Sắc ký lỏng hiệu năng cao là gì? 8 loại Detector thường được sử dụng

Sắc ký lỏng hiệu năng cao (High Performance Liquid Chromatography: HPLC) được ứng dụng rộng rãi trong đa số các lĩnh vực như phân tích thuốc, kiểm nghiệm thực phẩm, phân tích mẫu sinh học, phân tích môi trường,.... Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc thông tin về phương pháp sắc ký lỏng hiệu năng cao.
1 Khái niệm
Sắc ký lỏng hiệu năng cao (High Performance Liquid Chromatography: HPLC) được ứng dụng rộng rãi trong đa số các lĩnh vực như phân tích thuốc, kiểm nghiệm thực phẩm, phân tích mẫu sinh học, phân tích môi trường,... Các chất phân tích bằng HPLC rất đa dạng, ví dụ đối với thuốc, HPLC có thể phân tích được hầu hết các chất có tác dụng dược lý khác nhau như thuốc kháng sinh, thuốc tim mạch, thuốc hạ huyết áp, corticoid, ... Trong kiểm nghiệm thuốc, HPLC không những sử dụng để định tính, định lượng, xác định tạp chất của thuốc dạng nguyên liệu (hóa dược, dược liệu) phục vụ sản xuất, chế phẩm bảo chế dùng trực tiếp trên người, mà còn phân tích các chất trong dịch sinh học phục vụ đánh giá sinh dược học của thuốc hoặc phục vụ công tác hóa pháp. Do quá trình tách và phát hiện các chất trong mẫu phân tích xảy ra ở nhiệt độ bằng hoặc cao hơn một chút so với nhiệt độ môi trường xung quanh, nên phương pháp này lý tưởng cho các hợp chất kém bền với nhiệt. HPLC với kỹ thuật phát hiện không phá hủy chất phân tích cũng cho phép thu các phân đoạn sắc ký để tiếp tục dùng trong các phép phân tích khác. Các kỹ thuật phát hiện khác nhau (các loại detector khác nhau) không chỉ cho phép phân tích các hợp chất có độ nhạy cao mà còn có tính chọn lọc cao.
2 Nguyên lý của sắc ký lỏng hiệu năng cao
2.1 Nguyên tắc
HPLC là một kỹ thuật sắc ký được sử dụng để tách các hợp chất trong hỗn hợp với mục đích định tính, định lượng và tinh chế các thành phần của hỗn hợp. Trong đó, các chất phân tích di chuyển qua cột chứa các hạt pha tĩnh dưới tác động của pha động lỏng. Các chất phân tích có thể tách khỏi nhau trong hỗn hợp do mỗi chất có cấu trúc khác nhau, tính chất khác nhau do đó ái lực của nó với pha tĩnh dưới tác động của pha động khác nhau. Với các chất cùng nhóm có cấu trúc khá tương đồng hoặc đồng phân đối quang cần tác động vào hệ tách gồm pha tĩnh và pha động để có thể thu được kết quả tách đáp ứng yêu cầu.
Theo phân loại, HPLC được gọi là sắc ký lỏng, sắc ký cột và sắc ký rửa giải do pha động là chất lỏng, pha tĩnh được nhồi trên cột và chất phân tích được phát hiện khi rùa giải ra khỏi pha tỉnh. Cơ chế tách tùy thuộc bản chất pha tĩnh, pha động và chất phân tích. Các cơ chế tách bằng HPLC có thể là phân bố, hấp phụ, loại theo cỡ (rây phân tử), trao đổi ion, ái lực, sắc ký lỏng siêu tới hạn, tách đồng phân đối quang.

2.2 Thiết bị
Thiết bị HPLC được sản xuất bởi các hãng sản xuất có công nghệ cao ở các nước phát triển như Agilent, Thermo Scientific, Waters, Perkin Elmer của Mỹ, Merck của Đức; Shimadzu, Jasco, Hitachi của Nhật Bản,... Tùy yêu cầu là phân tích hay điều chế mà HPLC có một số cấu hình tiêu biểu như sau:
- Cấu hình dùng trong phân tích có 1 kênh dung môi;
- Cấu hình dùng trong phân tích có ≥ 2 kênh dung môi;
- Cấu hình dùng trong phân tích có bộ phận dẫn xuất hóa sau cột;
- Cấu hình dùng trong điều chế có bộ phận hứng mẫu ở mỗi phân đoạn.
Ngoài ra, còn có thể có hệ thống HPLC kết nối thiết bị thử độ hòa tan, kết nối thiết bị chiết mẫu tự động để tự động hóa trong quá trình phân tích.
HPLC thường sử dụng các loại pha tĩnh khác nhau chứa trong các cột, bơm cao áp đẩy pha động và các thành phần mẫu qua cột tới bộ phận phát hiện (detector). Detector được kết nối với bộ phận xử lý số liệu có khả năng xác định thời gian lưu đặc trưng của các thành phần mẫu và diện tích píc phản ánh lượng chất phân tích đi qua detector, đồng thời có thể cung cấp thông tin bổ sung liên quan đến chất phân tích như dữ liệu phổ UV - Vis, nếu là detector UV - DAD hoặc UV - PDA. Thời gian lưu của chất phân tích thay đổi tùy thuộc vào lực tương tác của nó với pha tĩnh, thành phần và tốc độ dòng của pha động và kích thước cột.
Mẫu cần tách và phân tích được đưa vào dòng chảy của pha động qua cột, với một thể tích nhỏ. Các thành phần của mẫu di chuyển qua cột với các vận tốc khác nhau, phụ thuộc vào bản chất hóa học của nó, vào bản chất của pha tĩnh (cột) và vào thành phần của pha động. Thời gian mà chất phân tích rửa giải được gọi là thời gian lưu. Thời gian lưu được xác định trong các điều kiện cụ thể được coi là đặc điểm nhận dạng của một chất phân tích nhất định.
2.3 Pha tĩnh và cột sắc ký
Pha tĩnh dùng trong HPLC là các hạt nhỏ có hình dạng hạt khác nhau như không đồng đều, hình cầu, pha tĩnh đơn khối xốp. Hạt pha tĩnh có thể được chế tạo dạng tro xốp bề mặt, xốp toàn phần và được nhồi vào cột hình trụ có chất liệu là thép không gỉ hoặc polymer. Các thông số quan trọng của cột sắc ký gồm bản chất pha tĩnh, kích cỡ hạt, đường kính trong của cột và độ xốp của hạt nhồi.

2.3.1 Kích cỡ hạt
Cột sắc ký chứa các hạt có kích cỡ khác nhau, giá trị “đường kính hạt” là giá trị trung bình. Hạt pha tĩnh có nhiều kích cỡ khác nhau từ 3 - 10 pm, có thể nhỏ tới 1,7 km, trong đó hạt 5 km là loại được dùng phổ biến trong phân tích. Các hạt nhỏ hơn có diện tích bề mặt lớn hơn và có khả năng phân tách tốt hơn, nhưng áp suất cần thiết cho vận tốc tuyến tính tối ưu của pha động sẽ tăng theo nghịch đảo của bình phương đường kính hạt. Điều này có nghĩa là khi giảm kích thước các hạt pha tĩnh mà giữ nguyên kích thước của cột, sẽ tăng gấp đôi hiệu quả tách, nhưng áp suất cần tăng lên hệ số bốn. Các hạt có kích cỡ lớn hơn (20 - 75 pm) được sử dụng trong HPLC điều chế và cho các ứng dụng khác như chiết pha rắn. Nếu hạt pha tĩnh có cỡ hạt khác nhau nhiều sẽ làm píc mở rộng dẫn tới hiệu lực tách bị hạn chế.
2.3.2 Đường kính trong của cột (Innerdiameter: I.D)
Đường kính trong của cột HPLC là một thông số quan trọng ảnh hưởng đến độ nhạy khi phát hiện và độ chọn lọc phân tách trong rửa giải gradient. Nó cũng xác định lượng chất phân tích có thể được nạp vào cột. Các cột lớn thường được sử dụng trong sản xuất như tinh chế nguyên liệu làm thuốc, tinh chế chất chuẩn. Cột ID nhỏ cải thiện độ nhạy một cách rõ rệt và tiêu thụ dung môi thấp hơn. I.D của cột phân tích có những loại sau:
- Cột ID lớn (trên 10 mm) được sử dụng để tinh chế nguyên liệu do dung lượng chịu tải lớn.
- Cột dùng trong phân tích (4 mm hoặc 4,6 mm) là loại cột phổ biến nhất, mặc dù các cột nhỏ hơn như 3,5 mm; 3,9 mm thậm chí 2,1 mm đang ngày càng trở nên phổ biến. Chúng được sử dụng trong phân tích định lượng mẫu thường qui và thường sử dụng với detector UV - Vis.
- Cột hẹp lòng (Narrow - bore columns) (1 - 2 mm) được sử dụng cho các ứng dụng khi mong muốn có độ nhạy cao hơn với detector UV - Vis, detector huỳnh quang hoặc sắc ký lỏng - khối phổ.
- Cột mao quản (dưới 0,3 mm) hầu như chỉ được sử dụng với detector khối phổ. Chúng thường được làm từ các mao quản silica nung chảy, thay vì ống thép không gi.
Sử dụng các hạt pha tĩnh kích cỡ nhỏ hơn nhồi trong cột có I.D nhỏ hơn đòi hỏi phải sử dụng áp suất hoạt động cao hơn và sẽ thu được hiệu lực tách tốt hơn (độ phân giải cao hơn).
2.3.3 Kích thước lỗ xốp (Pore Size)
Pha tĩnh có dạng xốp sẽ có diện tích bề mặt lớn hơn. Các lỗ xốp nhỏ có diện tích bề mặt lớn hơn trong khi kích thước lỗ lớn hơn có động học tốt hơn, đặc biệt là đối với các chất phân tích có cấu trúc lớn hơn. Ví dụ, protein chỉ nhỏ hơn một chút so với lỗ xốp và có thể đi vào lỗ xốp nhưng không dễ dàng đi ra.
2.3.4 Bản chất pha tĩnh
Rất nhiều chất mang biến đổi hoá học được chế tạo từ polymer, silica bằng cách gắn thêm các hydrocarbon mạch dài. Bề mặt của chất mang, ví dụ như các nhóm silanol của silica được phản ứng với các thuốc thử silan khác nhau tạo thành các dẫn xuất silyl có liên kết cộng hóa trị, che phủ một số lượng khác nhau các vị trí hoạt động trên bề mặt chất mang. Bản chất của các pha liên kết là tham số quan trọng để xác định các tính chất tách của hệ sắc ký.
Trong phân tích thuốc, sắc ký lỏng pha đảo (Reversed Phase Liquid Chromatogaphy - RP - LC) được dùng phổ biến, trong đó sự phân tách về nguyên tắc cơ bản dựa trên sự phân bố phân tử các chất giữa pha động và pha tĩnh. Các loại cột hay sử dụng trong HPLC - RP là C8, C18 trong Dược điển Mỹ (USP) ký hiệu là L7 và L1, còn trong Dược điển Anh và Dược điển Việt Nam ký hiệu là pha tĩnh A và C. Ngoài ra, pha tĩnh loại biến đổi hoá học đặc biệt, ví dụ dẫn xuất của celulose hoặc amylose, protein hoặc peptid, cyclodextrin,... dùng để tách các đồng phân đối quang (sắc ký đối quang) cũng đã được chế tạo và sử dụng.
Chiều dài và đường kính trong của cột, kích thước hạt pha tĩnh ảnh hưởng tới sự tách do đó qui cách cột được qui định trong các chuyên luận riêng đối với từng phép phân tích. Trong các chuyên luận dược điển không nếu tên thương mại của cột cụ thể nhưng có thể có gợi ý trong 5 phụ lục (USP), còn trong phụ lục (USP), còn trong các bài báo tên thương mại của cột thường được nêu cụ thể. Có thể thay đổi qui cách cột về chiều dài, đường kính trong hoặc đường kính hạt pha tĩnh trong phân tích theo chuyên luận dược điển nhưng phải nằm trong giới hạn qui định. Một số loại cột HPLC loại C18 hay dùng trong phân tích thuốc được trình bày ở Bảng 2.1.
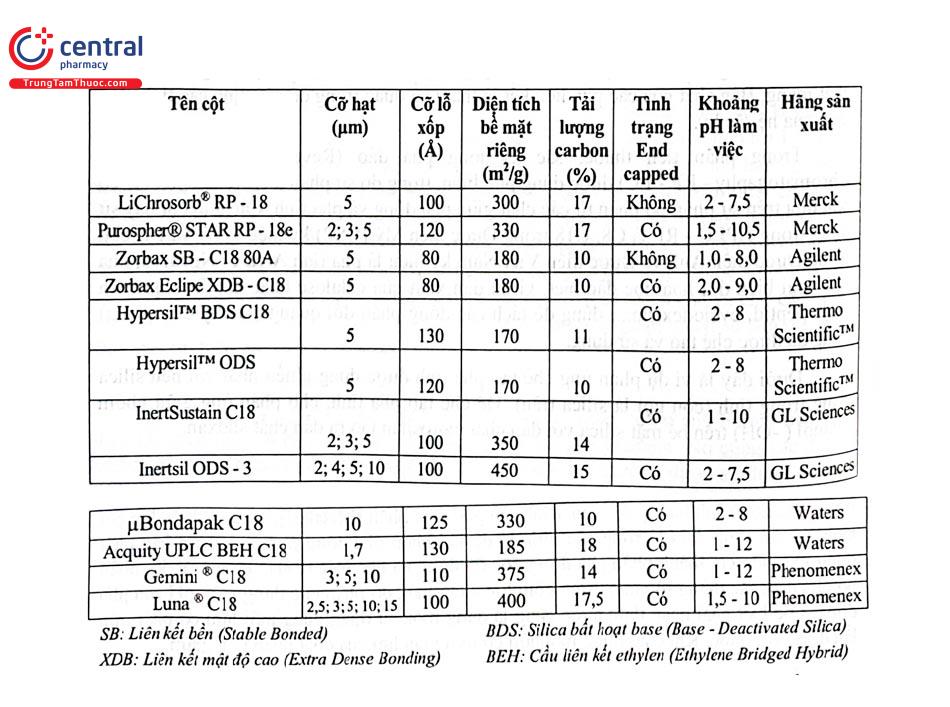
Đánh giá tải lượng của cột HPLC trong phân tích có thể dùng thông số Dung lượng hấp thu của cột. Đó là số mili đương lượng gam (mEg) chất phân tích hấp thu vào 1 g chất hấp thu (pha tĩnh). Chất phân tích nạp vào cột thường có khối lượng khoảng vài phần trăm dung lượng hấp thu của cột để đảm bảo cân bằng phân bố xảy ra nhanh. ,
Trong HPLC, có thể dùng cột bảo vệ lắp trước cột phân tích để lưu giữ chất cản trở của mẫu trước khi vào cột, tăng tuổi thọ cột tách. Cột bảo vệ có chiều dài nhỏ hơn hoặc bằng 15% chiều dài cột phân tích, đường kính trong bằng hoặc nhỏ hơn cột tách, chất nhồi giống pha tĩnh của cột sắc ký.
Bản chất pha tĩnh sẽ quyết định khả năng lưu giữ chất phân tích. Trong sắc ký phân bố pha đảo, đối với tách hỗn hợp 6 chất phân tích có độ phân cực giảm dần bằng 3 loại pha tĩnh có mạch carbon giảm dần trong ví dụ dưới đây cho thấy các cột có pha tĩnh có mạch carbon càng dài thì khả năng lưu giữ càng cao.

2.4 Pha động
Dung môi hoặc hỗn hợp dung môi pha động được qui định trong mỗi qui trình phân tích. Pha động có thể có thành phần không đổi và di chuyển theo tốc độ hằng định trong phép phân tích (chế độ đẳng dòng) hoặc theo chế độ rửa giải gradient hay chương trình dung môi tức là thành phần, tỷ lệ, tốc độ dòng dung môi thay đổi trong quá trình phân tích. Rửa giải gradient sẽ được qui định cụ thể thời gian, tỷ lệ thành phần và lưu lượng pha động ở các thời điểm sắc ký. Việc lựa chọn các thành phần pha động, chất phụ gia (như muối hoặc acid) và điều kiện gradient phụ thuộc vào bản chất của cột và thành phần mẫu. Điều kiện pha động thường được lựa chọn sau khi khảo sát với một loạt thành phần, tỷ lệ khác nhau thực hiện với mẫu để xác định điều kiện mang lại sự phân tách tốt nhất với thời gian phân tích hợp lý.
Pha động ảnh hưởng đến quá trình rửa giải chất phân tích (thời gian lưu), hiệu lực cột tách, sự mở rộng dải (độ rộng của píc) do đó khi chọn pha động phải chú ý tới bản chất dụng môi hữu cơ, thành phần pha động, tốc độ dòng và đặc biệt là pH. Trong sắc ký phân bố pha đảo, các thông số quan trọng gồm lực dung môi, độ phân cực dung môi, pH của pha động. Đặc biệt phải nhận thức nước cũng là dung môi, sử dụng dụng môi hữu cơ phải trộn lẫn được với nước. Khi tăng dung môi hữu cơ thời gian lưu của chất phân tích giảm.
