năm có 17.073 trường hợp mới mắc UTP, trong đó 12.958 nam và 4.115 nữ và là loại ung thư đứng hàng đầu ở nam giới. Tỷ lệ mắc theo tuổi là 40,2/100.000 dân ở nam và 10,6/100.000 ở nữ (20,21).
3 Một số đột biến gen phổ biến trong ung thư phổi không tế bào nhỏ
Sự hiểu biết sâu hơn về sinh bệnh học của ung thư phổi không tế bào nhỏ (NSCLC) đã mở ra hướng nghiên cứu các phân tử nhỏ, đó là các đột biến gen đích, đóng vai trò quan trọng trong tiến trình ung thư phổi di căn. Đột biến EGFR, KRAS và ALK (anaplastic lymphoma kinase) là những đột biến thường gặp ở bệnh nhân NSCLC, và sự hiện diện của một đột biến thay cho một đột biến khác có thể ảnh hưởng đến đáp ứng với liệu pháp điều trị nhắm đích. Do đó, xét nghiệm các đột biến này và liệu pháp điều trị phù hợp được chấp nhận rộng rãi như thực hành chuẩn (8). EGFR biểu hiện trên bề mặt tế bào và chiếm một tỷ lệ đáng kể trong NSCLC. Các nghiên cứu ban đầu với thuốc ức chế tyrosine kinase (TKIs) như gefitinib (Iressa) và Erlotinib (Tarceva) đã chỉ ra hoạt tính sinh học và lâm sàng trong một nhóm nhỏ bệnh nhân ung thư phổi. Nghiên cứu sâu hơn đã chứng minh tỷ lệ đáp ứng cao nhất đối với các TKI này được quan sát thấy ở những bệnh nhân bị đột biến sinh dưỡng trong miền EGFR-TK, đặc biệt đột biến mất đoạn ở exon 19, L858R ở exon 21 và G719X ở exon 18 (23). Ngược lại, đột biến T790M ở exon 20 được ghi nhận có liên quan đến đề kháng TKI mắc phải (24).
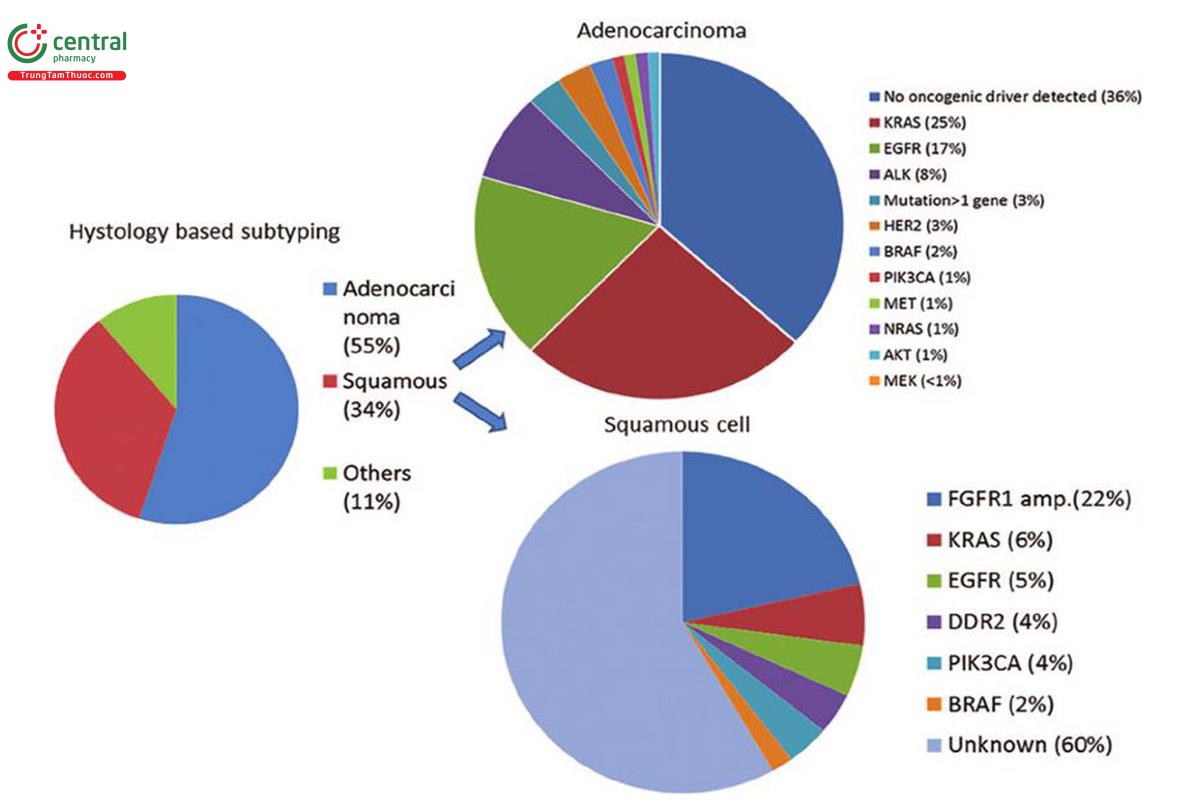
Nhìn chung, việc kích hoạt đột biến EGFR thường được quan sát thấy nhiều hơn ở những bệnh nhân mắc ung thư biểu mô tuyến và không có tiền sử hút thuốc trước đó, cũng như ở phụ nữ và những người gốc Châu Á (26). Dựa trên phân loại mới ung thư biểu mô tuyến do Hiệp hội quốc tế về nghiên cứu ung thư phổi, Hiệp hội lồng ngực Mỹ và Hiệp hội hô hấp châu Âu đề xuất, các nghiên cứu ghi nhận đột biến gen EGFR chiếm khoảng 50,5% trong số ung thư biểu mô tuyến phế quản được phẫu thuật cắt bỏ (4). Những đột biến này chiếm ưu thế ở týp vi nhú (micropapillary) và týp lepidic (trước đây gọi là ung thư týp biểu mô tiểu phế quản phế nang). Các nghiên cứu cũng cho thấy đột biến EGFR được quan sát thấy ở khoảng 50% người châu Á và 10% ở người không phải gốc châu Á (27).
Đột biến KRAS cũng chủ yếu được quan sát thấy ở ung thư biểu mô tuyến, chiếm 25% trường hợp. Tuy nhiên, ít gặp trong quần thể bệnh nhân châu Á, thường gặp ở bệnh nhân hút thuốc. Điều quan trọng nhất, bệnh nhân có đột biến KRAS dường như có tiên lượng kém hơn và đề kháng với EGFR-TKIs nhiều hơn, mặc dù mức độ ảnh hưởng đến lựa chọn điều trị vẫn chưa rõ rang (7,28).
Cuối cùng, sự hợp nhất giữa EML4 (echinoderm microtubule-associated proteinlike 4) và ALK được phát hiện thấy ở khoảng 2-7% bệnh nhân ung thư biểu mô tuyến (26). Sự kết hợp này và sự tái sắp xếp các ALK khác thường gặp ở những người không hút thuốc hoặc những người hút thuốc nhẹ và ở những người mắc ung thư biểu mô tuyến. Vì đột biến EGFR và ALK là những đột biến loại trừ lẫn nhau, những bệnh nhân có đột biến tái sắp xếp ALK sẽ không có lợi từ các TKI nhắm đích EGFR. Các trường hợp này được chỉ định bằng thuốc ức chế ALK như crizotinib (Xalkori), ceritinib (Zykadia), brigatinib (Alunbrig) (23).
3.1 Đột biến KRAS (Kristen Rat Sarcoma)
KRAS là một phần của họ gen tiền ung thư (proto-oncogenes) (KRAS, NRAS và HRAS xảy ra ở người) và mã hóa một protein G có vai trò quan trọng trong việc kiểm soát các con đường dẫn truyền tín hiệu điều hòa sự tăng sinh, biệt hóa và sống sót của tế bào (28).
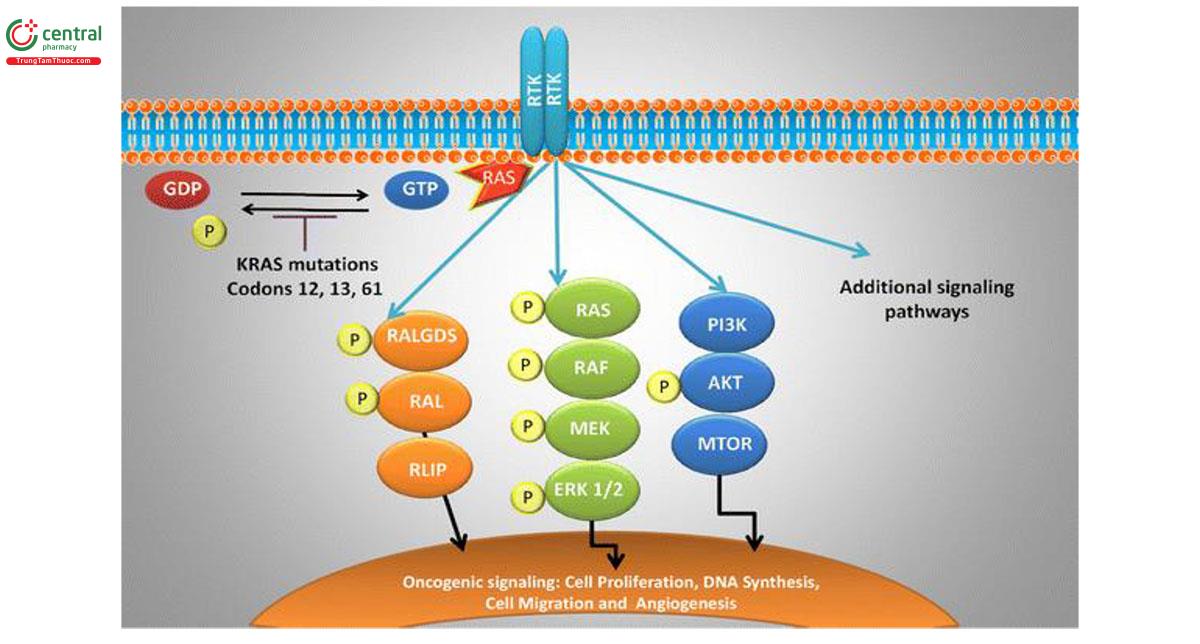
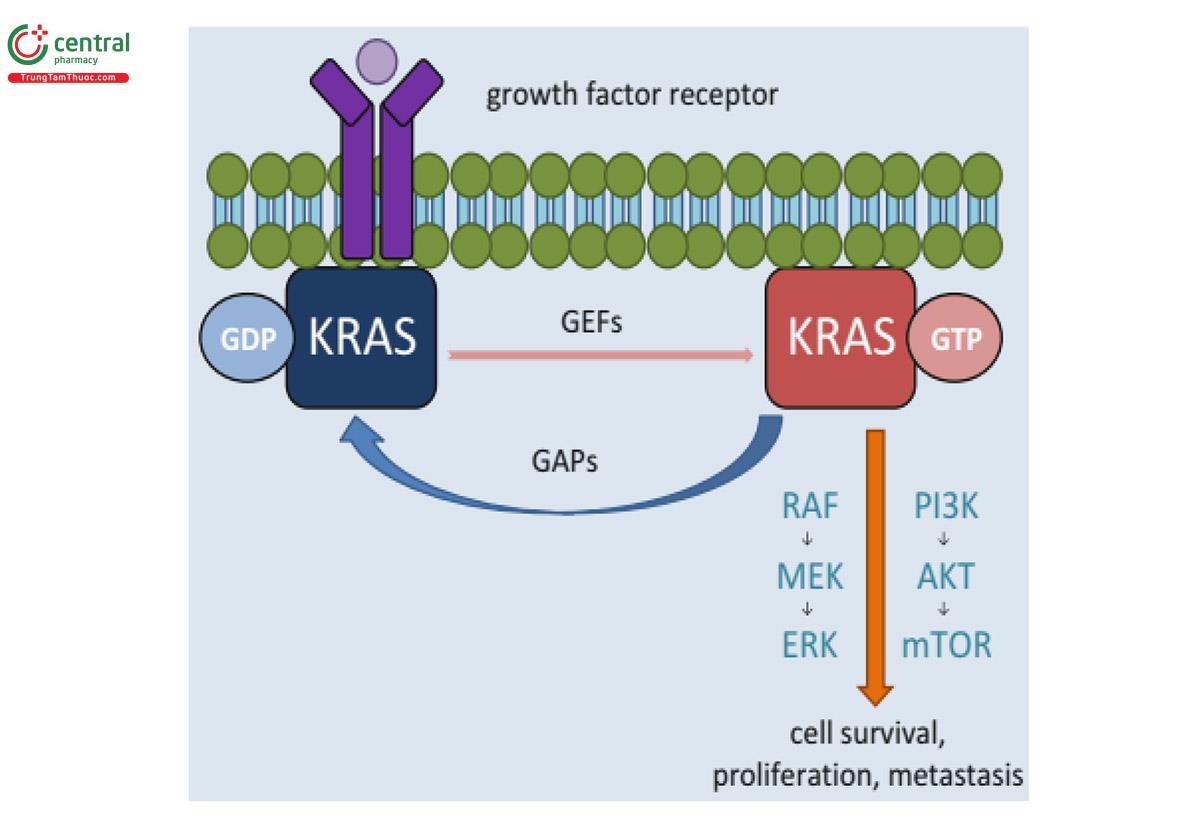
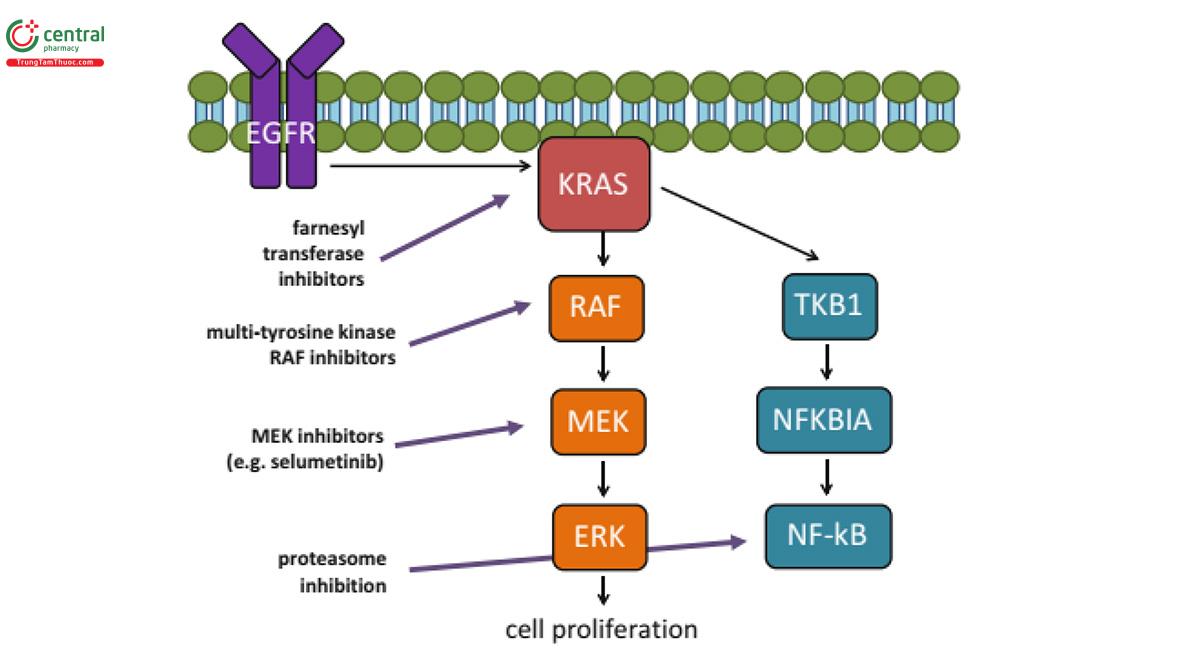
Các protein Ras là guanosine diphosphate (GDP) được liên kết và bất hoạt trong các tế bào im lặng (quiescent cells) bình thường. Có một sự chuyển đổi sang dạng liên kết guanosine triphosphate (GTP) được kích hoạt sau khi kích hoạt các thụ thể của yếu tố tăng trưởng ngược dòng (upstream growth factor receptors). Ras-GTP được kích hoạt sau đó liên kết và kích hoạt một số con đường xuôi dòng (downstream pathways) bao gồm protein kinase được hoạt hóa bằng mitogen (MAPK: mitogen-activated protein kinase), con đường RAS/RAF/MEK/MAPK và con đường PI3-K (đích của rapamycin động vật có vú (mTOR: mammalian target of rapamycin) (29)
KRAS đóng một vai trò quan trọng trong việc truyền tín hiệu xuôi dòng gây ra bởi một loạt các thụ thể của yếu tố tăng trưởng bao gồm cả EGFR và hoạt hóa hình thành protein, phá vỡ nhu cầu truyền tín hiệu qua trung gian của yếu tố tăng trưởng. Kích hoạt đột biến làm thay đổi hoạt động GTPase của protein, cản trở sự bất hoạt của RAS-GTP hoạt động thành GDP, dẫn đến tăng tín hiệu thông qua nhiều con đường thúc đẩy tăng trưởng xuôi dòng. Dòng thác truyền tín hiệu RAS/RAF/MEK/MAPK đóng vai trò trung tâm ở nhiều bệnh nhân ung thư phổi, với ít nhất một đột biến trong con đường được xác định ở 132/188 khối u, trong đó phổ biến nhất là đột biến ở KRAS (30)
Kích hoạt đột biến gen KRAS gây ung thư là sự thay đổi gây ung thư phổ biến nhất ở ung thư biểu mô tuyến phế quản (ADC) xảy ra ở khoảng 25-40% trường hợp, trong khi đột biến HRAS và NRAS rất hiếm gặp. Sự khác biệt về tỷ lệ đột biến KRAS ở ADC phổi rất có thể liên quan đến các quần thể bệnh nhân khác nhau, vì đột biến KRAS phổ biến hơn ở dân phương Tây so với dân châu Á và thường gặp hơn ở nam giới và những bệnh nhân hút thuốc (5).