2.4.1 Dung môi
Các dung môi hữu cơ hay dùng trong RP LC là methanol, acetonitril, tetrahydrofuran; còn trong sắc ký pha thuận (Normal - Phase Liquid Chromatography NP - LC) hay sử dụng dung môi n - hexan cùng một tỷ lệ nhỏ một trong các dung môi cloroform, isopropanol, methylen clorid. Các dung môi này phải đạt độ tinh khiết dùng cho HPLC. Các hãng sản xuất dung môi dùng cho HPLC nổi tiếng là Merck - Đức, Prolabo - Pháp; Aldrich - Mỹ; Baker - Mỹ; Scharlau - Tây Ban Nha; ...Các muối hoặc acid pha Dung dịch đệm ít nhất phải đạt độ tinh khiết phân tích.
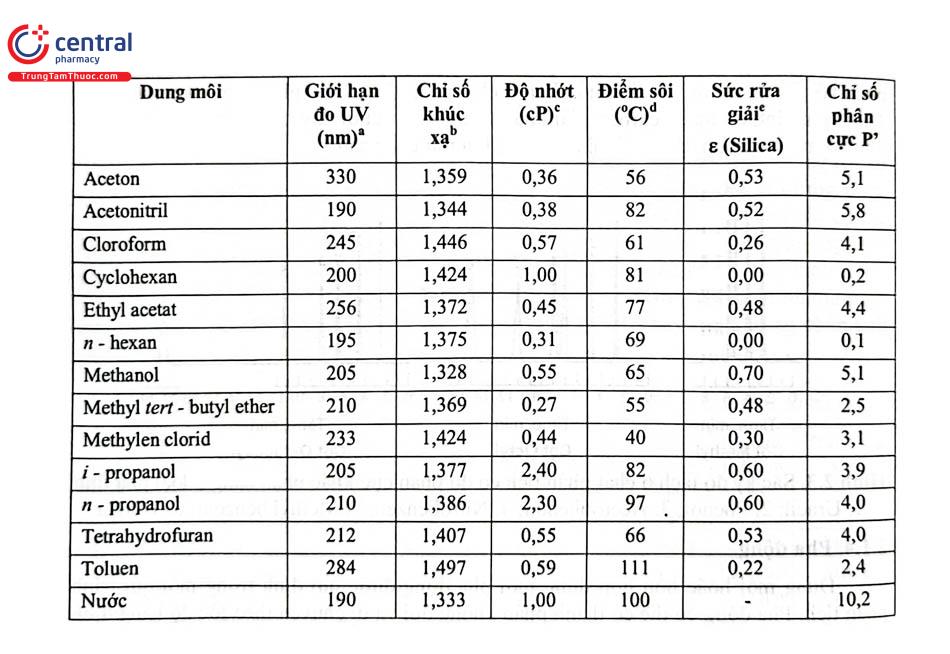
a. Bước sóng tại đó độ hấp thụ của dung môi là 1,0 AU trong cuvet 10 - mm. Giá trị giới hạn đo UV (UV cutoff); chú ý đối với phép phân tích phát hiện bằng detector UV; dung môi hữu dụng phụ thuộc vào bước sóng để phát hiện mẫu thử.
b. Chỉ số khúc xạ: phát hiện bằng detector RI; giá trị thấp tốt hơn.
c. Độ nhớt: ảnh hưởng đến áp suất cột, độ nhớt thấp tốt hơn.
d. Điểm sôi: Ảnh hưởng tới hiệu năng bơm, dung môi có điểm sôi cao phù hợp hơn.
e. Sức rửa giải (eluent strength) của dung môi (ε ): lựa chọn dung môi có ε thích hợp để tối ưu giá trị k (1 ≤k<10).
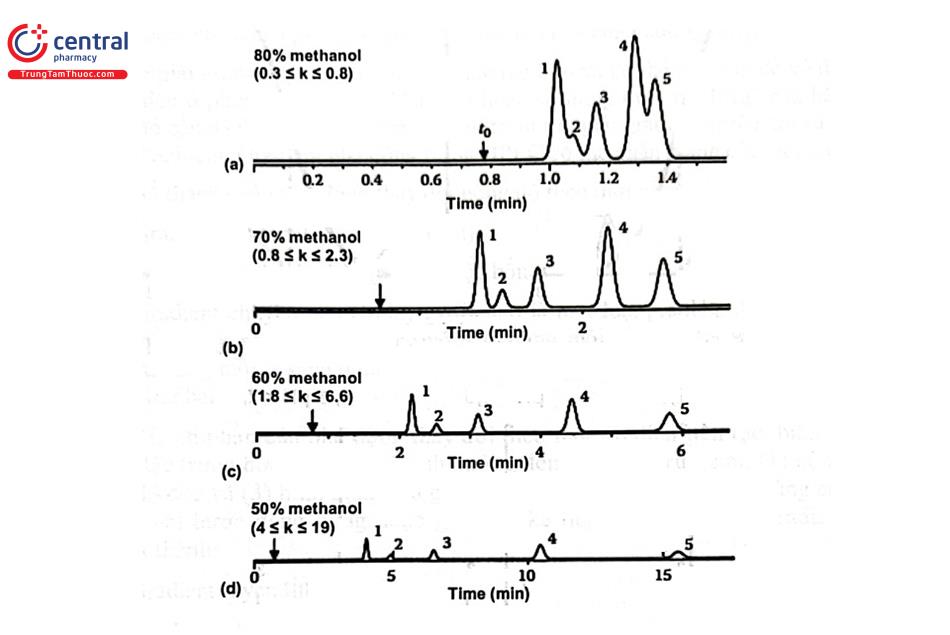
Dung môi và tỷ lệ dung môi có ảnh hưởng tới độ chọn lọc, độ phân giải và thời gian phân tích khi tách các chất trong hỗn hợp. Hình 2.4 minh họa sự ảnh hưởng tỷ lệ thành phần pha động khi tách hỗn hợp 5 chất.
2.4.2 pH
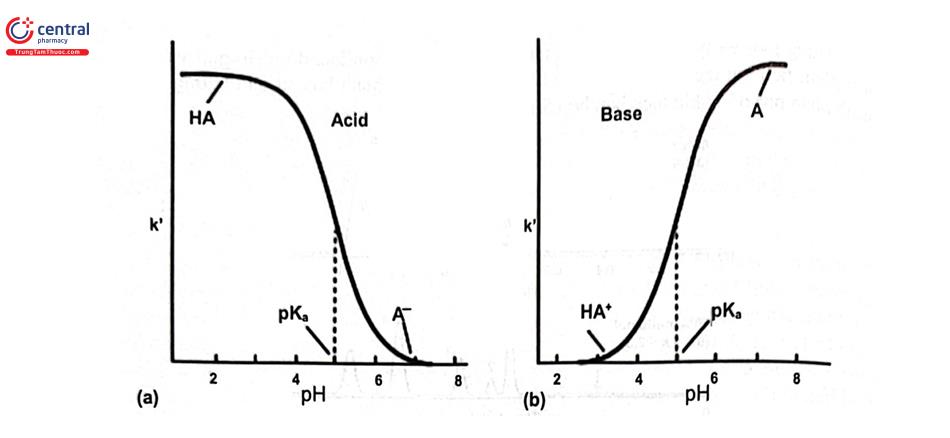
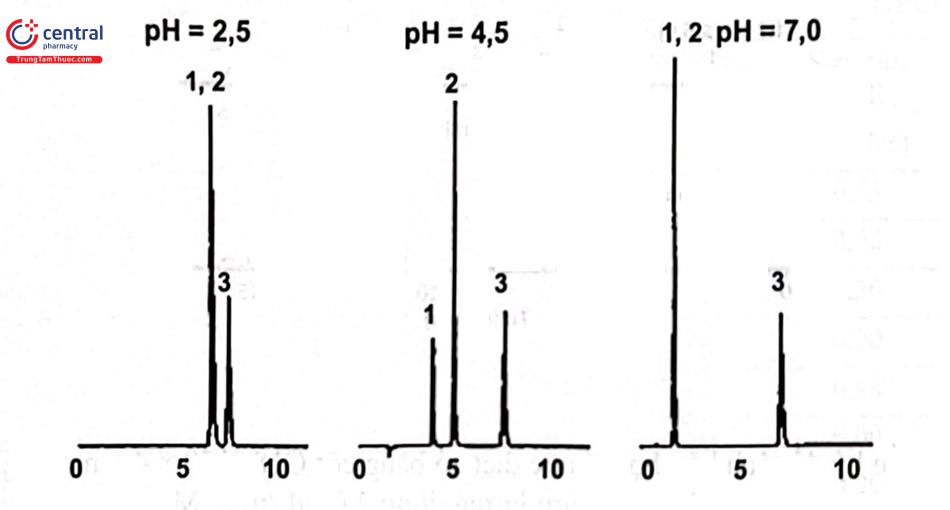
pH của pha động có ảnh hưởng tới các chất dễ ion hóa (acid hữu cơ, base hữu cơ). Trong quá trình tách bằng LC -RP cần hạn chế tối đa sự ion hóa của chất phân tích do đó cần kiểm soát pH chặt chẽ vì khi thay đổi 0,1 đơn vị pH sẽ có ảnh hưởng rõ rệt tới thời gian lưu của chất phân tích. Pha động sử dụng dung dịch đệm cho độ tái lặp pH tốt hơn. Lựa chọn dung dịch đệm có pH = pKa ± 1 (có dung lượng đệm tốt hơn) và nồng độ đệm không nên lớn hơn 100 mM vì có thể ảnh hưởng tới độ bền vật liệu của thiết bị, mặt khác tăng độ nhớt pha động sẽ gây áp suất cao. Hình 2.5 minh họa sự phụ thuộc của hệ số dung lượng (khả năng lưu giữ) của các chất có bản chất acid yếu và base yếu theo pH và Hình 2.6 là ví dụ ảnh hưởng của pH pha động tới khả năng tách các chất có bản chất acid yếu và trung tính. Khi sử dụng hệ đệm cần kiểm tra độ tan chất phân tích trong pha động. Các dung dịch đệm hay dùng trong RP - LC là acid và muối phosphat, acetat, format, ...
2.4.3 Rửa giải đẳng dòng và gradient
Chế độ rửa giải đẳng dòng đơn giản, các điều kiện sắc ký được giữ không đổi trong suốt quá trình phân tích. Tuy nhiên, nhiều mẫu phức tạp chứa các hợp chất khác nhau nhiều về độ lưu giữ, do đó nếu sử dụng chế độ rửa giải đẳng dòng khó thành công trong tách sắc ký. Khi điều chỉnh điều kiện đẳng dòng có thể lưu giữ được các chất có ái lực kém với pha tĩnh và rửa giải được các chất bị lưu giữ mạnh trong mẫu. Bởi nếu sử dụng chế độ đẳng dòng có thể các chất ái lực kém với pha tĩnh có thể không tách được do rửa giải ra cùng dung môi, còn các chất bị lưu giữ mạnh thì thời gian lưu dài, gây ra hiện tượng mở rộng dải và làm giảm độ nhạy của phương pháp phân tích.
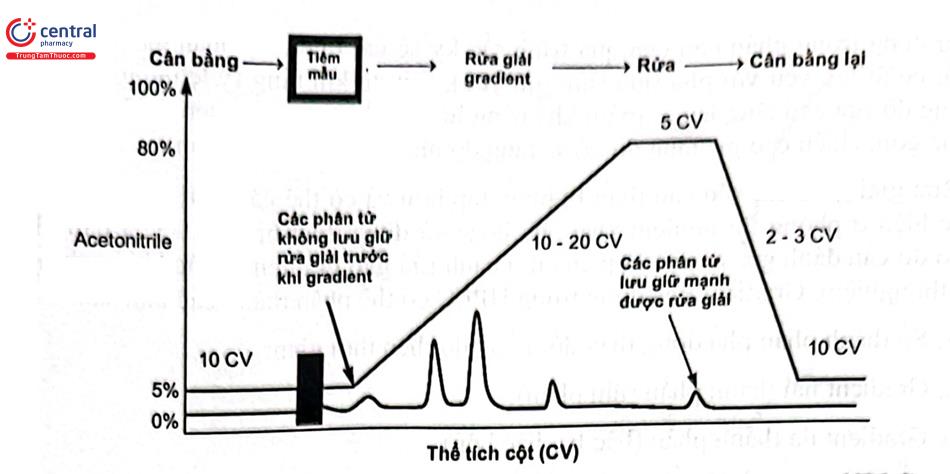
Trong HPLC, rửa giải gradient dung môi với thành phần của pha động thay đổi trong quá trình sắc ký bằng cách trộn hai hoặc nhiều thành phần. Pha động yếu hơn được sử dụng trong phần đầu của quá trình sắc ký sẽ cải thiện độ phân giải do lưu giữ các chất có ái lực yếu với pha tĩnh (lưu giữ yếu), sau đó khi tăng tỷ lệ dung môi mạnh thì cường độ rửa giải tăng lên sẽ giảm khả năng lưu giữ kèm với thu hẹp dải hơn nên thu được píc gọn, chiều cao píc tăng do đó sẽ tăng độ nhạy của phương pháp.
Rửa giải gradient yêu cầu thiết bị phức tạp hơn và có thể gặp vấn đề về độ tái lặp do thực hiện ở phòng thí nghiệm khác và/ hoặc sử dụng thiết bị HPLC của hãng khác nhau do đó cần đánh giá sự phù hợp của qui trình rửa giải gradient trước khi sử dụng tại phòng thí nghiệm. Gradient pha động trong HPLC có thể phân thành các loại sau:
1. Số thành phần pha động thay đổi nồng độ theo thời gian:
a. Gradient hai thành phần (nhị phân);
b. Gradient đa thành phần (bậc ba, bậc bốn);
c. Gradient chuyển tiếp (Relay gradients) là một loại gradient đa thành phần đặc biệt bao gồm một số bước tiếp theo với các dung môi khác nhau, trong đó pha động thay đổi từ dung môi A sang dung môi B ở bước đầu tiên, từ dung môi B sang dung môi C ở bước thứ hai, ...
2. Thành phần của pha động thay đổi theo một gradien liên tục, biên dạng của nó được đặc trưng bởi ba thông số ảnh hưởng đến khả năng rửa giải: (1) nồng độ ban đầu, (2) độ dốc và (3) hình dạng của gradient. Chương trình gradient cũng có thể bao gồm một vài bước đẳng dòng hoặc gradient kế tiếp. Theo đó, các gradient có thể được chia thành:
a. Gradient tuyến tính;
b. Gradient phi tuyến;
c. Gradient bậc, bao gồm một số bước đẳng dòng kế tiếp với nồng độ ngày càng tăng của một hoặc nhiều thành phần rửa giải mạnh của pha động;
d. Gradient phân đoạn, bao gồm một số bước kế tiếp nhau, thường là tuyến tính với các độ dốc khác nhau hoặc các khoảng giữ đẳng dòng (thường xảy ra ở cuối hoặc khi bắt đầu rửa giải, nhưng cũng có thể ở giữa các bước gradient tuyến tính);
e. Gradient ngược với nồng độ giảm của thành phần rửa giải mạnh của pha động thường được sử dụng để khôi phục các điều kiện ban đầu trước khi tiêm mẫu tiếp theo.
3. Kiểu sắc ký và loại pha động:
a. Sắc ký pha đảo: Có thể gradient các điều kiện dung môi, pH, nồng độ thuốc thử cặp ion (kiểu RP - IP);
b. Sắc ký trao đổi ion: Có thể gradient các điều kiện cường độ ion, dung môi, pH;
c. Sắc ký pha thuận: Gradient nồng độ của một hoặc nhiều dung môi phân cực;
d. Sắc ký tương tác thân nước: Gradient nồng độ của nước hoặc dung môi hữu cơ phân cực hơn.
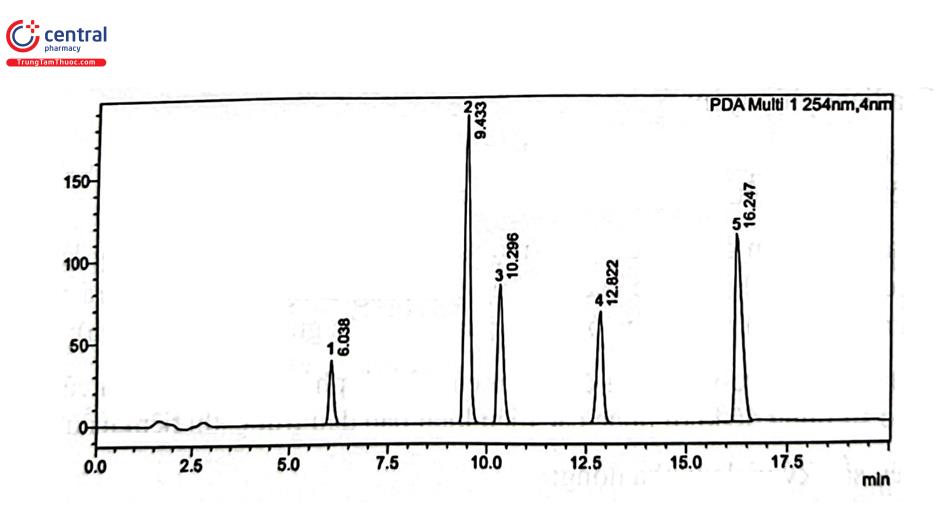
Sắc ký pha đảo có khả năng tách được hỗn hợp chất phân tích không phân cực, phân cực, thậm chí cả các hợp chất ion dựa trên sự khác biệt về tính kỵ nước và/hoặc kích thước của chất phân tích. Nếu phân tích theo chế độ đẳng dòng, thời gian phân tích sẽ rất dài, do đó thực hiện chế độ gradient pha động sẽ thu được kết quả đáp ứng yêu cầu với thời gian hợp lý. Thông thường chế độ gradient phân đoạn hay được áp dụng vì cho phép cân bằng hệ thống trước mỗi lần phân tích do đó đảm bảo tính ổn định quá trình phân tích. Trong kiểm nghiệm thuốc, gradient pha động hay được sử dụng trong phép thử tạp chất, kiểm nghiệm thành phẩm thuốc đa thành phần và kiểm nghiệm dược liệu. Hình 2.8 minh họa áp dụng chế độ gradient pha động kiểu tuyến tính trong tách 5 thuốc thuộc nhóm kháng histamin H1.