Adenocarcinoma ở những người không hút thuốc đã được báo cáo có chứa đột biến KRAS trong khoảng 0-15% trường hợp. Ngoài ra, đột biến KRAS rất hiếm hoặc không gặp trong ung thư biểu mô tế bào vảy (SCC: Squamous cell carcinomas) và ung thư tế bào nhỏ (small cell carcinomas). Phân tích bộ gen toàn diện của 188 SCC Chỉ Xác định được 1 đột biến KRAS ở codon 61. Đột biến KRAS trong Adenocarcinoma bao gồm các thay thế acid amin đơn tại các điểm tới hạn (hotspots) nằm chủ yếu ở codon 12 nhưng hiếm gặp hơn ở codon 13 và 61. Các đột biến phổ biến nhất trong KRAS là chuyển đổi từ G sang T (~84%) ở những người hút thuốc trong khi ở những người không hút thuốc có nhiều khả năng chuyển từ G sang A (2).
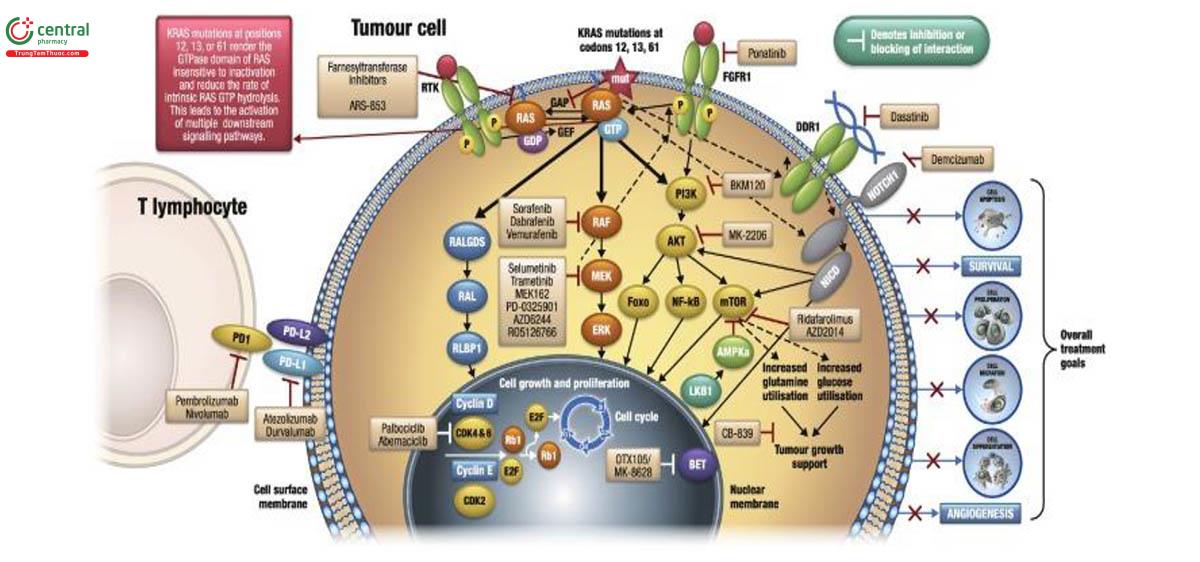
Biểu đồ tế bào u và tế bào lympho T minh họa các chiến lược điều trị mới cho NSCLC. AMPK, protein kinase hoạt hóa AMP; BET, bromodomain và miền cuối; CDK, kinase phụ thuộc cyclin; DDR1, thụ thể miền discoidin 1; ERK, kinase điều hòa tín hiệu ngoại bào; FGFR1, thụ thể yếu tố tăng trưởng nguyên bào sợi; FOXO, forkhead box O3; Protein kích hoạt GAP, GTPase; GEF, yếu tố trao đổi nucleotide guanine; MEK, MAPK / ERK kinase; mTOR, đích rapamycin của động vật có vú; NICD, miền nội bào Notch; NF, yếu tố nhân; PD1, thụ thể chết theo chương trình-1; PD-L1/2, phối tử của thụ thể chết theo chương trình 1/2; PI3K, phosphatidylinositol-4,5-bisphosphate 3- kinase; RAF, fibrosarcoma phát triển nhanh; RALGDS, kích thích phân ly nucleotide guanine RAL; RAS, virus sarcoma chuột; Rb, u nguyên bào võng; RTK, thụ thể tyrosine kinase.
Để phù hợp với vai trò các thay đổi KRAS như các đột biến điều khiển, chúng không xảy ra liên quan đến các đột biến EGFR, mặc dù có một số ít các trường hợp ngoại lệ. Một phân tích tổng hợp cho thấy các khối u đột biến KRAS kháng với các chất ức chế tyrosine kinase (TKIs), do đột biến KRAS dẫn đến kích hoạt hình thành các con đường hạ lưu của EGFR (31). Có bằng chứng cho thấy các protein đột biến KRAS khác nhau có ý nghĩa lâm sàng khác nhau. Điều thú vị là, sử dụng dữ liệu từ thử nghiệm BATTLE (prospective phase II Biomarker-integrated Approaches of Targeted Therapy for Lung cancer Elimination), đột biến KRAS dạng G12C hoặc G12V dự báo khả năng sống sót tiến triển ngắn hơn so với các đột biến KRAS khác hoặc các loại KRAS hoang dã. Hơn nữa, các thay thế acid amin khác nhau có liên quan đến các con đường hoạt hóa khác nhau (PI3-K, MEK với Gly12Asp và Ral với Gly12Cys hoặc đột biến Gly12Val) do cấu tạo protein khác nhau từ các đột biến chuyên biệt liên quan đến khả năng thay đổi các chất trung gian protein xuôi dòng. Điều này nhấn mạnh rằng việc sử dụng phù hợp các liệu pháp nhắm đích và thiết kế thử nghiệm lâm sàng cần đánh giá cẩn thận ý nghĩa lâm sàng và điều trị của các thay đổi di truyền chuyên biệt trong ung thư phổi. Tần số cao của đột biến KRAS trong ung thư phổi khiến nó trở thành mục tiêu điều trị lý tưởng nhưng không may là kết quả các thử nghiệm lâm sàng các tác nhân nhắm đích nói chung chưa mang lại các kết quả mong muốn (23).
3.2 Đột biến EGFR (Epidermal growth factor receptor)
Sự thay đổi của thụ thể yếu tố tăng trưởng biểu bì (EGFR: epidermal growth factor receptor) có liên quan đến sinh bệnh học của nhiều loại u, trong đó bao gồm NSCLC. EGFR mã hóa một tyrosine kinase xuyên màng với miền (domain) liên kết với phối tử ở ngoại bào và một thành phần nội bào chứa một miền tyrosine kinase. Liên kết của yếu tố tăng trưởng biểu bì - phối tử dẫn đến sự đồng hợp tử hoặc dị hợp tử với các thành viên khác trong gia đình EGFR và kích hoạt miền tyrosine kinase (9).
Sự truyền tín hiệu được kích hoạt bởi EGFR xảy ra thông qua các đường truyền tín hiệu PI3K/AKT/mTOR, RAS/RAF/MAPK và JAK/STAT. EGFR có liên quan đến việc điều chỉnh các chức năng gây ung thư như tăng sinh tế bào, sống sót, biệt hóa, tân tạo mạch, xâm lấn và di căn. Kích hoạt đột biến EGFR dẫn đến kích hoạt tyrosine kinase và chuyển dạng tế bào sinh ung thư của các tế bào biểu mô phổi trong ống nghiệm (33).
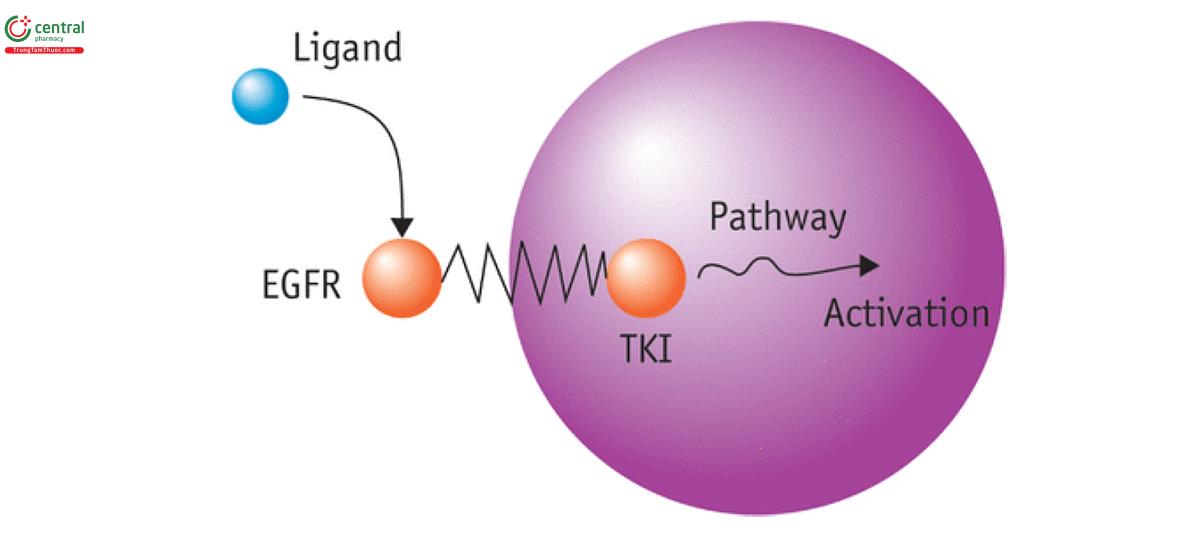
Một mô hình chuột biến đổi gen với biểu hiện rõ ràng của các đột biến EGFR phổ biến nhất cho thấy sự phát triển của nhiều ADC phổi nhạy cảm với sự ức chế phân tử nhỏ. Các cơ chế khác của việc tăng tín hiệu EGFR bao gồm tăng biểu hiện protein hoặc tăng số lượng bản sao gen. Đột biến EGFR hoạt hóa được ghi nhận ở 10-15% bệnh nhân phương Tây không chọn lọc và 30-40% dân châu Á. Sự khác biệt về tỷ lệ lưu hành được báo cáo của các đột biến khác nhau có thể một phần liên quan đến các quần thể bệnh nhân khác nhau nhưng cũng phụ thuộc vào độ nhạy của các kỹ thuật phân tích đột biến được sử dụng trong các nghiên cứu khác nhau (34).
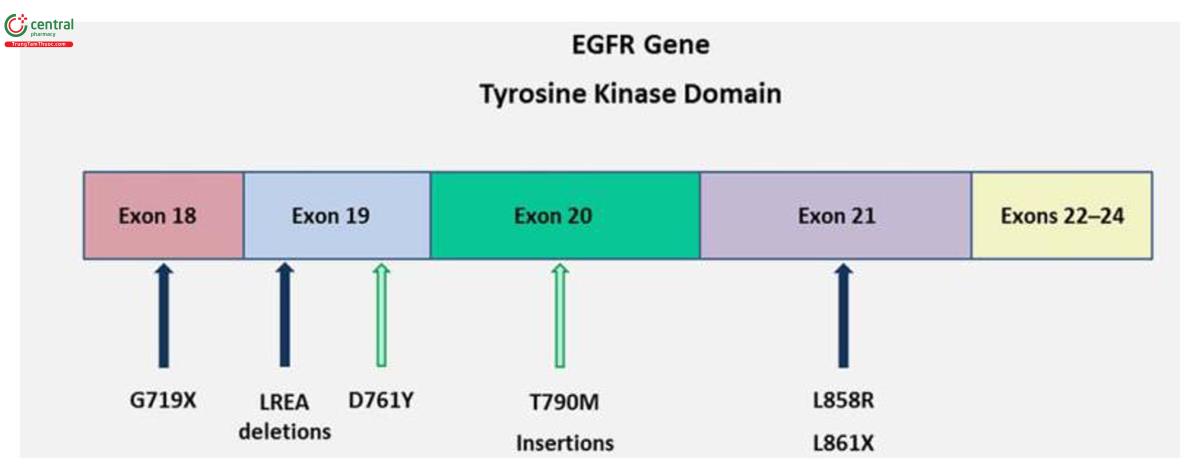
Trong NSCLC, đột biến EGFR xảy ra ở bốn exon đầu tiên của miền tyrosine kinase nội bào, phổ biến nhất là đột biến mất đoạn (frame deletions) ở exon 19 (~45%), trong đó có hơn 20 biến thể, phổ biến nhất là delE746-A750. Các đột biến EGFR phổ biến tiếp theo là đột biến sai nghĩa (missense mutations), đặc biệt là L858R, một đột biến điểm nucleotide đơn lẻ ở exon 21 dẫn đến thay đổi acid amin duy nhất từ leucine thành Arginine ở codon 858 (~40%) (35). Tuy nhiên, chúng ta đã tìm thấy trong quần thể dân Australia, đột biến kích hoạt ở exon 18 chiếm 14% các trường hợp có đột biến EGFR ở bệnh nhân ung thư phổi giai đoạn sớm và đột biến L858R bao gồm chỉ 29% của đột biến EGFR có trong theo dõi đoàn hệ này. Ngoài ra còn có một loạt các đột biến ít phổ biến hơn, bao gồm cả đột biến lặp đoạn (in frame duplications) hoặc đột biến thêm đoạn (insertions) ở exon 20 (~5-10%), trong đó có nhiều biến thể có liên quan đến kháng với TKI của EGFR (36)
Trong ung thư phổi, hầu hết các đột biến EGFR xảy ra ở ung thư biểu mô tuyến phế quản mặc dù chúng cũng có thể được phát hiện ở ung thư biểu mô tuyến-vảy (adenosquamous carcinomas). Đột biến EGFR gặp phổ biến hơn nhưng không chỉ là phát hiện duy nhất ở những bệnh nhân nữ, trẻ tuổi và không có tiền sử hút thuốc. Đột biến EGFR xảy ra rất hiếm ở các trường hợp mẫu bệnh phẩm mô học của ung thư biểu mô vảy (SCC) thuần nhất. Tuy nhiên, phân tích bộ gen toàn diện của 188 SCC ghi nhận xác định đột biến EGFR ở 2 trường hợp, cả hai đều có đột biến L861G. Mặc dù đột biến gen EGFR rất hiếm gặp ở SCC, nhưng đột biến biến thể III liên quan đến miền ngoại bào của EGFR, tăng số lượng bản sao chép và tăng biểu hiện protein quá mức gặp phổ biến hơn ở SCC so với ADC [38].
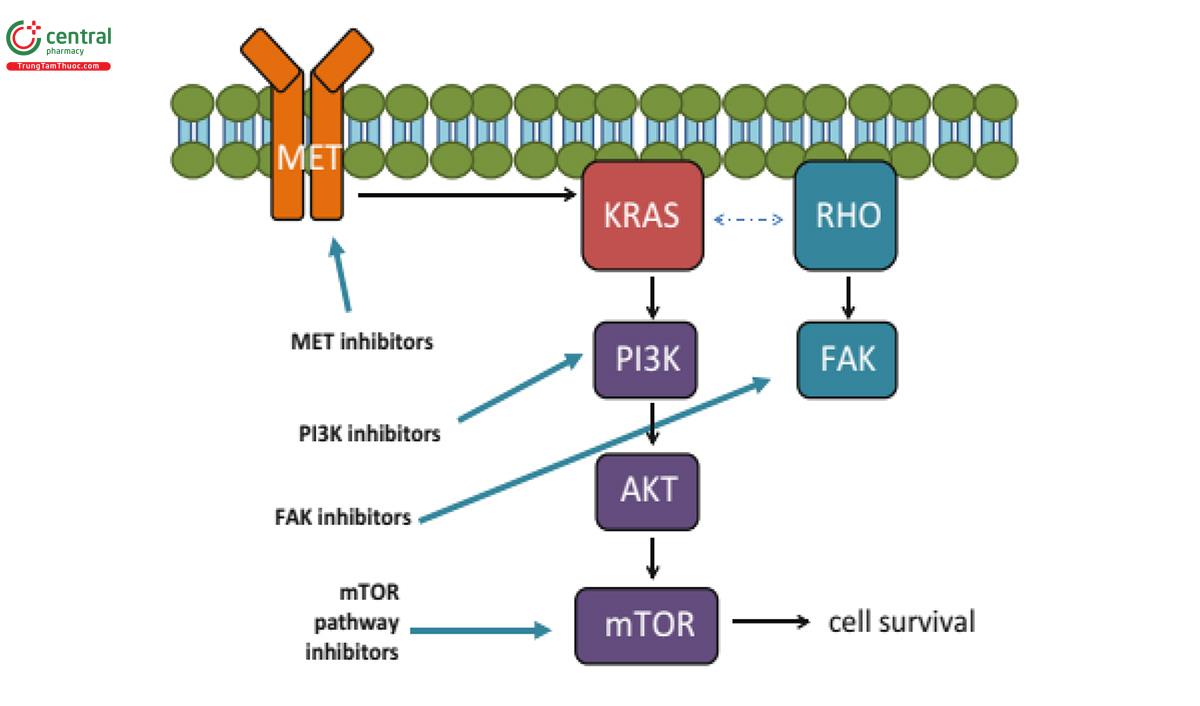
Đột biến EGFR thứ phát phát triển hoặc được chọn lọc như bản sao ở những bệnh nhân xuất hiện đề kháng với EGFR - TKIs, phổ biến nhất là đột biến điểm hoạt hóa T790M ở exon 20, thay thế cho một Methionine khổng lồ (bulkier) đối với threonine gây cản trở liên kết với TKI đảo ngược. T790M được phát hiện thấy trong khoảng 50% các loại u từ những bệnh nhân phát triển đề kháng TKI mắc phải. Một cách thú vị, các nhà nghiên cứu đã quan sát thấy đột biến ở exon 20 bao gồm đột biến T790M liên quan đến đề kháng với liệu pháp EGFR TKI đã được phát hiện thất ở 29% bệnh nhân bị đột biến gen EGFR trong một nghiên cứu đoàn hệ. Kích hoạt các con đường xuôi dòng qua cầu nối ức chế EGFR cũng có thể góp phần vào sự kháng thuốc EGFR-TKI bao gồm kích hoạt con đường PI3K thông qua khuếch đại MET (31,38).
3.3 Đột biến ALK (Anaplastic lymphoma kinase)
ALK là một thụ thể tyrosine kinase bất thường khác, gặp ở nhiều loại bệnh lý ác tính khác nhau. Mặc dù vai trò của ALK đối với bệnh ung thư ở người từ lâu đã ghi nhận sự hợp nhất NPM-ALK ở những bệnh nhân ung thư lympho không Hodgkin (non-Hodgkin lymphoma), sự hợp nhất EML4- ALK đã được ghi nhận trong y văn lần đầu tiên ở bệnh nhân NSCLC bởi Soda và cs vào năm 2007 như là một đột biến điều khiển tiềm tàng mới sinh ung thư (novel potential oncogenic driver mutant kinase). Khoảng 3-7% các khối u phổi chứa phức hợp ALK (26).