2.5 Các điều kiện khác
Ngoài yếu tố pha tĩnh và pha động, quá trình tách sắc ký còn chịu ảnh hưởng của các yếu tố khác như điều nhiệt cột tách, thông thường khi tăng nhiệt độ cột thì hiệu quả tách tốt và nhanh hơn. Tuy nhiên, phải chú ý tới chất kém bền với nhiệt, nhiệt độ cột tách thường không lớn hơn 60°C
Tiêm mẫu có thể dùng syring tiêm tay vào vòng tiêm mẫu có thể tích cố định hoặc tiêm mẫu tự động. Trong tiêm mẫu tự động, buồng chứa mẫu có thể được kiểm soát nhiệt độ để tránh phân hủy mẫu. Lượng mẫu có thể ảnh hưởng tới quá trình tách, tiêm mẫu với thể tích nhỏ, nồng độ thấp có thể thu được độ phân giải tốt hơn nhưng sẽ ảnh hưởng tới giới hạn phát hiện và độ đúng của phép phân tích.
3 Detector
HPLC có ưu điểm nổi bật so với các quá trình tách thông thường đó là quá trình tách và phát hiện diễn ra đồng thời do có nhiều loại detector để lựa chọn phù hợp với chất phân tích.
HPLC có nhiều loại detector khác nhau, bao gồm detector hấp thụ tử ngoại - khả kiến kể cả detector mảng diod, detector huỳnh quang, detector khúc xạ ánh sáng, detector điện hoá, detector khối phổ, detector tán xạ ánh sáng bay hơi hoặc các loại detector đặc biệt khác. Trong định lượng bằng HPLC, để phân tích mẫu thử có khoảng hàm lượng khác nhau, dải tuyến tính động học rộng (dải nồng độ mà đáp ứng detector tỷ lệ với nồng độ chất phân tích, ví dụ, 10 khi phát hiện bằng detector UV) các thành phần chính và vi lượng (tạp chất) có thể được xác định trong một lần phân tích duy nhất trong khoảng nồng độ rộng. Đối với phương pháp qui định, phải phát hiện được các píc > 0,1% đáp ứng của hoạt chất (= 100%), do đó yêu cầu khoảng tuyến tính ít nhất 100/0,1 = 103.
Sử dụng detector nào trong phép phân tích sẽ được qui định trong chuyên luận riêng khi thực hiện theo Dược điển hoặc qui trình phân tích đã được phê duyệt. Trong xây dựng phương pháp phân tích sẽ căn cứ vào bản chất chất phân tích, mức nồng độ và nền mẫu mà lựa chọn detector phù hợp.
3.1 Detector UV - Vis
Detector UV - Vis sử dụng phân tích các chất có khả năng hấp thụ UV - Vis như chất có cấu trúc alken, alkin, hợp chất thơm, hợp chất có nối đôi giữa C và O, N hoặc S. Ưu điểm của detector UV - Vis là có độ nhạy khá cao (10- g/ml), dễ vận hành, ít phụ thuộc vào thay đổi của điều kiện phân tích (nhiệt độ). Nhược điểm của nó là chỉ phân tích các chất hấp thụ UV - Vis. Có một số chất khả năng hấp thụ UV kém nhưng vẫn được phát hiện bằng detector UV ở bước sóng thấp như Artemisinin, ginsenosid, ... Khi đó, phải chú ý tới ảnh hưởng của dung môi hữu cơ và dung dịch đệm tới tín hiệu khi phát hiện.
Dung môi pha động cần được đặc biệt chú ý vì đặc tính của chúng thường phải nằm trong giới hạn hẹp. Các dung môi sử dụng làm pha động phải có độ hấp thụ A < 0,2AU ở bước sóng phát hiện các chất phân tích trong mẫu. Độ hấp thụ của dung môi thấp hơn có thể đồng nghĩa với tăng độ chính xác của phép định lượng và thu được kết quả tốt hơn khi rửa giải gradient; tuy nhiên, độ hấp thụ cao hơn có thể được chấp nhận đối với phép phân tích đăng dòng.

Detector UV - Vis được sử dụng rộng rãi trong phân tích do có ưu điểm sau:
- Độ nhạy cao (đối với các mẫu hấp thụ UV - Vis);
- Khoảng tuyến tính rộng (> 105);
- Thích hợp với cả cuvet đo thể tích nhỏ để giảm thiểu sự mở rộng dải;
- Ít bị ảnh hưởng bởi sự thay đổi tốc độ dòng và nhiệt đội
- Độ tin cậy cao;
- Không phá hủy cấu trúc của mẫu;
- Đáp ứng khác nhau đối với các chất tan khác nhau;
- Tương thích với rửa giải gradient;
- Dễ dàng lựa chọn được bước sóng phát hiện;
- Dễ vận hành thiết bị;
- Dịch vụ bảo dưỡng, hiệu chuẩn quốc tế.
Detector UV - Vis có thể tiến hành ở các chế độ bước sóng cố định và bước sóng thay đổi. Hiện nay, đa số detector UV của HPLC là bước sóng thay đổi và mảng diod (DAD/PDA). Trong quá trình phát triển phương pháp phân tích thường lựa chọn bước sóng có hấp thụ cực đại để phát hiện chất phân tích, còn nếu phân tích đa thành phần ở một bước sóng sẽ ưu tiên đặt bước sóng phát hiện ở cực đại của chất có nồng độ thấp và/hoặc đáp ứng detector kém. Khi sử dụng chế độ DAD/PDA, các tín hiệu từ các diod quang riêng lẻ (512 hoặc 1024 diod là phổ biến) được xử lý để tạo ra phổ của chất phân tích. Vì quang phổ được tạo ra tại cùng thời điểm (so với bước sóng đơn, theo dõi bằng detector bước sóng thay đổi), DAD có thể góp phần định tính píc.
Detector DAD/PDA có những ưu điểm sau:
- Có thể thu thập dữ liệu ở một hoặc nhiều bước sóng trên sắc ký đồ;
- Có thể thu thập phổ của một hoặc nhiều chất phân tích trong sắc ký;
- Có khả năng xác định độ tinh khiết píc.
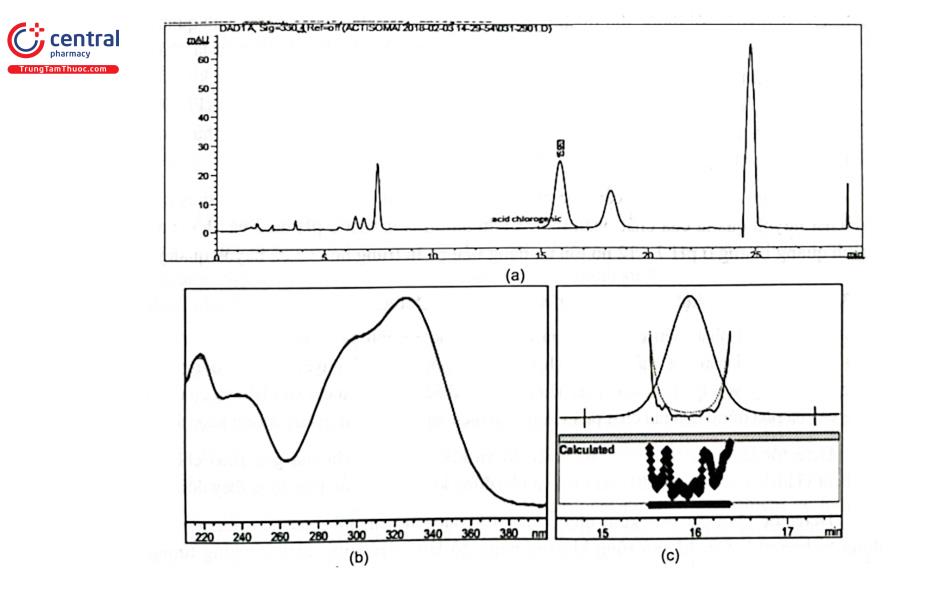
Ví dụ: Tách acid chlorogenic trong cao actiso bằng cột Inertsil ODS 3 (250 x 4,6 mm, 5 um), pha động gồm acetonitril - dung dịch acid phosphoric 0,1% chạy theo chương trình gradient dung môi; detector UV - DAD đặt ở chế độ scan từ 210 nm - 400 nm và bước sóng cố định 330 nm. Kết quả cho thấy píc của acid chlorogenic được rửa giải ở thời gian 15,93 phút, có bước sóng hấp thụ cực đại 328 nm. Píc ứng với acid chlorogenic trong mẫu thử đạt độ tinh khiết píc (Peak purity) là 999,751 và hệ số phù hợp (Match Index) của phổ ứng với pic acid chlorogenic trong mẫu thử so với píc acid chlorogenic trong mẫu chuẩn là ~ 1000,0. (Hình 2.9).
3.2 Detector huỳnh quang (Fluorescence detector: FLD)
Detector này rất nhạy và chọn lọc đối với các chất phân tích phát huỳnh quang khi bị kích thích bởi bức xạ UV. Các thành phần mẫu không phát huỳnh quang không tạo ra tín hiệu đo, do đó bước làm sạch mẫu có thể được đơn giản hóa. Đối với một số chất, detector huỳnh quang có độ nhạy gấp hàng trăm lần so với detector UV - Vis và là một trong những detector HPLC nhạy nhất.
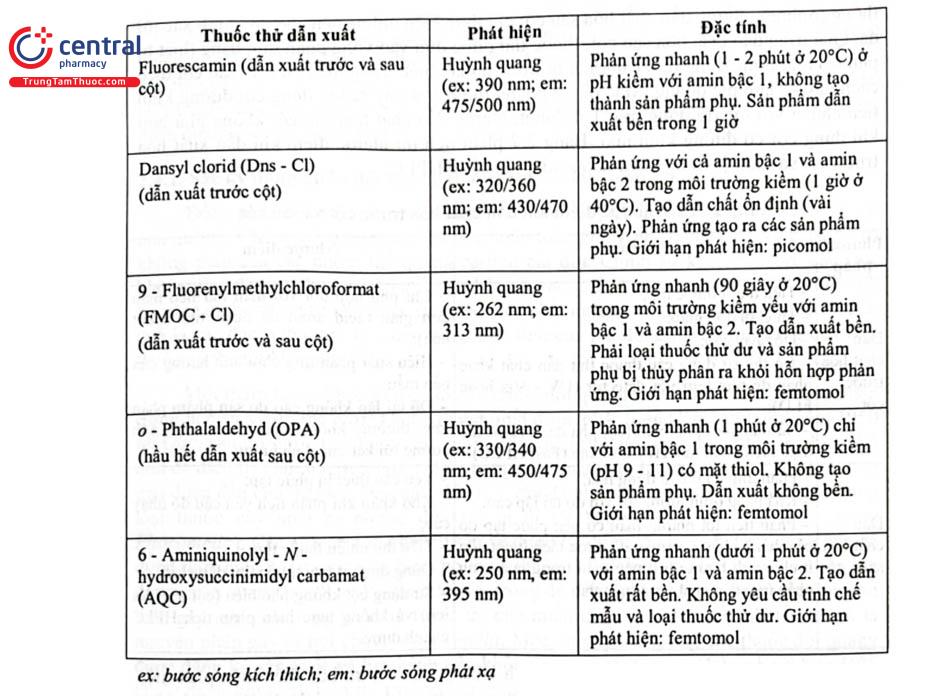
Những hợp chất phát hiện trực tiếp bằng detector huỳnh quang trong phân tích HPLC là các hydrocarbon thơm đa vòng. Đối với những chất không phát huỳnh quang có thể cho dẫn xuất với thuốc thử thích hợp tạo sản phẩm có khả năng phát huỳnh quang và được phân tích bằng HPLC với detector huỳnh quang. Ví dụ điển hình nhất là phân tích các acid amin hoặc các chất trong cấu trúc có nhóm amin bậc 1, bậc 2 sau khi dẫn chất với thuốc thử o - Phthalaldehyde (OPA), 9 - Fluorenylmethylchloroformat (FMOC - CI), ninhydrin, ... Nội dung này sẽ được làm rõ hơn trong phần chuẩn bị mẫu thử bằng cách dẫn xuất hóa ở phần sau.
Khi phân tích bằng FLD cần đặc biệt chú ý pH vì khi pH thay đổi có thể thay đổi tính chất huỳnh quang của chất phân tích. Ví dụ anilin là cation ở pH acid và không có huỳnh quang nhưng ở pH 7 - 12 nó tồn tại dạng tiểu phân trung hòa và có huỳnh quang.
3.3 Detector khúc xạ ánh sáng (Refractive Index Detector: RID)
Detector RI phát hiện các chất dựa trên sự khúc xạ ánh sáng. Đây là detector vạn năng đáp ứng với hầu hết chất phân tích nhưng độ nhạy kém. Nguyên lý hoạt động của detector theo nguyên lý đo vi sai. Tín hiệu đo là sự khác nhau giữa chỉ số khúc xạ của pha động và của pha động hoà tan chất phân tích. Tín hiệu này có thể có giá trị dương hoặc âm.
Detector khúc xạ rất nhạy với nhiệt độ và khó sử dụng khi rửa giải theo chế độ gradient vì khó so sánh trị số RI của mẫu và pha động khi thành phần pha động thay đổi.
Detector khúc xạ vừa kém nhạy, lại có khoảng tuyến tính hẹp nên phạm vi sử dụng bị hạn chế. Có thể mở rộng khoảng nồng độ làm việc bằng cách sử dụng tương quan logarithm y = ax. RID thường được sử dụng trong phân tích các chất có phân tử lượng lớn như đường, polymer và đây là detector sử dụng phổ biến nhất trong sắc ký thẩm thấu gel hoặc sắc ký loại theo cỡ.
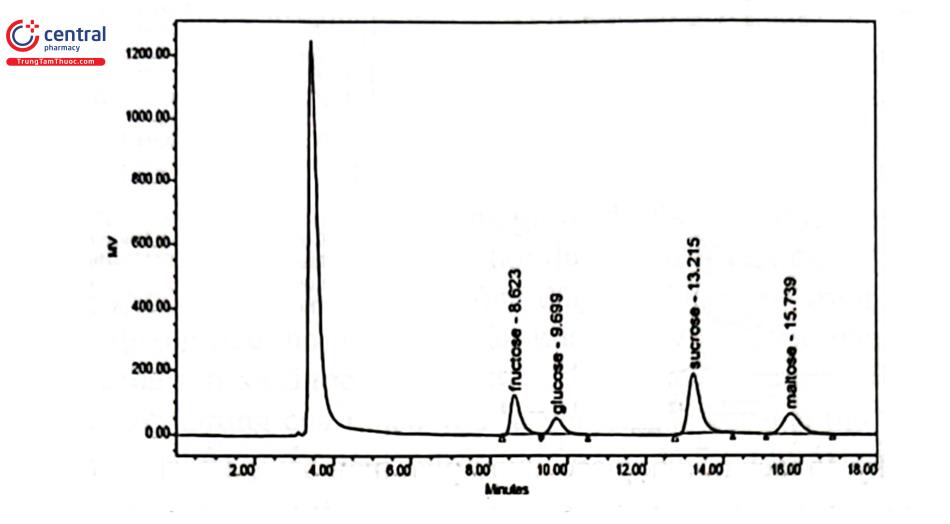
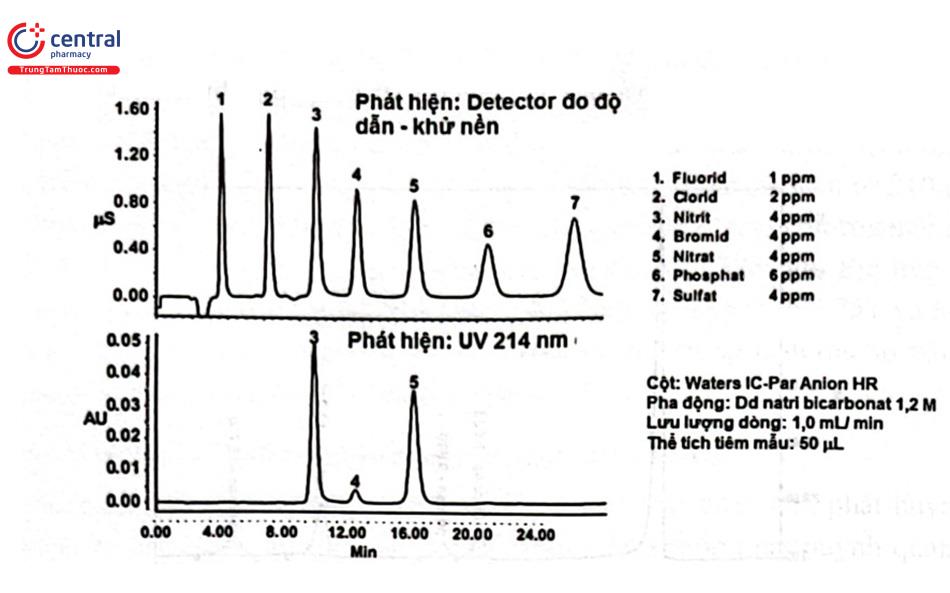
3.4 Detector độ dẫn điện (Conductivity Detector: CD)
Detector độ dẫn điện sử dụng để phát hiện các ion trong dung dịch. Áp một hiệu điện thế không đổi giữa các điện cực, khi các ion chất phân tích được rửa giải sẽ xuất hiện sự thay đổi dòng điện. Detector độ dẫn điện được sử dụng chủ yếu trong sắc ký ion để phân tích các chất có hoạt tính điện hóa như ion vô cơ và các chất hữu cơ phân tử lượng nhỏ, bao gồm acid hữu cơ và amin. Các ion kim loại có thể phân tích bằng phổ hấp thụ nguyên tử hoặc phổ phát xạ plasma. Tuy nhiên, khả năng phân tích đồng thời các ion âm là một ưu điểm đặc biệt của sắc ký ion.
Detector độ dẫn điện có độ nhạy cao nhưng rất dễ bị ảnh hưởng bởi tác động của sự thay đổi nhiệt độ (nhiệt độ dung dịch thay đổi 1 độ C gây ra sự thay đổi độ dẫn điện khoảng 2%). Có nhiều phương pháp khác nhau để tránh sự thay đổi nhiệt độ đã được sử dụng như kiểm soát nhiệt độ cột tách hay dùng tế bào đo nhiệt độ không đổi.
Độ dẫn điện của pha động cao sẽ tạo ra nhiễu nền lớn. Hai biện pháp để hạn chế ảnh hưởng này là: sử dụng pha động có độ dẫn điện thấp thì sẽ dùng phương pháp không khử nền (non - suppressor) hoặc khi độ dẫn điện của dịch rửa giải sau khi tách trên cột bị giảm sẽ dùng phương pháp khử nền (suppressor). Phương pháp khử nền làm giảm dẫn điện thay cation bằng H+ hoặc thay thế anion bằng OH - trong dịch rửa giải bằng nhựa hoặc màng trao đổi ion. Nhựa trao đổi ion (được nhồi vào cột ngắn) hoặc màng trao đổi ion được nối vào sau cột phân tích có nhiệm vụ loại các ion của pha động, nhưng giữ lại các ion chất phân tích. Phương pháp không khử rất dễ dàng và kinh tế bởi chỉ đơn giản là một detector độ dẫn được kết nối với hệ thống sắc ký lỏng thông thường.
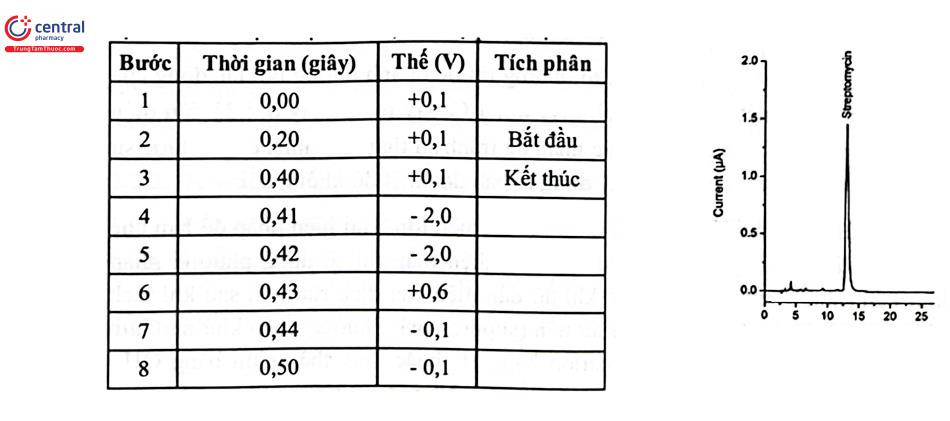
3.5 Detector điện hóa (Electrochemical Detector: ECD)
ECD được sử dụng để phát hiện các chất có phản ứng oxy hóa - khử và phát hiện dòng điện tạo ra bởi các phản ứng này. ECD có độ chọn lọc cao vì điện áp cần thiết để gây ra phản ứng oxy hóa - khử phụ thuộc vào chất phân tích trong thành phần mẫu thử. ECD cũng có độ nhạy cao và thường được sử dụng để phân tích các chất có nguồn gốc sinh học.
Đ Trong kiểm nghiệm thuốc, ECD được áp dụng phân tích các chất có hấp thụ UV kém như dược chất thuộc nhóm aminoglycosid (gentamycin, Kanamycin, Amikacin, streptomycin) và nhóm macrolid (erythromycin, Azithromycin). Ví dụ định lượng Streptomycin theo USP, sử dụng cột tách amin bậc 4 (4 mm × 25 cm; 10 um), cột bảo vệ cùng loại (4 mm × 5 cm), pha động dung dịch NaOH 70 mM, điện cực làm việc: vàng, điện cực so sánh: bạc - bạc clorid; chế độ phân cực dương. Chương trình thế như sau:
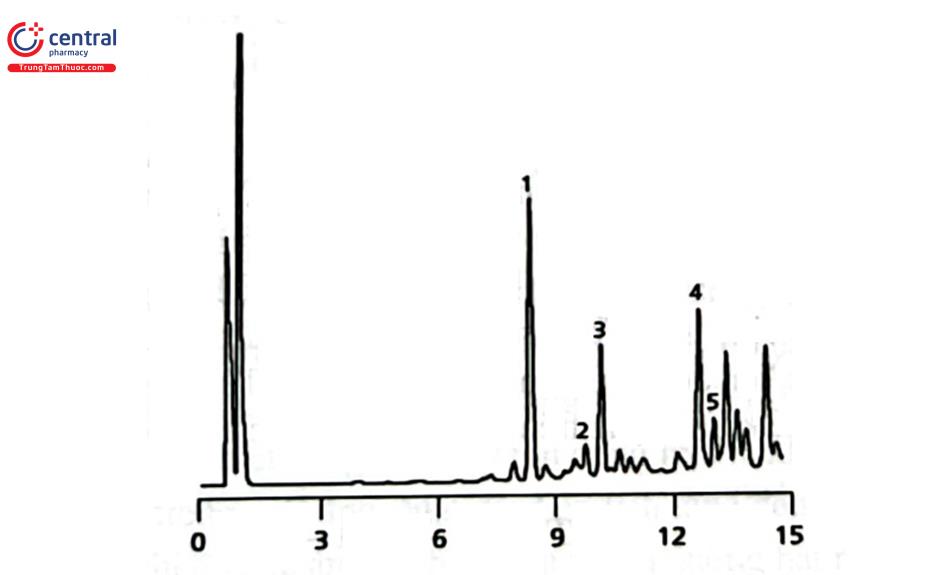
3.6 Detector tán xạ ánh sáng bay hơi (Evaporative Light Scattering Detector: ELSD)
ELSD nguyên tử hóa các chất rửa giải từ cột tách, chiếu ánh sáng vào các hạt tạo thành và phát hiện ánh sáng tán xạ thu được. Về mặt lý thuyết, ELSD có thể phát hiện bất kỳ thành phần không bay hơi nào. ELSD có độ nhạy cao hơn khoảng 10 lần so với detector RI, nhưng có độ nhạy hơi thấp đối với các thành phần có phân tử thấp do kích thước nhỏ. ELSD được sử dụng chủ yếu để phát hiện các thành phần không hấp thụ UV. Cần chú ý không thể sử dụng các muối không bay hơi làm dung môi rửa giải.
Nguyên tắc phát hiện duy nhất của ELSD liên quan đến quá trình phun sương, bay hơi và phát hiện các hạt chất tan không bay hơi còn lại. Bên trong bộ phận phun sương, dịch rửa giải từ cột sắc ký đi qua một kim và trộn với khí nitơ để tạo thành đám sương nhỏ (aerosol) chứa các giọt phân bố đồng nhất. Kích thước của mỗi giọt phụ thuộc vào tốc độ dòng khí. Tốc độ dòng khí tối ưu sẽ cho độ nhạy (S/N) cao nhất. Sau đó, pha động bay hơi trong một ống bằng thép không gỉ (drift tube) đã được gia nhiệt, để lại một lớp sương mịn ưu sẽ tạo ra đường nền ổn định nhất mà không ảnh hưởng đến độ nhạy. Quá trình phát hiện xảy ra bên trong tế bào quang học, nơi các hạt mẫu không bay hơi làm gián đoạn chùm ánh sáng laser. Ánh sáng tán xạ được phát hiện bởi một diod quang. Quá trình phát hiện bằng ELSD chịu ảnh hưởng của nhiều yếu tố nên có thể kết quả thu được không đáp ứng yêu cầu do đó có thể dùng phương pháp chuẩn nội để hạn chế sai số khi định lượng.
ELSD được ứng dụng nhiều trong phân tích các hợp chất thiên nhiên không phát hiện được bằng detector UV và/ hoặc có phân tử lượng lớn như carbohydrat, glycosid, các polymer,... Ví dụ định lượng các terpen lacton trong cao Ginkgo Biloba sử dụng ELSD 2000 (Alltech Associates) với điều kiện hoạt động của detector là bộ phận phun sương ở chế độ dừng (không chia dòng aerosol); nhiệt độ drift tube 110°C, tốc độ khí nitơ 3,1 L/phút. Cột tách Alltima C18 (100 mm x 4,6 mm, 3 um); pha động gồm dung dịch acid trifluoroacetic 0,1% trong nước và acid trifluoroacetic 0,1% trong methanol, chạy chế độ gradient. (Hình 2.13).
3.7 Detector phun sương tích điện (Charged Aerosol Detector: CAD)
Giống như ELSD, CAD nguyên tử hóa các chất rửa giải từ cột tách tạo thành các tiểu phân chất phân tích. Điểm khác biệt của CAD là dùng khí nitơ tích điện nên chất phân tích trong các giọt nhỏ sẽ được ion hóa và được phát hiện bằng tín hiệu điện. Corona® CAD® có khả năng phát hiện các thành phần có độ nhạy cao hơn ELSD, độ nhạy ít phụ thuộc vào thành phần. Yêu cầu về pha động đối với CAD tương tự như đối với ELSD.
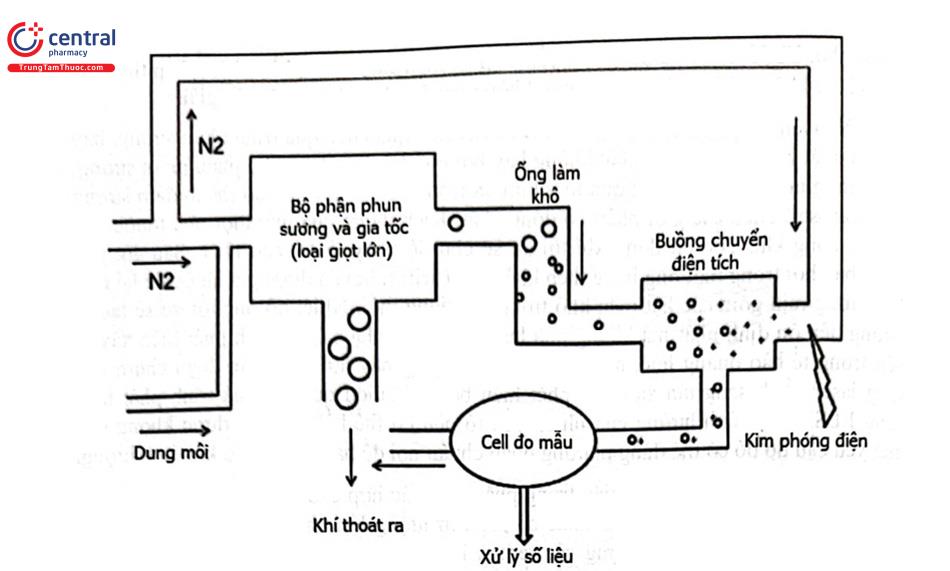
CAD cũng được ứng dụng phân tích các chất như đối với ELSD nhưng có độ nhạy cao hơn. Trong kiểm nghiệm dược phẩm, CAD hay được sử dụng phân tích các chất nhũ hóa như chất hoạt động bề mặt nhóm ethoxylat, polyethylen glycol, polysorbat, lecithin, hydroxypropylmethyl cellulose, ...
3.8 Detector khối phỗ (Mass spectrometry Detector: MSD hay MS)
Detector MS phát hiện chất phân tích bằng kỹ thuật đo trực tiếp tỷ số khối lượng và điện tích (m/z) của ion được tạo thành trong pha khí từ phân tử hoặc nguyên tử của mẫu. Tỷ số này được biểu thị bằng đơn vị khối lượng nguyên tử (amu - Atomic mass unit) hoặc bằng dalton (Da). 1 amu = 1 Da và bằng khối lượng của nguyên tử hydro. Cũng có thể sử dụng đơn vị m/z là Thomson. Đây là một đơn vị không sử dụng phổ biến trong các tài liệu khoa học liên quan đến lĩnh vực khối phổ. Tên gọi này ám chỉ Joseph John Thomson, người đã đo tỷ lệ khối lượng trên điện tích của các electron và ion.
Trong quá trình phân tích, mẫu phân tích ở thể lỏng từ bộ phận sắc ký lỏng đi tới giao diện kết nối của bộ phận MS được loại dung môi thành các phân tử trung hòa điện tích ở thể khí bay hơi, sau đó được nạp vào buồng ion hóa của thiết bị (ion source) bằng các thiết bị chuyển mẫu phù hợp để hình thành các ion hoặc tiểu phân mang điện tích ở pha khí. Hỗn hợp các ion hoặc tiểu phân mang điện (gọi chung là các ion) được gia tốc và chuyển đến bộ phận phân tích khối (mass analyzer) để chia tách thành từng loại tùy thuộc vào tỉ lệ khối lượng/điện tích (m/z) của chúng. Dưới tác động của điện trường và từ trường ở bộ phận phân tích khối, các ion có khối lượng khác nhau sẽ chuyển động có hướng với tốc độ và/ hoặc quỹ đạo chuyển động khác nhau. Tốc độ, quỹ đạo chuyển động của ion phụ thuộc vào bản chất và cường độ điện trường của bộ phận phân tích khối; điện tích và khối lượng của ion. Do vậy, sau khi đi qua qua bộ phận phân tích khối, các ion trong hỗn hợp sẽ được phân tách riêng thành từng loại. Mỗi loại ion được phân tách sẽ được đưa đến detector ghi nhận thành từng vạch phổ tương ứng trên bộ phận ghi phổ đồ. Cường độ tín hiệu vạch phổ trên phổ đồ phụ thuộc vào số lượng ion được ghi nhận ở bộ phận phát hiện. Quá trình phân tích khối và phát hiện được thực hiện trong môi trường chân không (áp suất khoảng 10-5 đến 10-8 Torr).
Phổ đồ được biểu diễn dạng vạch thẳng đứng có độ cao tỉ lệ với cường độ và có vị trí trên trục hoành tương ứng với tỉ số m/z của mỗi ion. Cường độ trục tung là cường độ tương đối. Thường chọn vạch mạnh nhất làm vạch cơ bản và quy cho nó có cường độ 100%. Cường độ các vạch khác được tính ra % so với vạch cơ bản. Các vạch được sắp xếp theo giá trị m/z từ thấp đến cao.
Giai đoạn ion hóa rất quan trọng trong phân tích khối phổ. Có nhiều phương pháp ion hóa khác nhau như ion hóa bằng va chạm điện tử, ion hóa hóa học, ion hoá áp suất khí quyển (API - Atmospheric Pressure Ionization), Trong đó, API là kỹ thuật ion hoá quan trọng nhất của sắc ký lỏng - khối phổ (LC - MS), có khoảng 99% các thiết bị LC - MS model mới sử dụng nguồn API. Có 2 kiểu API là ion hoá phun sương điện tử (ESI: Electron Spray Ionization) và ion hoá hoá học ở áp suất khí quyển (APCI - Atmospheric Pressure Chemical Ionization). Ion hóa phun sương điện tử được dùng chủ yếu trong phân tích những hợp chất không bền nhiệt, có tính phân cực như dược phẩm.