Nhiều biến thể phức hợp EML4-ALK khác nhau đã được mô tả trong NSCLC, điển hình với nhiều vị trí hòa hợp tại EML4 nhưng với vị trí hòa hợp cố định trong ALK. Các phức hợp EML4- ALK thường được phát hiện thấy ở những bệnh nhân hút thuốc lá nhẹ (<10 bao-năm) hoặc không bao giờ hút thuốc, và có xu hướng gặp ở độ tuổi trẻ hơn. Sự sắp xếp lại gen sinh ung thư EML4- ALK cũng phát hiện thấy ở nhiều nhóm chủng tộc khác nhau. Trong nghiên cứu đoàn hệ châu Á, một số nghiên cứu đã xác định tỷ lệ chuyển dịch sinh ung thư dao động 2,3-6,7% và không có sự khác biệt đáng kể so với nhóm không hút thuốc. Mặt khác, tỷ lệ sắp xếp lại EML4-ALK được ghi nhận có tỷ lệ thấp ở nhóm người da trắng, chiếm khoảng 1-3%. Điều thú vị là, theo một nghiên cứu được thực hiện trên nhóm các mẫu bệnh phẩm NSCLC thu thập từ Italia và Tây Ban Nha, tỷ lệ mới mắc các đột biến này được phát hiện tương tự với đoàn hệ châu Á, ở mức 7,5% (23,39).
Trong hầu hết các trường hợp, các phức hợp EML4-ALK không chồng lấp với các đột biến gây ung thư khác của EGFR hoặc KRAS. Sự hiện diện của phức hợp EML4-ALK liên quan đến đề kháng các EGFR-TKI. Mặc dù mối quan hệ giữa EML4-ALK và MET chưa được đánh giá đầy đủ, nhưng crizotinib, một loại thuốc ban đầu được phát triển như một chất ức chế MET, gần đây đã được Cơ quan Quản lý Thực phẩm và Dược phẩm Mỹ (FDA) phê chuẩn để điều trị NSCLC dương tính với EML4-ALK và bây giờ có thể được kê đơn như là một điều trị đầu tay (first-line treatment) (30,40).
Trong nghiên cứu gần đây của Shaw và cs, bệnh nhân NSCLC có dương tính với ALK khi điều trị bằng crizotinib có liên quan đến khả năng sống sót được cải thiện so với nhóm chứng không dùng crizotinib (crizotinib-naive controls). Thật không may, như với các loại thuốc điều trị ung thư nhắm đích khác, cuối cùng cũng phát hiện có đề kháng với crizotinib (41). Các đột biến kháng mới liên tục được xác định. Choi và cs báo cáo 4374G -> A và 4493C -> A ở gene ALK (42), sau đó Sasaki cs đã báo cáo đột biến F1174L tại miền ALK kinase. Với phân tích nền tảng NGS (giải trình tự gene toàn bộ), gần đây đã phát hiện ra nhiều hình thức hợp nhất ALK mới hơn, chẳng hạn như C2orf44-ALK trong ung thư đại trực tràng (43). Các biến thể hợp nhất khác của sự chuyển dịch ung thư phổi sẽ được tạo ra hiệu quả hơn và với những nỗ lực của kỹ thuật NGS sẽ áp dụng trong các quần thể người khác nhau trong tương lai gần.
4 Ứng dụng lâm sàng
Ung thư phổi cho đến nay vẫn là nguyên nhân phổ biến nhất gây tử vong liên quan đến ung thư trên toàn thế giới với gần 1,6 triệu ca tử vong vào năm 2012 hoặc chiếm tỷ lệ tử vong gần 20% do ung thư nói chung. Trong thập kỷ qua, những tiến bộ nghiên cứu phân tử đã mang lại những đột phá lớn trong hiểu biết mới, các phương pháp chẩn đoán và điều trị ung thư phổi, đặc biệt đối với ung thư phổi không tế bào nhỏ (NSCLC) là loại mô học chiếm gần 85% tổng số ung thư. Ngược lại, điều trị ung thư phổi tế bào nhỏ vẫn dựa trên hóa trị liệu, với các chất gây độc tế bào mới, chủ đạo là các thuốc platinum-etoposide (27,33).
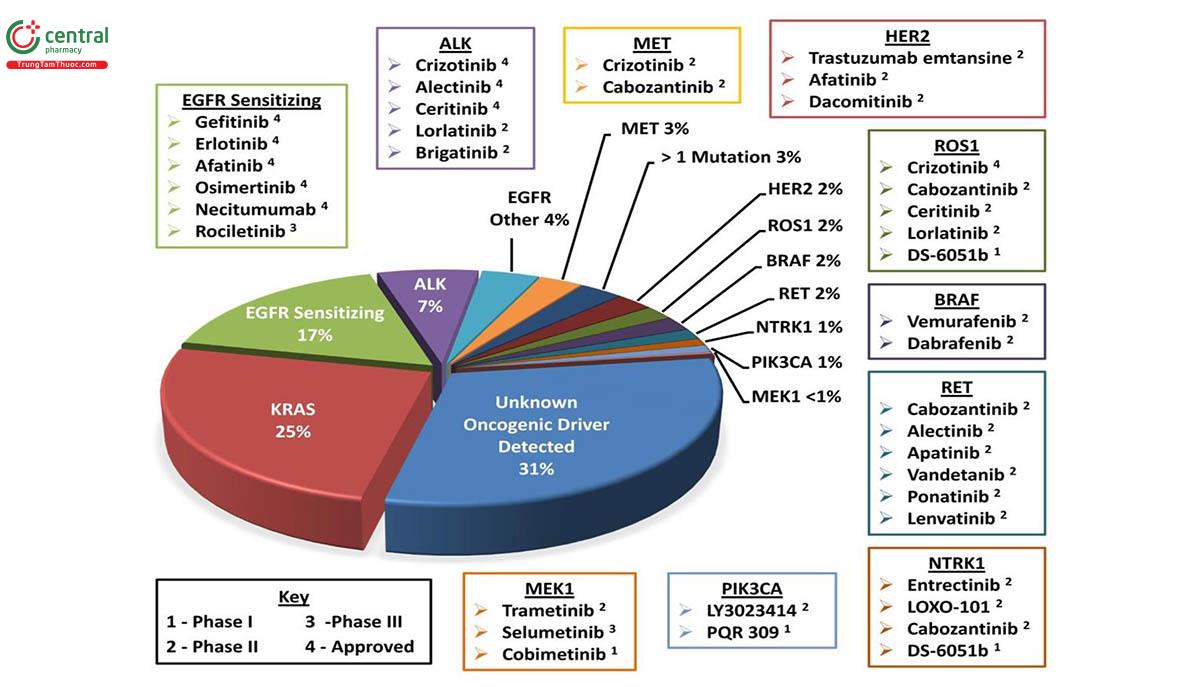
Ghi chú: (1) Phase I; (2) Phase II, (3) Phase III; (4) Đã được FDA chứng nhận điều trị
Tuy nhiên, kết quả điều trị ung thư phổi tiếp tục gây thất vọng. Ngay cả với sự kết hợp tối ưu của phẫu thuật, xạ trị và hóa trị, chỉ <15% bệnh nhân có thể được chữa khỏi. Hóa trị được sử dụng thường xuyên ở hầu hết bệnh nhân ung thư phổi và các liệu pháp khu trú (ví dụ: phẫu thuật, xạ trị) là những lựa chọn tiêu chuẩn khi bệnh ở giai đoạn sớm và giai đoạn muộn nhưng còn khu trú (2,34,45).
Nhiều thập kỷ nghiên cứu chuyên sâu đã đưa đến việc xác định các con đường phân tử có tầm quan trọng trung tâm trong phát triển ung thư và phát triển khối u. Các thuốc đã được nghiên cứu phát triển để nhắm vào các cơ chế này, từ đó cải thiện kết quả điều trị và tiện lượng ở bệnh nhân ung thư phổi. May mắn thay, một số tác nhân nhắm đích gần đây đã được đánh giá trong điều trị ung thư phổi, có kết quả đáng khích lệ và sẽ tiếp tục được nghiên cứu thêm (3,31,46).