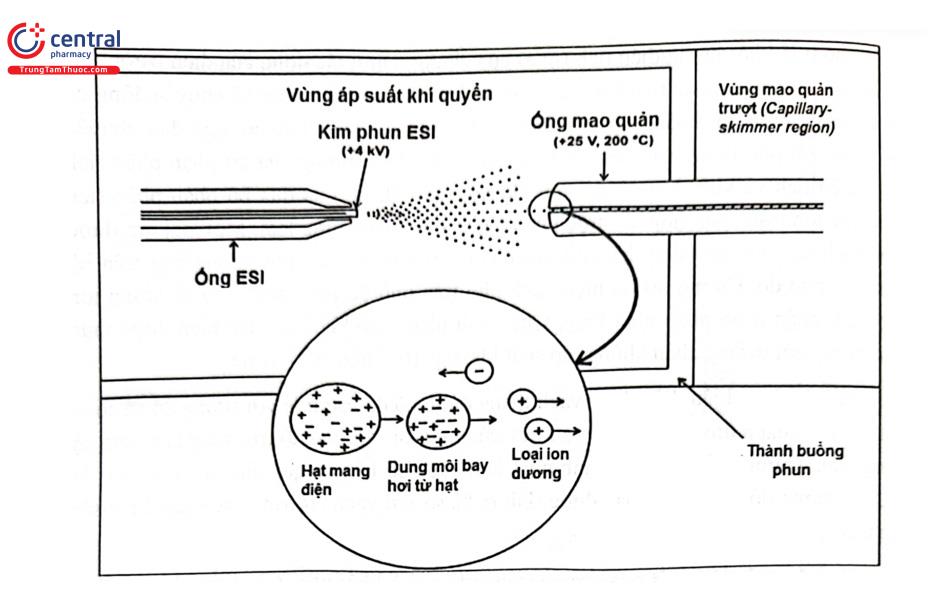
Kĩ thuật ion hóa phun điện tử bao gồm ba quá trình cơ bản là tạo thành các giọt mang điện tích; làm giảm kích thước của các hạt và phân nhỏ các hạt; quá trình hình thành pha hơi các ion. Tại đầu ống dẫn mao quản, dưới ảnh hưởng của điện thế cao và sự hỗ trợ của khí mang, mẫu được phun thành những hạt sương (giọt) nhỏ tích điện tại bề mặt. Khi ở xung quanh các hạt này tạo nhiệt năng làm bay hơi dung môi ra khỏi giọt sương làm mật độ điện tích tại bề mặt hạt sương gia tăng. Mật độ điện tích tăng đến một điểm giới hạn (giới hạn ổn định Rayleigh), khi đó hạt sương phân chia thành những hạt nhỏ hơn do lực đẩy lớn hơn sức căng bề mặt. Quá trình này được lặp lại nhiều lần để hình thành những hạt rất nhỏ mang điện tích cao (ion). Các ion chuyển thành thể khí bởi lực đẩy tĩnh điện và đi vào bộ phân tích khối. Quá trình ion hoá diễn ra bên ngoài vùng chân không của thiết bị. Trong kỹ thuật ESI, phân tử phải ở dạng chất điện ly tan trong dung dịch dùng để phun sương. Điều này phụ thuộc vào dung môi, pKa của chất điện ly và pH của dung dịch. Sơ đồ mô tả nguyên lý ion hóa bằng kỹ thuật ESI được trình bày ở Hình 2.15.
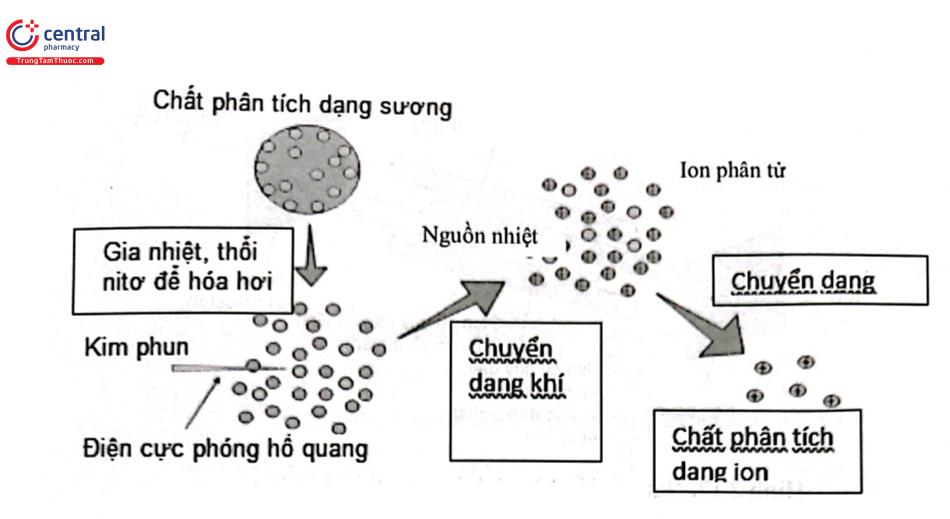
3.8.1 Ion hóa hóa học ở áp suất khí quyển (4tmospheric pressure chemical ionization: APCI)
APCI dựa trên phản ứng ion - phân tử ở pha khí, được ứng dụng chủ yếu phân tích các chất ít phân cực hoặc không phân cực, dễ bay hơi, có khối lượng phân tử dưới 1500 Da và thường tạo ion điện tích đơn. Chất phân tích trong dung môi pha động đi vào bộ phận phun sương, tạo ra lớp sương mỏng nhờ dòng khí nitrogen có tốc độ cao. Các giọt sương sau đó được làm nóng để bay hơi dung môi. Một điện cực (corona needle) tích điện và phóng hồ quang xúc tác cho chuỗi các phản ứng ion - phân tử giữa các thành phần như hoạt chất cần phân tích (M), tiểu phân dung môi pha động (S) và các tiểu phân khí mang (N2). Cấu tạo của M, điện thế của điện cực và các thành phần của pha động sẽ quyết định cơ chế chuỗi phản ứng ion - phân tử giữa M, S, Nz và các thành phần khác trong lớp sương mỏng theo hướng có hay không có sự trao đổi trao đổi proton và/ hoặc electron, từ đó quyết định loại ion sẽ hình thành khi ion hóa hoạt chất cần phân tích là [M+H]* hay [M - H] . Sơ đồ mô tả nguyên lý ion hóa bằng kỹ thuật APCI được trình bày ở Hình 2.16.
3.8.2 Bộ phân tích khối (mass analyzer)
Các ion hình thành ở nguồn ion hoá đi vào bộ phận phân tích khối. Bộ phận phân tích khối có nhiệm vụ tách các ion có tỷ lệ m/z khác nhau thành từng loại riêng biệt nhờ tác dụng của từ trường, điện trường trước khi đến bộ phận phát hiện và xử lý số liệu. Bộ phận phân tích khối (mass analyzer) của thiết bị khối phổ gồm 4 loại chính, có cấu tạo và nguyên lý hoạt động khác nhau, bao gồm: Bộ phân tích từ, bộ phân tích tứ cực, bộ phân tích thời gian bay và bộ phân tích cộng hưởng ion cyclotron. Trong đó, bộ phân tích khối kiểu tứ cực và tứ cực chập ba được sử dụng nhiều trong phân tích.
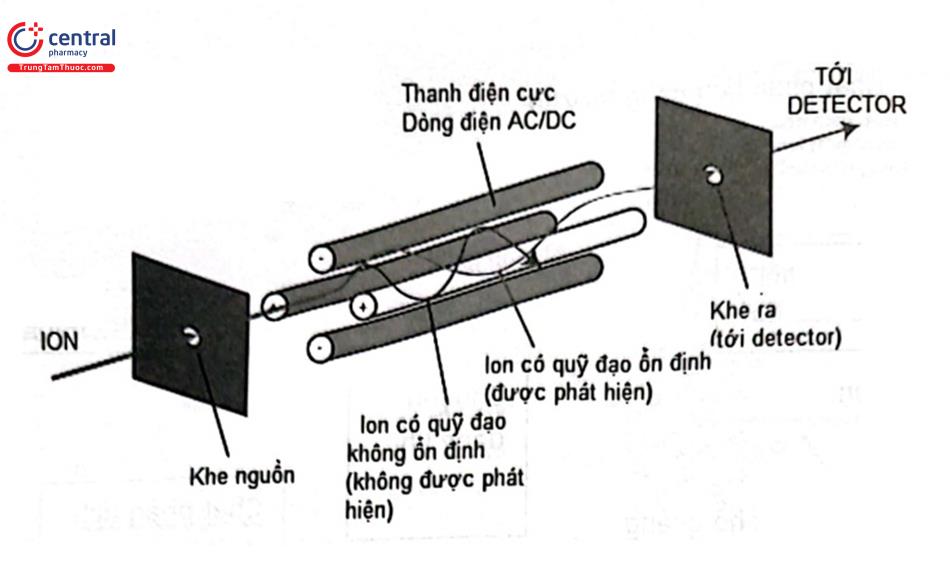
3.8.3 Bộ phân tích khối tử cực (Quadrupol mass analyser)
Gồm có 4 cực bằng kim loại đặt song song và sát nhau. Có một khoảng không giữa 4 cực để các ion bay qua. Dòng điện một chiều (DC) và điện thế xoay chiều cao tần được đặt vào từng cặp đối diện của tứ cực. Cả hai trường không làm tăng tốc dòng ion từ nguồn đi ra mà chỉ làm các ion dao động quanh trục trung tâm khi chuyển động. Các ion chuyển động theo hướng trục trung tâm và dọc theo các tứ cực, đồng thời dao động vuông góc với hướng trục trung tâm dưới ảnh hưởng của một trường điện thế tần số cao. Mỗi loại ion có tỷ số m/z khác nhau sẽ dao động xung quanh các trục với biên độ khác nhau. Chỉ có các ion có tỷ số m/z nhất định có biên độ dao động phù hợp với sự biến thiên tần số điện thế áp lên các cặp điện cực mới có thể chuyển động theo hướng trục trung tâm đi qua tứ cực để đến detector. Còn các ion khác có biên độ dao động tăng lên theo tần số biến thiên điện thế sẽ va đập vào thành các điện cực bị trung hòa điện tích và bị bơm chân không của thiết bị MS hút ra ngoài. Sơ đồ nguyên lý phân tách các ion của bộ phận phân tích phổ khối kiểu tứ cực được trình bày ở Hình 2.17.
3.8.4 Bộ phân tích tử cực kiểu chập ba (Triple quadrupole mass analyser)
Bộ phân tích tử cực kiểu chập ba còn gọi là khối phổ hai lần (LC - MS/MS) gồm 3 bộ tử cực nối tiếp nhau Q1, Q2 và Q3. Q1 sẽ tách các ion và lựa chọn một hoặc một số ion ban đầu (ion mẹ) đưa vào tứ cực Q2. Trong buồng Q2 với áp suất cao, ion mẹ bị phân mảnh do va chạm với các khí trơ có mặt trong buồng như nitơ, argon, heli. Nhờ va chạm này năng lượng động học của các ion chuyển thành nội năng nên chúng phân mảnh ion mẹ tạo ra các ion nhỏ hơn (ion con). Các ion con mới hình thành được dẫn đến Q3 phân tích khối tách riêng và đến bộ phận phát hiện. Các hợp chất cấu tạo khác nhau sẽ có cơ chế phân mảnh khác nhau và hình thành nên các ion con có số khối khác nhau. Nói cách khác, các ion ban đầu nếu có số khối giống nhau nhưng có cấu tạo khác nhau, khi phân mảnh sẽ tạo ra các ion con có số khối khác nhau tùy thuộc vào cấu tạo của ion mẹ. Do vậy, so với thiết bị MS tứ cực đơn, thiết bị MS kiểu tứ cực chập ba có độ đặc hiệu, độ nhạy cao hơn và nó còn cung cấp thêm các thông tin về cấu tạo của hợp chất.
3.8.5 Kỹ thuật ghi phổ
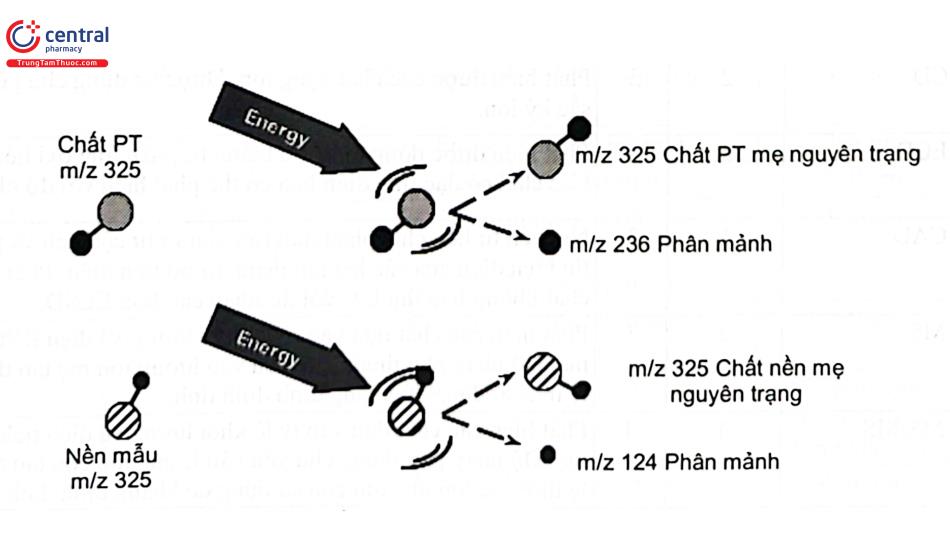
Trong phân tích khổ phối, việc xác định chính xác 1 ion rất quan trọng cho việc định danh chất phân tích. Một hợp chất xác định trong những điều kiện nhất định sẽ cho 1 ion có số khối xác định trên phổ đồ. Tuy nhiên, một ion có số khối xác định trên phổ đồ lại có thể xuất phát từ nhiều hợp chất khác nhau. Khi phân tích bằng LC - MS, nếu điều kiện sắc ký chưa đảm bảo việc phân tách các chất thì việc nhận định các ion thông số khối trên phổ đồ có thể bị ảnh hưởng. Đối với những trường hợp này, MS một lần (không có sự phân mảnh ion ban đầu) có thể cho kết quả không chính xác so với kỹ thuật khối phổ nhiều lần do tính chọn lọc khối không cao.
3.8.6 Kỹ thuật quét toàn phổ (Full Scan)
Khi thao tác với chế độ scan, MS sẽ nhận được tất cả các mảnh ion để cho phổ đồ toàn ion đối với tất cả các chất trong suốt quá trình phân tích. Thường dùng để định danh hay phân tích khi chất phân tích có nồng độ đủ lớn. Đối với MS tứ cực chập ba, chế độ Full scan MS thường được lựa chọn để khảo sát ion mẹ, chế độ Full scan MS/MS quét tất cả các ion con tạo thành thường được sử dụng để xác định ion con hình thành từ ion mẹ cho tín hiệu ổn định và bền nhất.
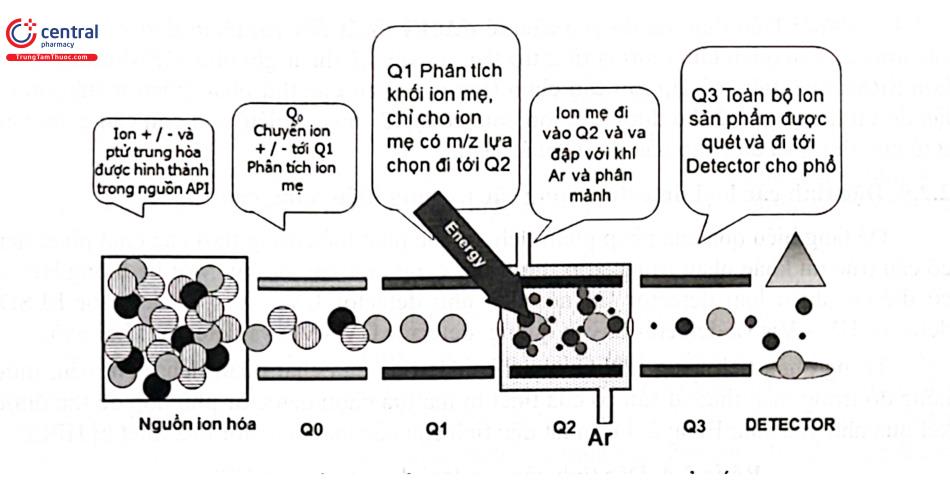
3.8.7 Kỹ thuật ghi phố chọn lọc ion (Selected Ion Monitoring - SIM)
Trong chế độ SIM, bộ phận phân tích khối chỉ cho phép ion có số khối xác định đi đến bộ ghi nhận tín hiệu. Do vậy, phổ đồ SIM chi cho tín hiệu của các ion đã được lựa chọn trước đó, không thể dùng để nhận danh hay so sánh với các thư viện có sẵn. Đối với MS tử cực chập ba, chế độ SIM thường được lựa chọn để khảo sát năng lượng phân mảnh khi đã biết ion mẹ.
3.8.8 Kỹ thuật ghi phổ phản ứng chọn lọc (SRM và MRM)
Đối với MS tứ cực chập ba, 2 kỹ thuật ghi phổ MS/MS có độ nhạy cao thường được sử dụng là SRM (Selected Reaction Monitoring) và MRM (Multiple Reaction Monitoring). Trong đó:
SRM: Cô lập ion cần chọn, sau đó phân mảnh ion cô lập đó, trong các mảnh ion tạo thành, cô lập 1 mảnh ion con cần quan tâm và đưa vào đầu dò để phát hiện.
- MRM: Trên thực tế, do yêu cầu về mặt kỹ thuật đối với phân tích vi lượng nên các ion con cần quan tâm thường từ 2 trở lên, do vậy kỹ thuật ghi phổ MRM thông dụng hơn SRM. Đầu tiên, cô lập ion cần chọn (ion mẹ) ở tứ cực thứ nhất, phân mảnh ion cô lập đó tại tử cực thứ 2 thu được các ion con, cô lập 2 (hoặc nhiều) ion con cần quan tâm ở tứ cực thứ 3 và đưa vào đầu dò để phát hiện.
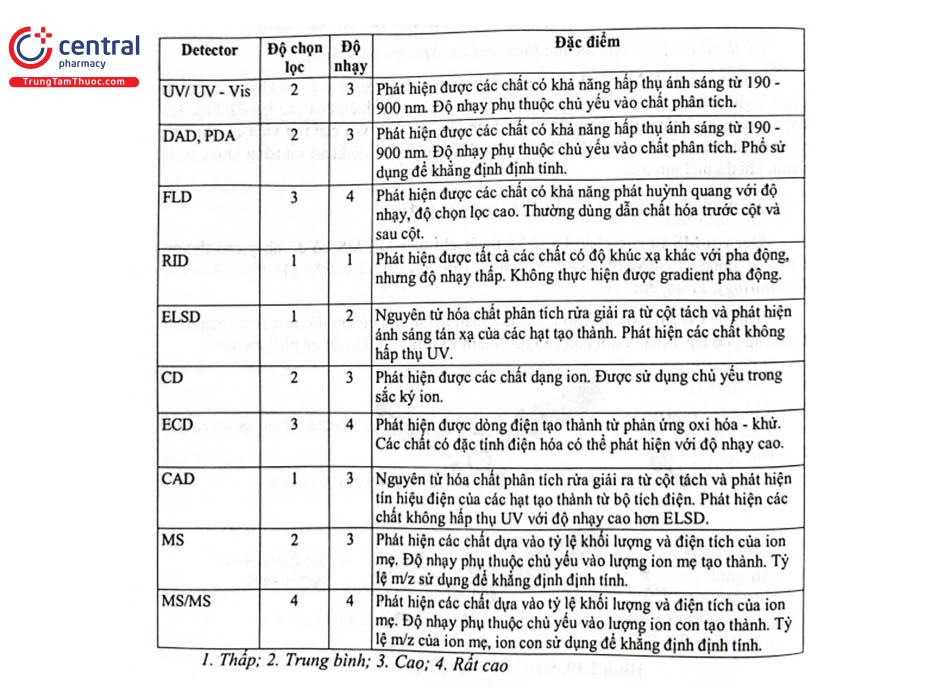
3.9 Đặc tính các loại detector trong sắc ký lỏng hiệu năng cao
Để tăng hiệu quả của phép phân tích, có thể phát hiện đồng thời các chất phân tích có cấu trúc rất khác nhau trong mẫu thử trong cùng một lần sắc ký. Một hệ thống HPLC có thể có nhiều loại detector lắp nối tiếp như detector UV - Vis và detector ELSD, detector UV - Vis và detector huỳnh quang, detector UV - Vis và detector khối phổ, ...
Trong phân tích bằng HPLC, tùy theo đặc tính của chất phân tích, nền mẫu, mức nồng độ trong mẫu thử, sự sẵn có của thiết bị mà lựa chọn detector phù hợp để thu được kết quả như yêu cầu. Bảng 2.4 tóm tắt đặc tính của các loại detector của thiết bị HPLC.
4 Dẫn chất hóa trong phân tích bằng sắc ký lỏng hiệu năng cao
Chuẩn bị mẫu phân tích trong HPLC được tiến hành qua các bước chính sau:
1. Mẫu thử: Lấy mẫu, bảo quản;
2. Làm sạch mẫu ngoài hệ thống (offline): Làm đồng nhất, hòa tan, lọc, ly tâm, kết tủa, thủy phân, chiết lỏng - lỏng, chiết lỏng dùng siêu âm, chiết lỏng siêu tới hạn, làm giàu;
3. Làm sạch trong hệ thống (online): Cột bảo vệ, chiết pha rắn online;
4. Dẫn chất hóa với thuốc thử thích hợp: trước cột, sau cột, online.
Đa số các phép phân tích HPLC đều phát hiện trực tiếp chất phân tích. Trong trường hợp, các chất phân tích có khả năng đáp ứng detector kém và (hoặc) nồng độ thấp có thể dẫn chất hóa (hay còn gọi là darector sẽ thuận lợi cho phát hiện và thu được là dẫn xuất hóa) với thuốc thử thích hợp tạo thành sản phẩm có khả năng đáp ứng cao với độ chính xác cao hơn. Ví dụ điển hình là phân tích các chất có nhóm amin bậc 1 và bậc 2 như các acid amin, sau khi dẫn chất hóa thu được dẫn chất có khả năng phát hiện tốt bằng detector UV - VIS hoặc huỳnh quang và tăng độ chọn lọc của phương pháp.
4.1 Dẫn chất hóa tạo sản phẩm hấp thụ UV - Vis
Sử dụng các chất đánh dấu tạo dẫn chất có khả năng hấp thụ UV - Vis là một trong các kỹ thuật dẫn xuất phổ biến. Các thuốc thử được lựa chọn để cho chất tạo thành không những có hấp thụ cực đại để tăng độ nhạy mà còn có độ chọn lọc cao, đồng thời phải giảm ảnh hưởng của sản phẩm phản ứng giữa nền mẫu và thuốc thử, cũng như nền mẫu. Bảng 2.5 trình bày một số thuốc thử thường dùng trong phản ứng dẫn chất acid
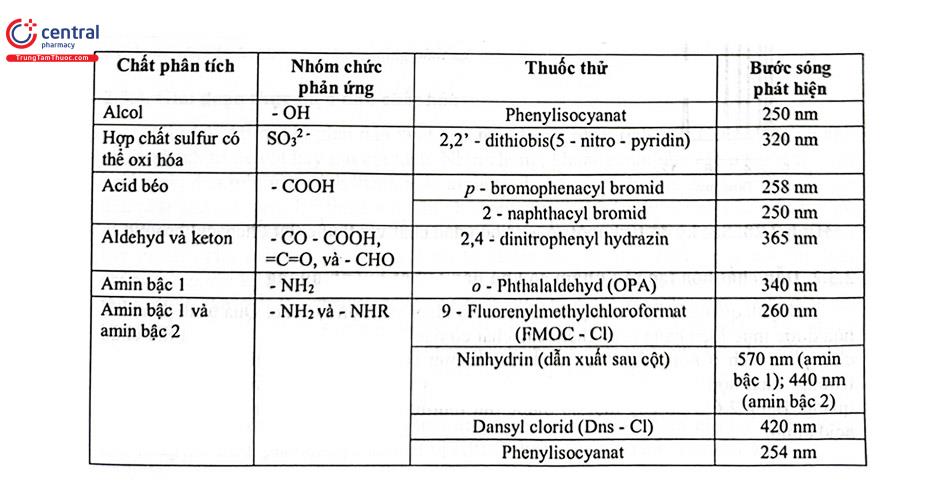
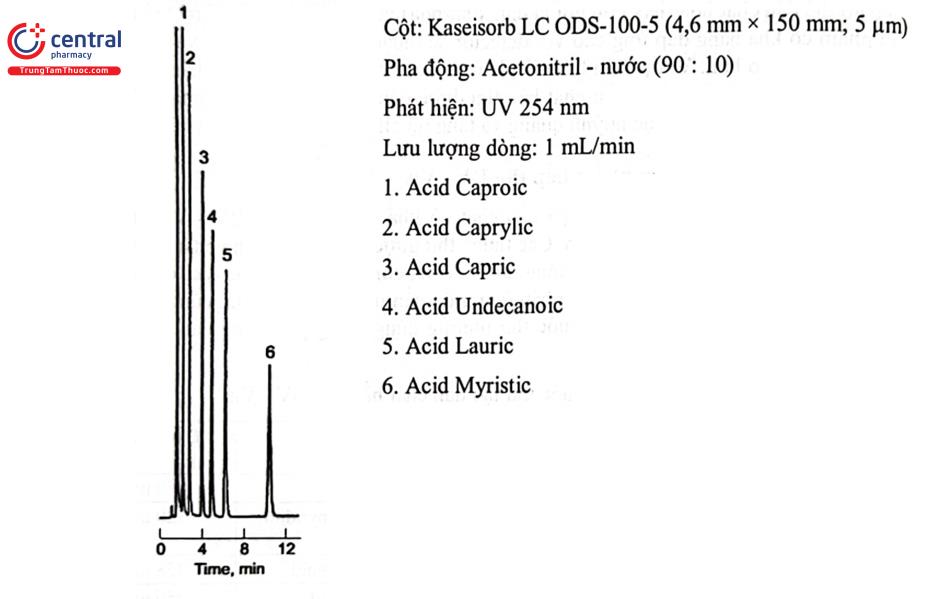
Ví dụ: Thuốc thử Phenacyl bromid thường được dùng dẫn chất các acid béo, acid mật, acid carboxylic trong rượu. Phenacyl bromid dễ dàng phản ứng với nhóm carboxy khi có mặt base để tạo thành este tương ứng bền và có thể phân tích bằng HPLC với detector UV ở bước sóng 254 nm.
Quá trình dẫn chất hóa được thực hiện bằng cách trộn khoảng 100 pg mẫu, 10 uL thuốc thử 12 mg/mL trong aceton và 10 FL triethylamin 10 mg/mL trong aceton, ủ dung dịch ở 50°C trong 2 giờ. Làm nguội dung dịch thu được đến nhiệt độ phòng và phân tích bằng HPLC.
4.2 Dẫn chất hóa tạo sản phẩm có khả năng phát huỳnh quang
Huỳnh quang là kỹ thuật phát hiện có độ nhạy và chọn lọc cao. Quá trình dẫn xuất hóa được thực hiện bằng cách thêm các chất có đặc tính huỳnh quang vào mẫu thử chứa chất phân tích ở nồng độ thấp và (hoặc) không có khả năng hấp thụ UV trong môi trường thích hợp, sản phẩm phản ứng được phân tích bằng HPLC với detector huỳnh quang. Bảng 2.6 trình bày một số thuốc thử thường dùng trong phản ứng dẫn xuất các acid amin.
4.3 Giai đoạn thực hiện dẫn chất hóa
Câu hỏi đặt ra khi phải dẫn xuất hóa mẫu trong phân tích HPLC là quá trình này sẽ tiến hành trước cột hay sau cột tách. Nhìn chung, khoảng một nửa các phép phân tích acid amin dựa trên việc tách thành acid amin tự do bằng sắc lý lỏng trao đổi ion rồi tạo dẫn chất sau cột tách. Kỹ thuật tạo dẫn chất sau cột có thể áp dụng cho các mẫu thử có một lượng nhỏ chất đệm (như các muối và urê) và thường cần từ 5 kg đến 10 ng mẫu thử protein cho một lần phân tích. Còn kỹ chúng bằng sắc ký lỏng pha đảo. Còn kỹ thuật tạo các dẫn chất trước cột rồi tách chúng bằng sắc ký lỏng pha đảo có độ nhạy rất cao chi can thử protein cho mỗi lần phân tích, nhưng lại có thể bị ảnh hưởng bởi các muối đệm có có thể chỉ cân từ 0,5 kg đến 1,0 kg mẫu trong mẫu thử. Mặt khác, kỹ thuật tạo dẫn chất trước cột còn có thể cho nhiều sản phẩm phản ứng khác nhau của cùng một acid amin, do đó gây trở ngại cho việc biện giải kết quả phân tích. So với kỹ thuật tạo dẫn chất trước cột, kỹ thuật tạo dẫn chất sau cột ít bị ảnh hưởng bởi các thay đổi trong quá trình định lượng hơn. Quá trình dẫn chất hóa có thể thực hiện trước cột (tiến hành phản ứng trong giai đoạn chuẩn bị mẫu hoặc dùng tính năng hút mẫu, pha loãng của thiết bị HPLC) hoặc sau khi tách ở cột sắc ký.
Kỹ thuật dẫn chất hóa trước cột có thể thực hiện ngoài hệ thống hoặc trong hệ thống (online), nhưng dẫn chất hóa sau cột chỉ thực hiện online nên cho độ chính xác tối đa. Quá trình dẫn chất hóa sau cột, thuốc thử được đưa vào vòng phản ứng bằng thiết phụ trợ như bơm, đồng thời cần thiết bị trộn và gia nhiệt. Tăng thể tích chết sau cột theo cách này sẽ làm píc doãng. Mặc dù sự doãng píc này ít xảy ra khi dùng cột đường kính tiêu chuẩn với tốc độ dòng trên 1 ml/phút, nhưng dẫn chất hóa sau cột không phù hợp khi dùng cột có đường kính nhỏ.
5 Một số kiểu sắc ký lỏng hiệu năng cao đặc biệt
Sắc ký lỏng hiệu năng cao có thể thực hiện dựa trên các đặc tính hóa lý, theo nhiều kỹ thuật khác nhau tuỳ thuộc vào đặc điểm cấu tạo của chất phân tích, bản chất pha tĩnh và thành phần pha động. Trong phạm vi tài liệu này sẽ phân tích cụ thể hai kiểu HPLC gần đây được phát triển và ứng dụng nhiều trong phân tích thuốc là sắc ký đồng phân quang học và sắc ký tương tác thân nước.
5.1 Sắc ký đồng phân đối quang (Chiral chromatography)
Đồng phân đối quang (enantiomers) là hai đồng phân không gian dạng ảnh và vật qua gương. Chúng có công thức hóa học hoàn toàn giống nhau, chỉ khác về cách bố trí không gian của các nhóm thế quanh carbon bất đối. Chính sự khác nhau về cấu trúc không gian đã làm cho mỗi dạng đồng phân có tương tác khác nhau với một thụ thể sinh học xác định (tương tự như phản ứng đặc hiệu chất mang - receptor của các enzym). Kết quả là các đặc tính dược lý - lâm sàng, độc tính của hai đồng phân đối quang của một thuốc có thể rất khác nhau.
Một thảm họa y khoa liên quan đến thuốc đối quang thalidomid đã được ghi nhận trong lịch sử y văn thế giới. Thuốc thalidomid được phát triển từ giữa những năm 1950 tại Đức, sau đó được bán tại 46 quốc gia trên khắp thế giới dưới dạng thuốc không kê đơn để điều trị những triệu chứng như chóng mặt, mất ngủ, buồn nôn cho thai phụ trong giai đoạn đầu của thai kỳ. Đến năm 1961, y văn đã ghi nhận hàng nghìn bà mẹ sử dụng loại thuốc này sinh ra những đứa trẻ bị dị tật bẩm sinh ở các chi (hội chứng Phocomelia). Các nghiên cứu chuyên sâu về dược lý, dược lực học và dược động học đã được thực hiện ngay sau thảm họa cho thấy, thuốc thalidomid là dạng racemic chứa các đồng phân quang học S và R - thalidomid, trong đó đồng phân hữu tuyền có tác dụng an thần gây ngủ còn đồng phân tả tuyền ức chế miễn dịch, gây độc đối với tế bào và là nguyên nhân gây ra hội chứng Phocomelia. Mặc dù vậy, số lượng các thuốc đối quang được đăng ký sản xuất và lưu hành vẫn không ngừng phát triển. Hiện có tới hơn 50% thuốc đang lưu hành trên thị trường là thuốc đối quang và trong đó có khoảng 40% là các thuốc đối quang đơn thành phần (chỉ chứa một đối quang tinh khiết - enantiopure). Sau thảm họa thalidomid, độ tinh khiết của các chất đồng phân đối quang sử dụng làm thuốc cho người dần được qui định kiểm soát chặt chẽ. Từ năm 1992, Cơ quan quản lý thuốc và thực phẩm Mỹ (US - FDA) và cơ quan quản lý thuốc châu u (EMA) đã yêu cầu các tính chất của mỗi đơn đồng phân phải được nghiên cứu riêng rẽ trước khi quyết định đưa thuốc ra thị trường dưới dạng một đồng phân đối quang hay là một racemic.
5.1.1 Cơ chế tách đồng phân đối quang
Phân tích đồng phân đối quang là một trong những chủ đề khó trong hóa phân tích do bản chất lý hóa quá gần nhau của hai đồng phân trong mỗi cặp đồng phân đổi quang. Trên thực tế, hai đồng phân đối quang cho cùng đáp ứng và không thể được tách riêng ở các điều kiện thông thường, ngay cả đối với các kỹ thuật tách hiện đại nhất như HPLC, sắc ký khí hay điện di mao quản. Đặc điểm duy nhất có thể lợi dụng để tách riêng hai đồng phân đối quang là khả năng tương tác khác nhau của chúng với chất có ái lực khác g với chat co ai tục khác biệt với mỗi đồng phân đối quang của một hỗn hợp racemic. Những chất này được gọi là các chất chọn lọc hoạt quang (chiral selector).
Sự khác biệt giữa các hằng số tạo phức KR và Ks là cơ sở hóa lý cho việc lưu giữ các đồng phân đối quang bởi một chất chọn lọc hoạt quang. Quá trình tách đồng phân này đạt được do sự khác nhau về năng lượng tương tác (∆G) của tác nhân chọn lọc hoạt quang với mỗi đồng phân. Trung bình ∆Gmin = 0,24 kcal/mol thì hệ số tách các đồng phân đối quang (a) có thể đạt 1,5.
Có nhiều mô hình tương tác giữa các đồng phân đối quang và chất chọn lọc đối quang đã được thảo luận nhưng đến nay, mô hình đáng tin cậy nhất là mô hình tương tác ba điểm của Easson - Stedman (Hình 2.21). Giữa chất chọn lọc đối quang và các đồng phân đối quang có 3 điểm tương tác, trong đó có ít nhất 1 trong 3 tương tác này đặc hiệu với 1 đồng phân đối quang nhất định. Sự khác nhau này là cơ sở cho phép tách được hai đồng phân đối quang.