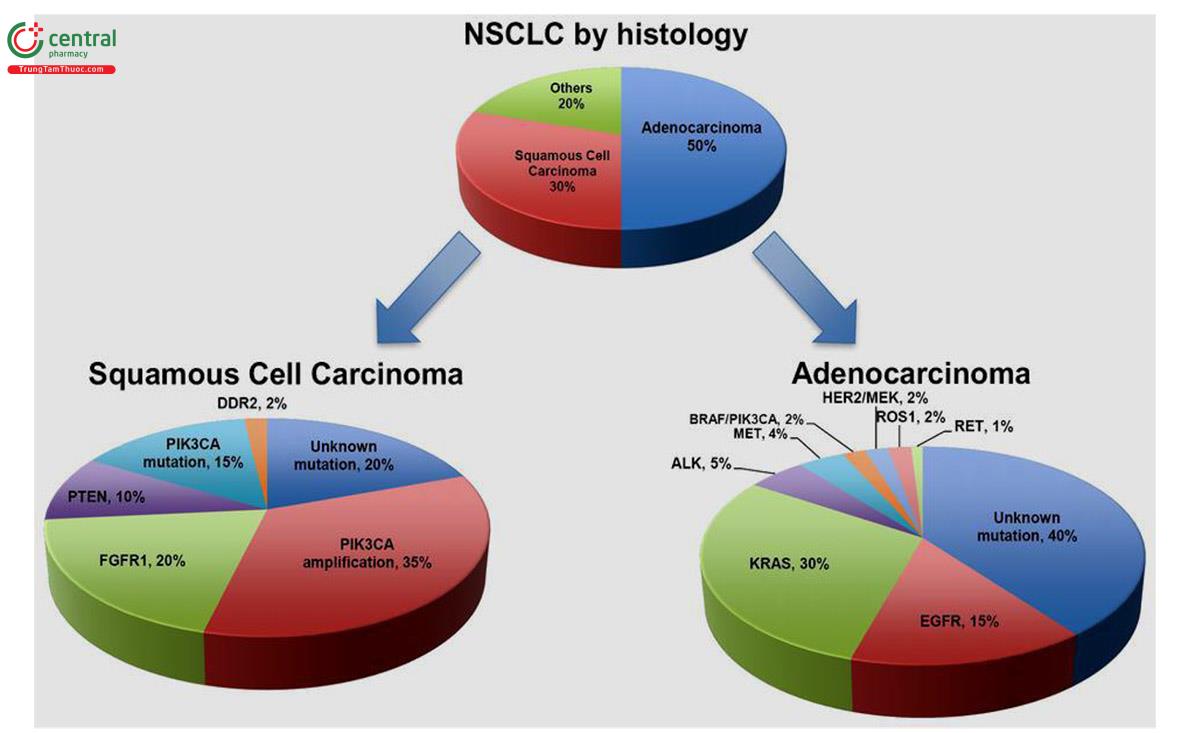
Sự kết hợp giữa việc dùng thuốc và xạ trị giúp gia tăng tác dụng của xạ trị, giảm di căn, và giữ lại được chức năng của mô chứa tế bào ung thư. Bên cạnh đó, các liệu pháp này cũng tồn tại hạn chế như phạm vi điều trị hẹp, phải điều trị nội trú, và đặc biệt là độc tính và các tác dụng phụ có hại cho sức khoẻ, ví dụ như các thuốc dẫn xuất platinum gây mệt mỏi, nôn mửa, và rối loạn chức năng thận, gan, giảm hồng cầu, bạch cầu, tiểu cầu, và đôi khi có thể ảnh hưởng đến chức năng tim mạch trong một số ít trường hợp (25,43,47). Chính vì thế việc phát triển các họ thuốc mới với tác dụng chọn lọc lên tế bào ung thư đồng thời có ít hoặc không có độc tính và tác dụng phụ lên tế bào lành (targeted therapy) đã được nghiên cứu hơn 30 năm nay. Một trong những thành quả đạt được là việc phát triển họ thuốc trị ung thư bằng con đường ức chế các enzym Tyrosine Kinase (Tyrosine Kinase Inhibitors - TKIs). Thuốc TKIs được đưa vào cơ thể bằng đường uống, do đó, bệnh nhân có thể không cần phải nhập viện điều trị (10,48).
.jpg)
Tyrosine Kinases (TKs) là một họ enzym được phát hiện lần đầu cách đây hơn 30 năm. Tính đến nay, khoảng 58 enzym TKs đã được định danh và nhiều nghiên cứu được tiến hành để tìm hiểu về hoạt động của họ enzym này. TKs đóng vai trò quan trọng trong các quá trình sinh trưởng, chuyển hóa, phân chia, và tồn tại của tế bào. Đột biến di truyền làm cho các enzym này gia tăng số lượng quá mức và có khả năng tự kích hoạt trong tế bào dẫn đến sự tăng trưởng và nhân đôi một cách mất kiểm soát của các tế bào liên quan. Đây là tiền đề của việc hình thành các tế bào ung thư. Một số nghiên cứu đã chỉ ra sự liên quan của enzym TKs với các loại ung thư như ung thư xương, ung thư phổi, và ung thư máu (23,31,46,49).
4.1 Đột biến KRAS
Vai trò của đột biến KRAS như một dấu ấn sinh học giúp dự đoán mức độ nhạy cảm với hóa trị hoặc với các EGFR - TKIs ở bệnh nhân NSCLC vẫn là một chủ đề gây tranh luận. Nhìn chung, hiệu quả điều trị của các phương pháp này ở bệnh nhân NSCLC có đột biến KRAS là kém nhưng có lẽ không có sự khác biệt đáng kể về hiệu quả so với những bệnh nhân ung thư phổi có đột biến gen KRAS loại hoang dã. Trong phác đồ điều trị bổ sung, những bệnh nhân NSCLC có đột biến gen KRAS và đột biến KRAS loại hoang dã (wild type) có được lợi ích tương tự với liệu pháp hóa trị dựa trên platinum (50).
Ở giai đoạn tiến triển, một nghiên cứu đoàn hệ tại Italia cho thấy, những bệnh nhân có đột biến KRAS (n = 60), được điều trị bằng hóa trị dựa trên platinum, có thời gian sống sót toàn bộ (OS) ngắn hơn so với những bệnh nhân có đột biến KRAS loại hoang dã (n = 187) (10,6 so với 14,4 tháng; HR: 1,41, 95% CI: 1,03 - 1,94; p = 0.032). Thời gian sống không bệnh (PFS) không khác biệt đáng kể giữa các nhóm nhỏ và trong trường hợp không có nhóm chứng, không thể làm sáng tỏ liệu thời gian sống sót toàn bộ (OS) ngắn hơn này là điểm riêng biệt do hiệu quả tiên lượng kém của KRAS hay không. Hơn nữa, kết quả của một nghiên cứu khác không hoàn toàn phù hợp với những phát hiện này (51,52).
Dữ liệu từ các thử nghiệm ngẫu nhiên các thuốc thế hệ thứ hai cho thấy lợi ích điều trị không khác biệt đáng kể giữa bệnh nhân có đột biến KRAS hoang dã và đột biến KRAS. Trong thử nghiệm TAILOR tại Italia (Tarceva Italian Lung Optimization tRial), ghi nhận những bệnh nhân NSCLCs có đột biến EGFR hoang dã và đột biến KRAS không dự đoán được lợi ích với Docetaxel hoặc erlotinib (53).
Đánh giá về khả năng tương tác điều trị âm tính giữa EGFR-TKIs và đột biến KRAS, hai phân tích gộp cho thấy bệnh nhân có đột biến KRAS có tỷ lệ đáp ứng với EGFR -TKI thấp hơn đáng kể so với bệnh nhân có đột biến KRAS loại hoang dã (54,55). Tuy nhiên, trong cả hai phân tích gộp, phân nhóm loại KRAS hoang dã, kể cả các đột biến gen EGFR, cản trở việc giải thích dữ liệu. Trong thử nghiệm TAILOR, hiệu quả vượt trội của docetaxel so với erlotinib là tương tự ở những bệnh nhân có đột biến KRAS và KRAS hoang dã. Xét thấy lợi ích của EGFR - TKIs là không đáng kể ở bệnh nhân NSCLC có đột biến EGFR hoang dã, không chắc chắn tình trạng đột biến KRAS sẽ ảnh hưởng đến lợi ích của EGFR - TKI trong nghiên cứu này (56).
4.2 Đột biến EGFR
Các thuốc EGFR-TKIs (gefitinib, erlotinib, afatinib) được ghi nhận có hiệu quả vượt trội về thời gian sống không bệnh (PFS: progression-free survival), tỷ lệ đáp ứng với điều trị (RR: response rates), độc tính tế bào (toxicity profile) và chất lượng cuộc sống (QoL: quality of life) so với phác đồ 2 hóa chất dựa trên platinum (IA). Chỉ có một nhóm nhỏ bệnh nhân được ghi nhận có sự cải thiện đáng kể về tỷ lệ sống sót toàn bộ khi chỉ định afatinib ở những bệnh nhân bị đột biến Del19 (đột biến mất đoạn tại exon 19). Kết quả so sánh trực tiếp hiệu quả của các EGFR - TKIs thế hệ thứ nhất, thứ hai và thứ ba ở những bệnh nhân chưa từng được điều trị trước đây cũng đã được báo cáo. Mặc dù lợi ích về chỉ số PFS đã được chứng minh đối với TKIs thế hệ thứ 3 như osimertinib (IA) và dacomitinib (IA). Nhưng chỉ có dacomitinib có hiệu quả trong việc tăng tỷ lệ sống sót toàn bộ, tuy nhiên các tác dụng phụ của các thuốc thế hệ thứ 3 này cũng cao hơn. Dữ liệu từ thử nghiệm FLAURA so sánh osimertinib với điều trị chuẩn vẫn còn chưa được đánh giá đầy đủ [9].