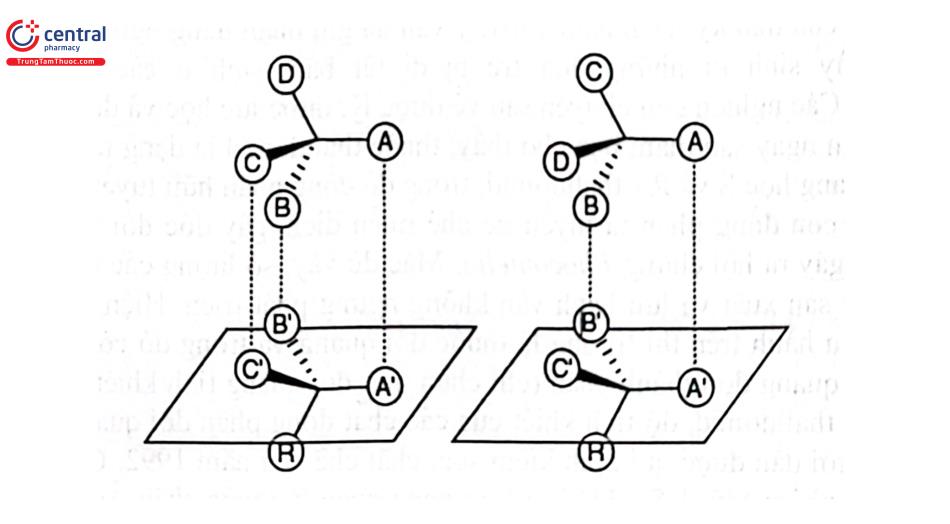
Tuy vậy, mô hình tương tác ba điểm chỉ có giá trị nếu tương tác của các phân tử đồng phân đổi quang với chất chọn lọc đối quang có thể xảy ra từ một phía. Hơn nữa, mô hình không phản ánh bản chất của sự tương tác (tức là ái lực hút hoặc đẩy giữa các hợp đồng phân không đối quang (diastereomeric). Để giải thích sự hình thành phức hợp diastereomeric giữa chất chọn lọc đối quang và các đồng phân đối quang bên cạnh mô hình tương tác 3 điểm cũng đã có nhiều mô hình khác được các tác giả đưa ra như mô hình tương tác van der Waal, liên kết n - a hay sự liên kết giữa phân tử đối quang và các “hốc” trên cấu trúc bề mặt của tác nhân hoạt quang là dẫn chất cyclodextrin (Hình 2.22).
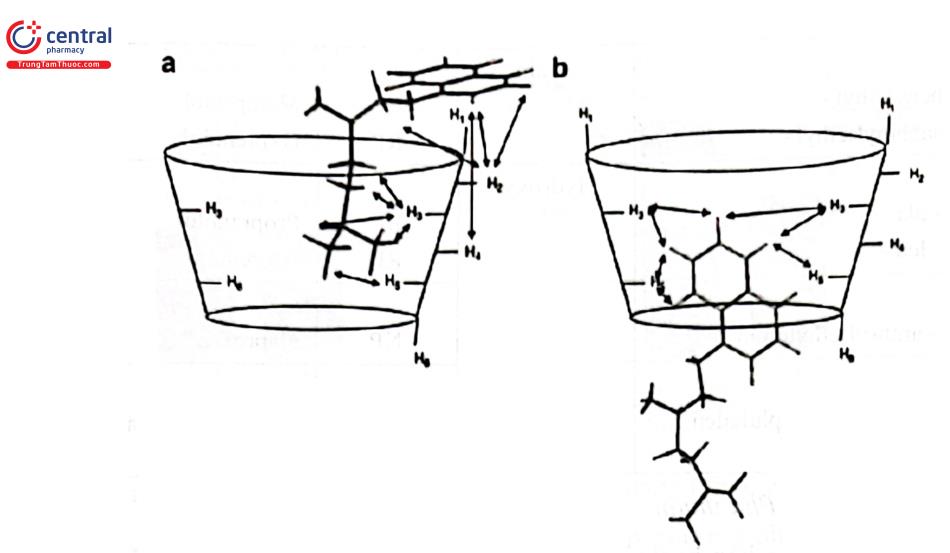
5.1.2 Các phương pháp tách đồng phân đối quang
Các tác nhân hoạt quang là thành phần quan trọng nhất của hệ thống phân tích chất đối quang. Chính vì vậy trong quá trình phát triển các kỹ thuật tách đồng phân đối quang, người ta luôn chú ý nghiên cứu, tìm kiếm các tác nhân này. Các chất chọn lọc đối quang, được thêm vào để tương tác với hỗn hợp đồng phân đối quang bằng trực tiếp và gián tiếp.
- Phương pháp gián tiếp: Hỗn hợp đồng phân đối quang (isomers) sẽ phản ứng với thuốc thử (thường là một chất đối quang, song chỉ chứa một đồng phân duy nhất chiral reagent) tạo thành một hỗn hợp chứa 2 đồng phân không hoạt quang (diasteromers). Các đồng phân này có thể được tách bởi phương pháp HPLC pha thuận hoặc pha đảo thông thường.
Quá trình phản ứng hóa học giữa dược chất và thuốc thử để hình thành nên hỗn hợp không đối quang phụ thuộc vào nhiều yếu tố như khả năng/ hiệu suất phản ứng giữa các nhóm chức; mức độ tinh khiết của thuốc thử; nồng độ hoặc tỷ lệ nồng độ giữa thuốc thử so với dược chất; nhiệt độ duy trì phản ứng để không xảy ra sự racemic hóa trong quá trình tạo dẫn xuất... Do vậy, hiện nay phương pháp này ít được sử dụng do hạn chế tìm ra dẫn xuất phù hợp và độ tin cậy của phương pháp bị ảnh hưởng bởi độ lặp lại của phản ứng tạo dẫn xuất.
- Phương pháp trực tiếp thêm đồng phân đối quang vào pha động:
+ Trong phương pháp này, thuốc thử là phối tử có tính liên kết chọn lọc với đồng + phân quang học (chiral ligand) tinh khiết và ion kim loại thích hợp được cho vào pha động HPLC để tạo thành một phức hợp đồng phân không đối quang tạm thời giữa chất phân tích và pha động. Hỗn hợp này sau đó có thể được tách bằng HPLC pha thuận hoặc pha đảo thông thường. Các chất thường được sử dụng để cho thêm vào pha động trong kỹ thuật này bao gồm các cyclodextrin và dẫn xuất; muối của một số kim loại hóa trị II (đồng, Kẽm, niken...) với một số acid amin như L - phenylalanin, L - isoleucin, ... hoặc một số protein như huyết thanh bò hay a - acid glucoprotein. Kĩ thuật này không đòi hỏi các cột sắc ký hoạt quang đắt tiền nhưng có một số nhược điểm như việc thêm liên tục chất đồng phân đối quang vào pha động thường tốn kém, phát hiện khó, hình dạng píc thường xấu (hệ số kéo đuôi lớn, số đĩa lý thuyết thấp,...).
- Phương pháp trực tiếp sử dụng cột pha tĩnh đối quang: Sử dụng một chất đồng phân đối quang tạo liên kết hóa học với pha tĩnh để tạo ra một pha tĩnh đối quang (Chiral Stationary Phase - CSPs). Pha tĩnh đối quang sẽ phản ứng với chất phân tích đệ tạo thành một phức hợp đồng phân không đối quang tạm thời (Hình 2.23). Lực liên kết của pha tĩnh với các đồng phân khác nhau nên khả năng lưu giữ chúng trên pha tĩnh khác nhau. Vì vậy các đồng phân này sẽ cho thời gian lưu khác nhau khi rửa giải sắc ký.
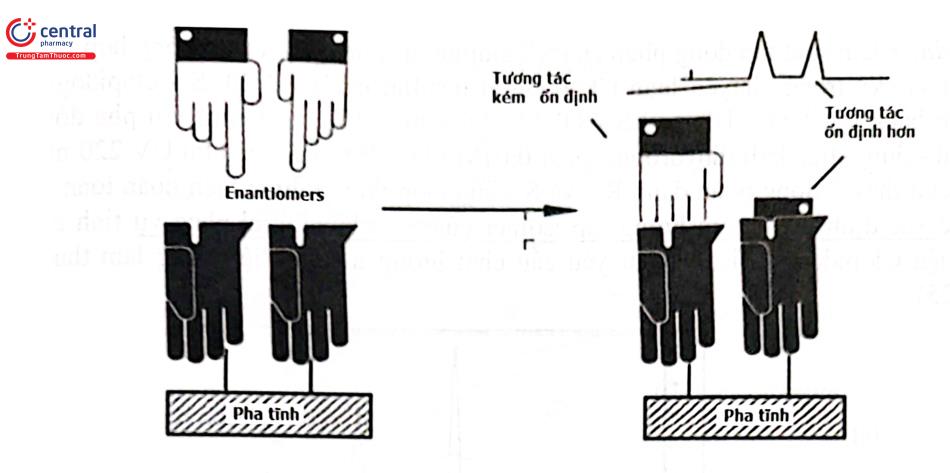
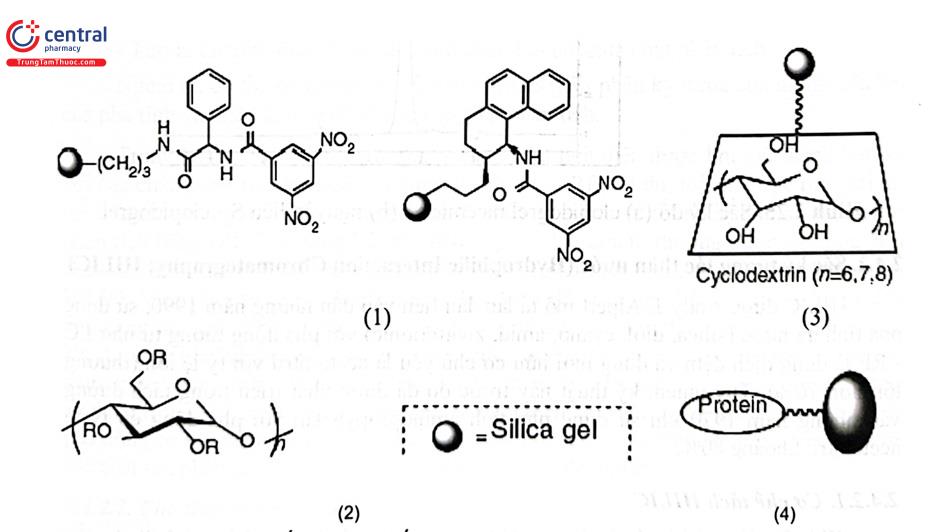
Phương pháp này được sử dụng rộng rãi hơn phương pháp thêm đồng phân đối quang vào pha động. Cho đến nay có hơn 100 loại cột HPLC có pha tĩnh hoạt quang đã được thương mại hóa. Tuy vậy, không có cột nào được coi là đa năng, có khả năng phân tách mọi chất bất đối. Ba yếu tố cần xem xét trong quá trình tiến hành tách là chất phân tích, pha tĩnh bất đối và pha động. Yếu tố để tách thành công một hỗn hợp racemic trên một cột pha tĩnh bất đối đã cho là nhận thức được cơ chế nhận diện chất bất đối. Cấu tạo một số nhóm pha tĩnh hoạt quang chủ yếu của cột HPLC dùng để tách các đồng phân đối quang được trình bày ở Hình 2.24.
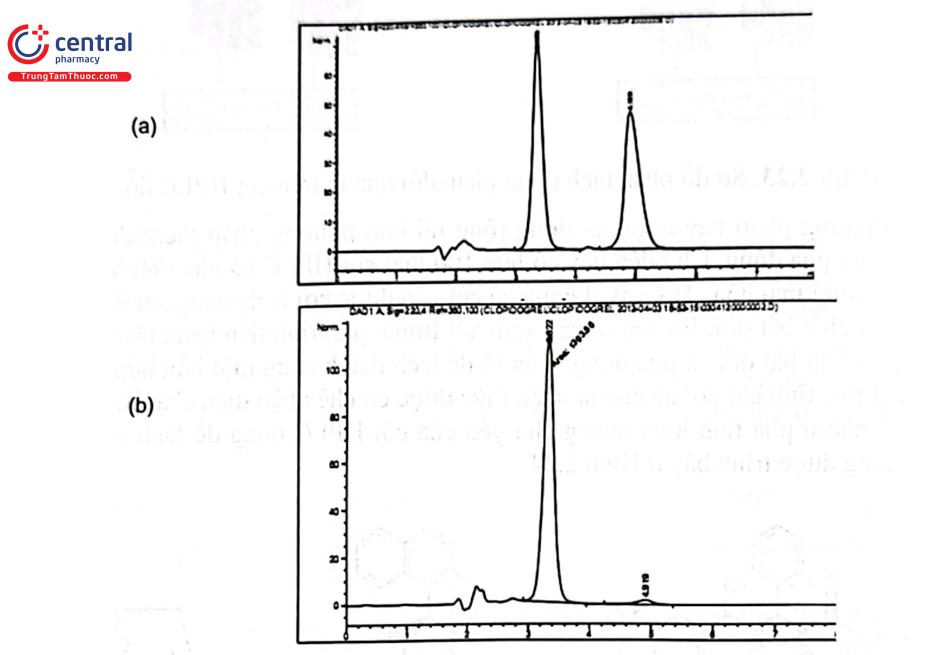
Ví dụ: Kiểm soát tạp đồng phân của S - Clopidogrel trong quá trình tổng hợp theo điều kiện sắc ký trong chuyên luận Clopidogrel bisulfat của USP 40. S - clopidogrel được tách bằng cột L57 - Ultron ES - OVM (4,6 mm × 15 cm; 5 km) với pha động acetonitril - dung dịch Kali dihydrophosphat 0,01M (25 : 75); phát hiện tại UV 220 nm. Kết quả cho thấy 2 đồng phân dạng R - và S - của clopidogrel được tách hoàn toàn do đó có thể xác định được hàm lượng tạp isomer của S - clopidogrel phục vụ tinh chế nguyên liệu Clopidogrel bisulfat đạt yêu cầu chất lượng nguyên liệu dùng làm thuốc (Hình 2.25).
5.2 Sắc ký tương tác thân nước (Hydrophilic Interaction Chromatography: HILIC)
HILIC được Andy J. Alpert mô tả lần đầu tiên vào đầu những năm 1990, sử dụng pha tĩnh ưa nước (silica, diol, cyano, amid, zwitterionic) với pha động tương tự như LC - RP là dung dịch đệm và dung môi hữu cơ chủ yếu là acetonitril với tỷ lệ lớn (thường lớn hơn 70%). Tuy nhiên, kỹ thuật này trước đó đã được phát triển trong tách đường vào những năm 1970 khi sử dụng pha tĩnh aminopropylsilan với pha động có tỷ lệ acetonitril khoảng 80%.
5.2.1 Cơ chế tách HILIC
Khi pha động (chủ yếu là dung môi hữu cơ chứa một lượng nhỏ nước) di chuyển qua pha tĩnh phân cực sẽ tạo thành một lớp chất lỏng giàu nước sát bề mặt pha tĩnh. Chất phân tích khi đi qua cột sắc ký sẽ được phân bố giữa pha động và lớp chất lỏng giàu nước này, chúng được rửa giải theo thứ tự tăng dần tính ưa nước (Hình 2.26).
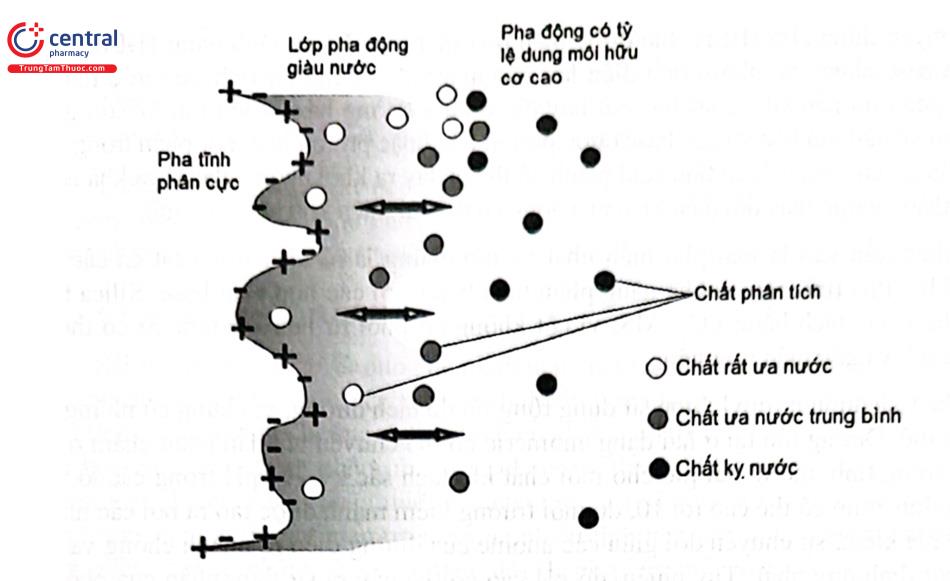
Cơ chế lưu giữ trong HILIC rất phức tạp, bao gồm sự kết hợp của:
- Sự phân bố thân nước của chất phân tích giữa lớp giàu nước và pha động;
- Liên kết hydro giữa các nhóm chức phân cực của chất phân tích và pha tĩnh;
- Tương tác tĩnh điện của các nhóm chức ion hóa của chất phân tích.
Ngoài ra, có thể có tương tác Van der Waals giữa phần kỵ nước của nhóm liên kết của pha tĩnh và phần không phân cực của chất phân tích.
Trong HILIC, các chất ưa nước, phân cực và tích điện được lưu giữ mạnh hơn so với các chất trung tính kỵ nước - ngược so với LC - RP. Ví dụ, toluen được rửa giải tại thể tích chết trong một chương trình HILIC điển hình, trong khi nó bị lưu giữ mạnh khi phân tích bằng cột C18 trong LC pha đảo. Ngược lại, uracil, thường được sử dụng làm chất xác định thể tích chết trong LC pha đảo lại cho thấy lưu giữ tốt trong tách bằng HILIC. Việc tăng tỷ lệ thành phần nước của pha động trong HILIC thường làm giảm khả năng lưu giữ. Thứ tự rửa giải chất tan giống như sắc ký pha thuận nên HILIC đôi khi được gọi là "sắc ký pha thuận trong nước"; một số nghiên cứu cho thấy có một số điểm tương đồng trong cơ chế tách của hai kỹ thuật này. Bề mặt của silica (thường được sử dụng trong HILIC) bị vô hiệu hóa do sự hiện diện của một lượng đáng kể nước trong pha động so với sắc ký pha thuận. Kết quả là ít gặp vấn đề về hình dạng píc hơn đối với các chất tan phân cực mạnh so với tách bằng sắc ký pha thuận.
5.2.2 Pha tĩnh trong HILIC
Một loạt các pha tĩnh phân cực sẵn có được dùng trong HILIC, bao gồm silica trần, aminopropyl, diol và zwitterionic (như sulfoalkylbetain); các pha liên kết có thể được chế tạo trên nền silica hoặc polyme. Tương tự như pha tĩnh trong HPLC, polyme có thể cho phép sử dụng cột trong phạm vi pH rộng hơn; tuy nhiên, hiệu năng của cột thấp hơn so với pha tĩnh nền silica. Một số công ty đã có thiết kế đặc biệt pha tĩnh nền silica chuyên dùng cho HILIC thu được hiệu quả tách tốt. Trong tách bằng HILIC, các chất tan base chứa các nhóm tích điện lưu giữ mạnh do chúng có tính ưa nước mạnh; đối với pha tĩnh nền silica, sự lưu giữ này được tăng cường bằng cách giữ lại ion giữa các nhóm silanol ion hóa và các base được proton hóa hoặc proton hóa một phần trong pha động. Ngược lại, các chất có tính acid mạnh có thể bị đẩy ra khỏi nhóm silanol và khả năng lưu giữ thấp, nhưng thay đổi điều kiện pha động có thể cải thiện khả năng lưu giữ.
Silica trần vẫn là loại phổ biến nhất và nói chung là rẻ nhất trong tất cả các vật liệu HILIC. Pha tĩnh này có khả năng phân tách tuyệt vời các hợp chất base. Silica trần có lợi thế trong tách bằng LC - MS, vì cột không có phối tử liên kết hữu cơ có thể bị “chảy máu” và gây hiệu ứng nền.
Pha tĩnh aminopropyl được sử dụng rộng rãi để tách đường, vì chúng có những ưu điểm cụ thể. Đường tồn tại ở hai dạng anomeric có thể chuyển hóa lẫn nhau chậm ở pH acid và trung tính, tạo ra hai pic cho mỗi chất khi tách sắc ký. Độ pH trong các lỗ xốp của pha tĩnh amin có thể cao tới 10, do môi trường kiềm mạnh được tạo ra bởi các nhóm base. Ở pH kiềm, sự chuyển đổi giữa các anome của đường diễn ra nhanh chóng và thu được một đỉnh duy nhất. Tuy nhiên, độ pH cao có thể gây ra sự thủy phân của phối tử liên kết và hòa tan silica do đó các pha tĩnh amin nền polyme sẽ thích hợp hơn. Pha tĩnh amin chủ yếu ứng dụng với đối tượng đường do có vấn đề độ ổn định và phản ứng có thể xảy ra của amin với aldehyd và xeton trong mẫu thử.
Pha tĩnh sulfoalkylbetain chuyên dụng để phân tách HILIC có chứa nhóm chức CH2 - N+ (CH3)2 - CH2 - CH2 - CH2 - SO3 - liên kết với silica hoặc polyme. Trong pha tĩnh zwitterionic, các nhóm ion chỉ được tách bằng một chuỗi alkyl ngắn, có thể quá gần nhau để tạo ra các tương tác độc lập với các chất tan cation hoặc anion. Trong thực tế, các điện tích của pha tĩnh hầu hết triệt tiêu lẫn nhau, chỉ còn lại các tương tác ion yếu. Bản chất phân cực của các nhóm này vẫn có hiệu quả trong việc tạo thành lớp nước để tách HILIC.
Các pha tĩnh diol, amid và cyano có các nhóm trung tính, thường được liên kết với silica. Một số tác giả cho rằng các pha tĩnh này cải thiện độ ổn định, khả năng tái lặp tốt hơn và thời gian cân bằng nhanh hơn so với silica trần. Các pha tĩnh này cũng được ứng dụng rộng rãi trong tách HILIC với nhiều đối tượng đa dạng.
5.2.3 Pha động trong HILIC B
Để tách các chất không ion hóa sử dụng pha động thường là hỗn hợp của acetonitril hoặc methanol và nước. Tỷ lệ nước thích hợp (thường là 5 - 15%) trong pha động cho phép duy trì một lớp nước đọng trên bề mặt pha tĩnh phân cực, ở đó có tiểu phân chất phân tích. HILIC tách các hợp chất bằng cách rửa giải các chất trong pha tĩnh ưa nước bằng pha động hữu cơ nồng độ cao, quá trình rửa giải được điều khiển bằng cách tăng tỷ lệ nước trong pha động.
Đối với các chất có thể ion hóa, việc lựa chọn đệm bị hạn chế do độ hòa tan kém của muối đệm vô cơ trong pha động có chứa tỷ lệ dung môi hữu cơ cao (ví dụ, muối phosphat thường được sử dụng trong HPLC pha đảo). Trong HILIC, acid fomic và Acid acetic được sử dụng trong pha động. Tuy nhiên, cần chú ý tới tính ổn định của silica trần vì bề mặt không được các phối tử liên kết bảo vệ. Độ pH của acid fomic và acid acetic tăng lên trong dung dịch có hàm lượng dung môi hữu cơ cao do các acid hầu như không phân ly nên cường độ ion của pha động thấp do đó có thể gây vấn đề về hình dạng píc và quá tải đối với các chất tan bị ion hóa. Trong trường hợp này, ưu tiên sử dụng đệm amoni fomat và amoni acetat; chúng hòa tan trong acetonitril ở nồng độ cao, duy trì cường độ ion hợp lý và cũng dễ bay hơi khi phân tích bằng LC - MS.
Trong HILIC, nếu thay acetonitril bằng methanol trong pha động sẽ cho đáp ứng cao hơn nhiều lần ở chế độ ion hóa dương. Nhưng ở chế độ ion hóa âm, đối với cùng chất phân tích pha động có chứa acetonitril hay methanol đều cho đáp ứng tương tự nhau.
5.2.4 Ứng dụng của HILIC
HILIC thường được sử dụng để phân tích chất chuyển hóa, acid amin, peptid, chất dẫn truyền thần kinh, oligosaccharid, carbohydrat, nucleotid và nucleosid,... Trong đó, phân tích các acid amin là ứng dụng phổ biến nhất của HILIC. Các acid amin nguồn gốc sinh học có trọng lượng phân tử lớn vì chúng là thành phần cơ bản của protein hoặc enzym nên chúng là một phần thiết yếu của thực vật, động vật và con người. Phân tích định lượng acid amin rất quan trọng trong nhiều lĩnh vực bao gồm kiểm nghiệm thuốc, kiểm nghiệm thực phẩm, theo dõi thay đổi thành phần môi trường trong sản xuất các chế phẩm sinh học, chẩn đoán lâm sàng và nghiên cứu y sinh. Các acid amin có tính phân cực cao và hầu hết có độ hấp thụ UV thấp. Do đó để phân tích, các chất này cần tạo dẫn xuất bằng thuốc thử thích hợp như OPA và FMOC để cải thiện khả năng lưu giữ khi tách bằng LC - RP và tăng cường độ nhạy khi phát hiện bằng detector UV hoặc detector huỳnh quang. Quá trình này tốn nhiều công sức, thời gian và có thể gây sai số. HILIC và ESI - MS là sự kết hợp hoàn hảo để phân tích các acid amin một cách trực tiếp và nhanh chóng. Sự phân tách HILIC dựa trên sự phân cực của chất phân tích, vì vậy chất phân tích càng phân cực thì khả năng lưu giữ trên cột HILIC càng mạnh.
Một ví dụ tách 14 acid amin bằng HILIC và phát hiện bằng ESI - MS của hãng Shimadzu với điều kiện sắc ký như sau: cột iHILIC - Fusion(+) (150 × 2,1 mm, 3,5 um) nhiệt độ 40°C; pha động A) acetonitril - nước - dung dịch amoni acetat 1 M, pH 5,75 (90:5:5); B) nước - acetonitril - amoni acetat 1 M, pH 5,75 (90:5:5); 0 - 0,5 min (90:10) A - B; 0,5 - 25 min, rửa giải gradient từ (90:10) A - B tới (60:40) A - B; lưu lượng: 0,3 mL/min; thể tích tiếm mẫu: 5 L. Kết quả được minh họa ở Hình 2.27 và Bảng 2.10.

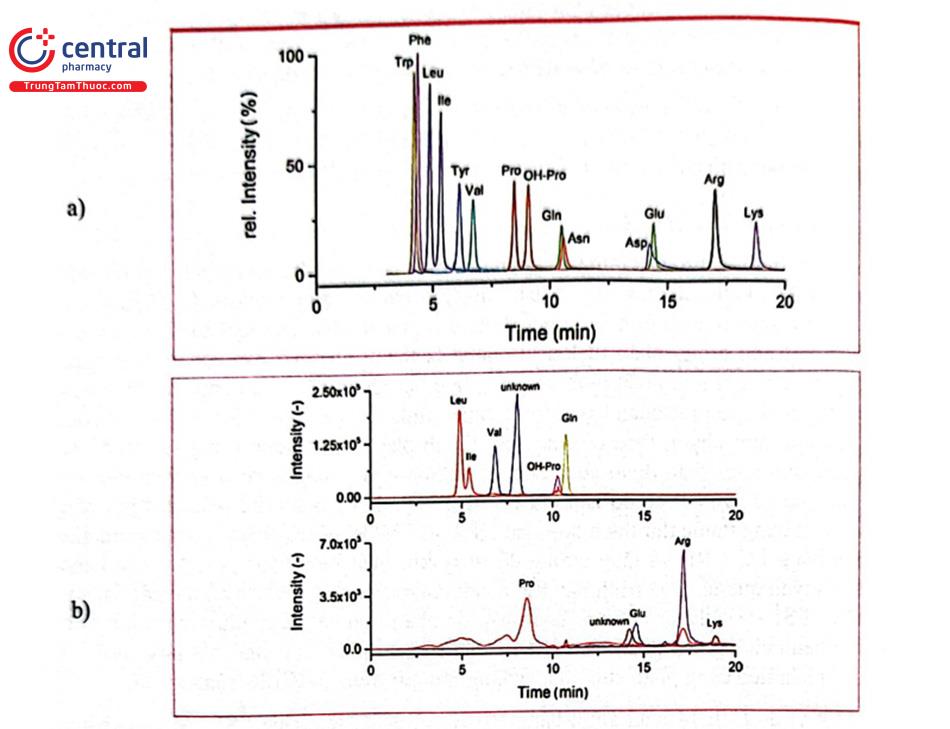
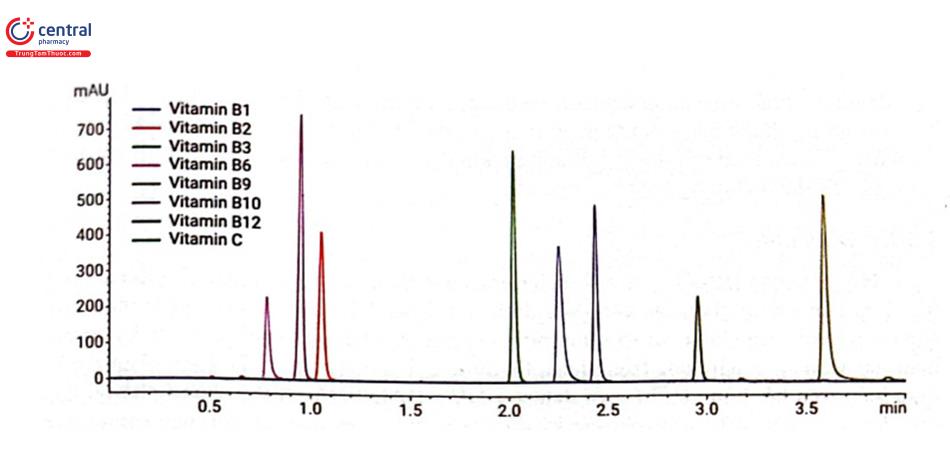
HILIC với khả năng tách tốt những chất có độ phân cực cao, đang được phát triển ứng dụng phân tích các vitamin tan trong nước. Vitamin tan trong nước rất cần thiết cho sự phát triển của cơ thể, chúng gồm các vitamin nhóm B (Bı, B, B3, B5, B6, B7, B9, B12), Biotin (vitamin H) và Vitamin C. Do đặc tính rất dễ bị ion hóa trong dung dịch nên các vitamin tan trong nước thường được phân tích bằng HPLC pha đảo tạo cặp ion vì khả năng lưu giữ kém của pha tĩnh kỵ nước. Yêu cầu của phương pháp là thêm chất hoạt động bề mặt dạng ion vào pha động của sắc ký pha đảo để tác động đến khả năng lưu giữ và tính chọn lọc của các hợp chất ion như các vitamin tan trong nước. Quá trình phân tích bằng sắc ký pha đảo tạo cặp ion sẽ phức tạp hơn, cân bằng hệ thống sắc ký chậm, thời gian rửa cột lâu và tốn kém hơn do phải sử dụng thuốc thử đắt tiền. Một công bố của hãng Agilent đã ứng dụng thành công HILIC tách 8 vitamin tan trong nước bằng cột Poroshell 120 HILIC-OH5 (2,1 × 100 mm, 2,7 km), pha động gồm A) 100 mM amoni acetat + 0,5% acid acetic B) acetonitril chạy chế độ gradient với acetonitril trong khoảng 50-87, phát hiện ở bước sóng 260 nm. (hình 2.28).
5.2.5 Ưu điểm của HILIC
HILIC có thể được coi là một kỹ thuật bổ sung cho HPLC pha đảo, do sự khác biệt về thứ tự rửa giải của các chất hòa tan và khả năng lưu giữ tốt của nó đối với các chất ưa nước, khó lưu giữ trong LC pha đảo. Ngoài ra, nó còn có những ưu điểm sau:
- Nâng cao độ nhạy khi được sử dụng kết hợp với MS. Tỷ lệ dung môi hữu cơ cao của pha động trong HILIC cho phép phun và solvat hóa hiệu quả trong MS chế độ phun điện. Độ nhạy có thể tăng tới 5 - 15 lần so với LC - MS pha đảo.
- Có thể tiêm trực tiếp dịch chiết từ cột chiết pha rắn C18, do các chất hòa tan thường được rửa giải với hỗn hợp có hàm lượng dung môi hữu cơ cao.
- Tốc độ pha động có thể tăng cao do độ nhớt của pha động thấp; độ nhớt của pha động chứa 90% acetonitril ở 20°C là 0,43 cP (trong HILIC) bằng khoảng một nửa của pha động chứa 10 - 30% acetonitril (0,90 - 0,86 cP) thường được sử dụng trong HPLC pha đảo. Ngoài ra, các pha động có độ nhớt thấp cho phép sử dụng các cột dài gấp đôi các cột pha đảo tương đương mà vẫn duy trì được áp suất ngược tương tự.
Tuy nhiên, điểm yếu của HILIC là hay gặp hiện tượng quá tải cột. Các píc bị quá tải trong HILIC thường ở dạng đổ đầu, trong khi kéo đuôi thường thu được trong LC pha đảo. Hiện tượng đổ đầu píc có thể là kết quả của tương tác giữa chất tan - chất tan trong pha tĩnh.
6 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Sắc ký lỏng hiệu năng cao”, Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 28 - 67. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược Điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
Phần nghiên cứu sau đây do Dược sĩ Hoàng Bích của Central Pharmacy biên soạn thêm, không nằm trong nội dung bài "Sắc ký lỏng hiệu năng cao là gì? 8 loại Detector thường được sử dụng" của Trường Đại học Dược Hà Nội biên soạn.
7 So sánh HPLC – UFLC – UPLC
HPLC (Sắc ký lỏng hiệu năng cao) | UFLC (Thuật ngữ của Shimadzu) | UPLC (Sắc ký lỏng hiệu năng cực cao) | |
| Tốc độ dòng chảy | 1mL/phút | Cao hơn HPLC (=2 mL/phút) | -0,6 mL/phút |
| Kích thước hạt | 3-5 μm | 2-3 μm | <2 μm |
| Áp suất | 4000 psi | 5000–6000 psi | Lên tới 15.000 psi |
| Tốc độ phân tích | Vừa phải | Nhanh | Rất nhanh |
| Độ phân giải | Tốt | Tốt | Xuất sắc |
| Giá | Vừa phải | Cao hơn một chút | Cao |
| Chuyên biệt theo nhà sản xuất | Không | Shimadzu | Waters |