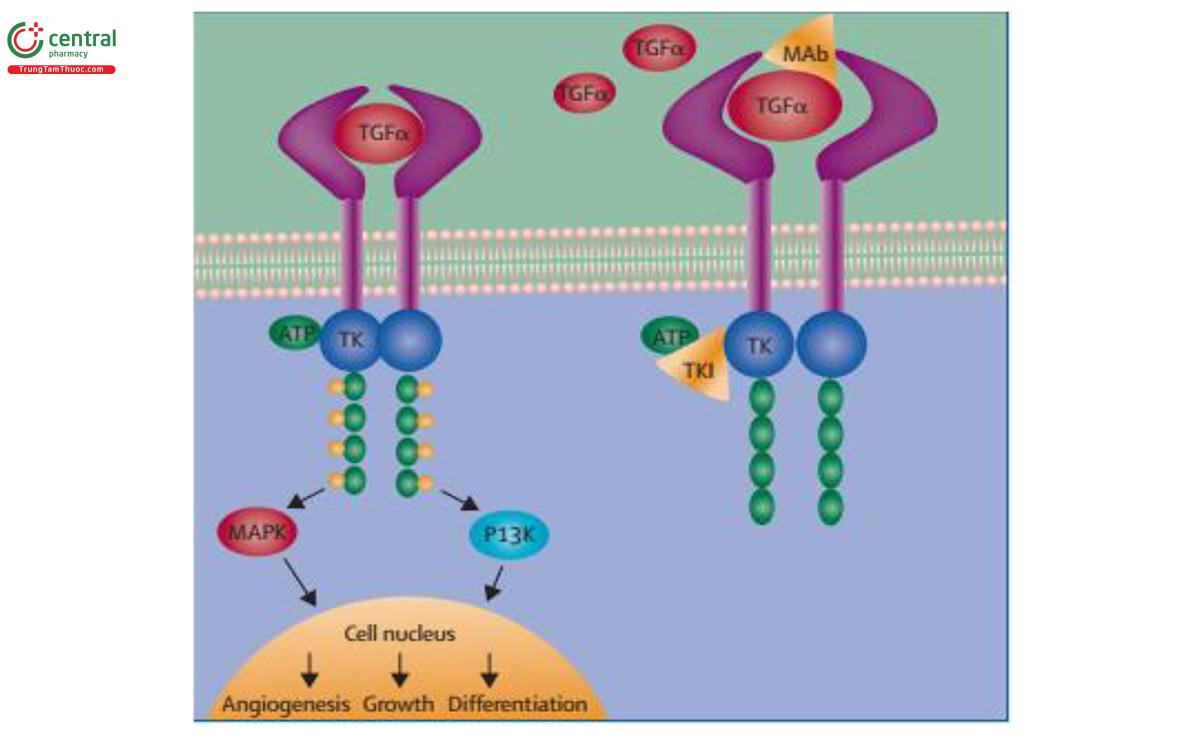
Ở các trường hợp bệnh tiến triển sau khi được điều trị bằng các thuốc TKIs, bệnh nhân có thể vẫn được hưởng lợi khi tiếp tục sử dụng EGFR - TKI, đặc biệt nếu lợi ích lâm sàng được duy trì do ngăn chặn sự phát triển của ung thư hoặc nếu có một sự lui bệnh có thể điều trị bằng các chiến lược tại chỗ (SART hoặc phẫu thuật) (IIA) (31).
Đột biến T790M tại Exon 20 của EGFR là cơ chế chính của tình trạng kháng thuốc mắc phải sau khi điều trị bằng EGFR - TKIs thế hệ thứ nhất hoặc thứ hai. Osimertinib đã được chứng minh có hiệu quả cao hơn so với hóa trị dựa trên Platinum (IA). Đối với những bệnh nhân có triệu chứng toàn thân tiến triển, trong khi đột biến T790M không phát hiện được hoặc những bệnh nhân mà bệnh tiến triển sau khi đã dùng osimertinib, hóa trị liệu dựa trên platinum vẫn là điều trị chuẩn (IIA). Phối hợp atezolizumab, Bevacizumab và hóa trị liệu đã được chỉ ra mang lại lợi ích đáng kể về chỉ số PFS ở một nhóm nhỏ bệnh nhân có đột biến gen EGFR (IIIA). Phối hợp EGFR- TKI với hóa trị dựa trên platinum không làm thay đổi chỉ số PFS và OS (10,23,28,48).
4.3 Đột biến ALK
Điều trị đầu tay bằng ALK - TKIs là điều trị được ưu tiên (IA). Crizotinib và ceritinib đã cho thấy sự cải thiện đáng kể thống kê về các chỉ số PFS và RR so với hóa trị trong các thử nghiệm ngẫu nhiên pha III (IA). Alectinib (I A) và brigatinib (IB) đã cho thấy sự cải thiện đáng kể về chỉ số PFS so với crizotinib và do đó, là các lựa chọn được ưu tiên hàng đầu. Các tác dụng phụ độ 3 - 5 cao hơn ở những bệnh nhân được điều trị bằng crizotinib. Điều quan trọng cần nhấn mạnh là brigatinib chưa được Cơ quan y tế Châu Âu chấp thuận điều trị (26,42).
Đối với những bệnh nhân xuất hiện đề kháng hoặc không dung nạp với crizotinib, các thuốc như ceritinib (IA), alectinib (IA) hoặc brigatinib (IIA) có thể được khuyến cáo. Ceritinib và alectinib đã cho thấy sự cải thiện đáng kể về chỉ số PFS và ít tác dụng phụ hơn so với liệu pháp hóa trị. Brigatinib đã cho thấy kết quả chỉ số PFS tốt hơn Crizotinib trong một thử nghiệm phase II có ALK dương tính dai dẳng (31,43,46).
5 Kết luận
Ung thư phổi là loại ung thư phổ biến nhất và là nguyên nhân hàng đầu gây tử vong liên quan đến ung thư trên toàn thế giới. Phần lớn các bệnh nhân mới được chẩn đoán ung thư phổi đều đã ở giai đoạn muộn, có di căn và không thể phẫu thuật cũng như không đáp ứng với các phương pháp điều trị. Khoảng 85% các trường hợp ung thư phổi thuộc nhóm không tế bào nhỏ, trong đó týp mô học phổ biến là ung thư biểu mô tuyến phế quản, đã được xác định có các đột biến gen. Các công nghệ giải trình tự gene đã giúp cho việc xác định các đột biến gen thúc đẩy tiến triển ung thư phổi. Việc xác định các đột biến gây ung thư phổi giúp xác định được các đích mới cho điều trị ung thư phổi không tế bào nhỏ (NSCLC) và tạo tiền đề phát triển các liệu pháp nhắm đích, như thuốc ức chế tyrosine kinase có thể được sử dụng để chống lại các thay đổi phân tử thúc đẩy hình thành ung thư.
Phát triển các liệu pháp nhắm đích không chỉ mang lại các lợi ích lâm sàng của các nghiên cứu phân tích gen. Các biomarker xác định được từ phân tích gen có thể được sử dụng để phát hiện ung thư phổi sớm, xác định tiên lượng bệnh nhân và đáp ứng với điều trị, cũng như theo dõi tiến triển bệnh. Các biomarker có thể được sử dụng để xác định quần thể bệnh nhân NSCLC sẽ được hưởng lợi nhiều nhất từ việc điều trị (liệu pháp nhắm đích hoặc hóa trị liệu), cung cấp các công cụ lâm sàng mà có thể được sử dụng để phát triển kế hoạch điều trị cá thể hóa.
6 Tài liệu tham khảo
1. Cooper WA, Lam DC, O'Toole SA, et al. (2013). “Molecular biology of lung cancer”. J Thorac Dis; 5 Suppl 305: S479-490.
2. Larsen JE, Minna JD. (2011). “Molecular biology of lung cancer: clinical implications”. Clin Chest Med; 32(4): 703- 740.
3. Lynch TJ, Bell DW, Sordella R, et al. (2004). “Activating mutations in the epidermal growth factor rece
4. Campbell JD, Lathan C, Sholl L, et al. (2017). “Comparison of Prevalence and Types of Mutations in Lung Cancers Among Black and White Populations”. JAMA Oncol; 3(6): 801-809.
5. Forbes S, Beare D, Boutselakis H, et al. (2017). “COSMIC: somatic cancer genetics at high-resolution”. Nucleic Acids Res; 45(Database issue): D777-D783.
6. Shea M, Costa DB, D R. (2016). “Management of advanced non-small cell lung cancers with known mutations or rearrangements: latest evidence and treatment approaches”. Ther Adv Respir Dis; 10(2): 113-129.
7. Alamgeer M, Ganju V, Watkins DN. (2013). “Novel therapeutic targets in non-small cell lung cancer”. Curr Opin Pharmacol; 13(3): 394-401.
8. Silvestri GA, Pastis NJ, Tanner NT, et al. (2016). “Clinical Aspects of Lung Cancer”. 940-964.e922.
9. da Cunha Santos G, Shepherd FA, Tsao MS. (2011). “EGFR mutations and lung cancer”. Annu Rev Pathol; 6: 49-69.
10. Hsu WH, Yang JC, Mok TS, et al. (2018). “Overview of current systemic management of EGFR-mutant NSCLC”. Ann Oncol; 29(suppl_1): i3-i9.
11. Alberg AJ, Brock MV, Samet JM. (2016). “Epidemiology of Lung Cancer”. 927-939.e925.
12. Travis WD, Brambilla E, Nicholson AG, et al. (2015). “The 2015 World Health Organization Classification of Lung Tumors: Impact of Genetic, Clinical and Radiologic Advances Since the 2004 Classification”. J Thorac Oncol; 10(9): 1243-1260.
13. de Groot PM,. Wu CC, Carter BW, et al. (2018). “The epidemiology of lung cancer”. Transl Lung Cancer Res; 7(3): 220-233.
14. Torre LA, Siegel RL, Ward EM, et al. (2016). “Global Cancer Incidence and Mortality Rates and Trends—An Update”. Cancer Epidemiol Biomarkers Prev; 25(1): 16-27.
15. Cao M, Chen W. (2019). “Epidemiology of lung cancer in China”. Thorac Cancer; 10(1): 3-7.
16. Yamaguchi T, Nishiura H. (2019). “Predicting the Epidemiological Dynamics of Lung Cancer in Japan”. J. Clin. Med; 8(3): 326-345.
17. Shin A, Oh C, Kim B, et al. (2017). “Lung Cancer Epidemiology in Korea”. Cancer Res Treat; 49(3): 616-626.
18. Siegel RL, Miller KD, Jemal A. (2018). “Cancer statistics, 2018”. CA Cancer J Clin; 68(1): 7-30
19. Nguyễn Bá Đức và CS. (2006). “Tình hình ung thư ở Việt Nam giai đoạn 2001- 2004 qua ghi nhận ung thư tại năm tỉnh thành Việt Nam”. Y học thực hành; 541: 9-17.
20. Nguyễn Văn Tình, Ngô Quý Châu, Nguyễn Văn Hưng. (2017). “Đặc điểm lâm sàng, cận lâm sàng và phân loại mô bệnh học ung thư biểu mô tuyến phế quản theo IASLC/ATS/ERS 2011”. Y học Việt Nam; 451(2017): 145 - 149.
21. Hưng NT. (2008). “Đặc điểm dịch tễ học mô tả ung thư cộng đồng dân cư Hà Nội giai đoạn 2001-2005”. Viện vệ sinh dịch tễ trung ương; Luận án tiến sĩ y học.
22. Bray F, Ferlay J, Soerjomataram I, et al. (2018). “Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries”. CA Cancer J Clin; 68: 394-424.
23. Stinchcombe TE. (2016). “Targeted Therapies for Lung Cancer”. Cancer Treat Res; 170: 165-182
24. Mayekar MK, Bivona TG. (2017). “Current Landscape of Targeted Therapy in Lung Cancer”. Clin Pharmacol Ther; 102(5): 757-764.
25. Rolfo C, Passiglia F, Ostrowski M, et al. (2015). “Improvement in lung cancer outcomes with targeted therapies: an update for family physicians”. J Am Board Fam Med; 28(1): 124-133.
26. Rossi A, Maione P, Sacco PC, et al. (2014). “ALK inhibitors and advanced non-small cell lung cancer (review)”. Int J Oncol; 45(2): 499-508.
27. Abdel Karim N, Kelly K. (2019). “Role of Targeted Therapy and Immune Checkpoint Blockers in Advanced Non- Small Cell Lung Cancer: A Review”. Oncologist.
28. Ferrer I, Zugazagoitia J, Herbertz S, et al. (2018). “KRAS-Mutant non-small cell lung cancer: From biology to therapy”. Lung Cancer; 124: 53-64.
29. Lindeman NI, Cagle PT, Aisner DL, et al. (2018). “Updated Molecular Testing Guideline for the Selection of Lung Cancer Patients for Treatment With Targeted Tyrosine Kinase Inhibitors: Guideline From the College of American Pathologists, the International Association for the Study of Lung Cancer, and the Association for Molecular Pathology”. Arch Pathol Lab Med; 142(3): 321-346.
30. Minguet J, Smith KH, Bramlage P. (2016). “Targeted therapies for treatment of non-small cell lung cancer--Recent advances and future perspectives”. Int J Cancer; 138(11): 2549-2561.
31. Putora PM, Schneider T, Rodriguez R, et al. (2012). “Targeted therapy in non-small cell lung cancer”. Breathe; 8(3): 206-215.
32. Naidoo J, Drilon A. (2016). “KRAS-Mutant Lung Cancers in the Era of Targeted Therapy”. In Lung Cancer and Personalized Medicine; DOI: 101007/97833192422318( Springer International Publishing Switzerland).
33. Carper MB, Claudio PP. (2015). “Clinical potential of gene mutations in lung cancer”. Clin Transl Med; 4(1): 33.
34. Chan BA, Hughes BG. (2015). “Targeted therapy for non-small cell lung cancer: current standards and the promise of the future”. Transl Lung Cancer Res; 4(1): 36-54.
35. Ellison G, Zhu G, Moulis A, et al. (2013). “EGFR mutation testing in lung cancer: a review of available methods and their use for analysis of tumour tissue and cytology samples”. J Clin Pathol; 66(2): 79-89.
36. Grigoriu B, Berghmans T, Meert AP. (2015). “Management of EGFR mutated nonsmall cell lung carcinoma patients”. Eur Respir J; 45(4): 1132-1141.
37. Wang R, Zhang Y, Pan Y, et al. (2015). “Comprehensive investigation of oncogenic driver mutations in Chinese non-small cell lung cancer patients”. Oncotarget; 6(33): 34300-34308
38. Saito M, Suzuki H, Kono K, et al. (2018). “Treatment of lung adenocarcinoma by molecular-targeted therapy and immunotherapy”. Surg Today; 48(1): 1-8.
39. Wang J, Liu Q, Yuan S, et al. (2017). “Genetic predisposition to lung cancer: comprehensive literature integration, meta-analysis, and multiple evidence assessment of candidate-gene association studies”. Sci Rep; 7(1): 8371.
40. Sholl LM. (2016). “The Molecular Pathology of Lung Cancer”. Surg Pathol Clin; 9(3): 353-378.
41. Shaw AT, Yeap BY, Solomon BJ, et al. (2011). “Effect of crizotinib on overall survival in patients with advanced non-small-cell lung cancer harbouring ALK gene rearrangement: a retrospective analysis”. Lancet Oncol; 12(11): 1004-1012.
42. Choi YL, Soda M, Yamashita Y, et al. (2010). “EML4-ALK Mutations in Lung Cancer That Confer Resistance to ALK Inhibitors”. N Engl J Med; 363(18): 1734-1739.
43. Sasaki T, Okuda K, Zheng W, et al. (2010). “The neuroblastoma associated F1174L ALK mutation causes resistance to an ALK kinase inhibitor in ALK translocated cancers”. Cancer Res; 70(24): 10038-10043
44. Schrank Z, Chhabra G, Lin L, et al. (2018). “Current Molecular-Targeted Therapies in NSCLC and Their Mechanism of Resistance”. Cancers; 10(7): 1-17.
45. El-Telbany A, Ma PC. (2012). “Cancer genes in lung cancer: racial disparities: are there any?”. Genes Cancer; 3(7-8): 467-480.
46. Onn A, Herbst RS. (2005). “Molecular targeted therapy for lung cancer”. The Lancet; 366(9496): 1507-1508
47. Sculier JP, Berghmans T, Meert AP. (2015). “Advances in target therapy in lung cancer”. Eur Respir Rev; 24(135): 23-29
48. Midha A, Dearden S, R M. (2015). “EGFR mutation incidence in non-small-cell lung cancer of adenocarcinoma histology: a systematic review and global map by ethnicity (mutMapII)”. Am J Cancer Res; 5(9): 2892-2911.
49. Wood SL, Pernemalm M, Crosbie PA, et al. (2015). “Molecular histology of lung cancer: from targets to treatments”. Cancer Treat Rev; 41(4): 361-375.
50. Shepherd FA, Domerg C, Hainaut P, et al. (2013). “Pooled analysis of the prognostic and predictive effects of KRAS mutation status and KRAS mutation subtype in early-stage resected non-small-cell lung cancer in four trials of adjuvant chemotherapy”. J Clin Oncol.; 31(17): 2173-2181
51. Marabese M, Ganzinelli M, Garassino MC, et al. (2015). “KRAS mutations affect prognosis of non-small-cell lung cancer patients treated with first-line platinum containing chemotherapy”. Oncotarget; 6(32): 34014-34022.
52. Macerelli M, Caramella C, Faivre L, et al. (2014). “Does KRAS mutational status predict chemoresistance in advanced non-small cell lung cancer (NSCLC)?”. Lung Cancer; 83(3): 383-388.
53. Garassino MC, Martelli O, Broggini M, et al. (2013). “Erlotinib versus docetaxel as second-line treatment of patients with advanced non-small-cell lung cancer and wild-type EGFR tumours (TAILOR): a randomised controlled trial”. Lancet Oncol; 14(10): 981-988.
54. Mao C, Qiu LX, Liao RY, et al. (2010). “KRAS mutations and resistance to EGFR-TKIs treatment in patients with non-small cell lung cancer: a meta-analysis of 22 studies”. Lung Cancer; 69(3): 272-278.
55. Linardou H, Dahabreh IJ, Kanaloupiti D, et al. (2008). “Assessment of somatic k-RAS mutations as a mechanism associated with resistance to EGFR-targeted agents: a systematic review and meta-analysis of studies in advanced non-small-cell lung cancer and metastatic colorectal cancer”. Lancet Oncol; 9(10): 962-972.
56. Rulli E, Marabese M, Torri V, et al. (2015). “Value of KRAS as prognostic or predictive marker in NSCLC: results from the TAILOR trial”. Ann Oncol; 26(10): 2079-2084.
57. Vyse S, Huang PH. (2019). “Targeting EGFR exon 20 insertion mutations in non-small cell lung cancer”. Signal Transduction and Targeted Therapy; 4(5): 1-10.
