Sắc ký là gì? 3 cách phân loại và ứng dụng của sắc ký

Sắc ký có tác động lớn đến tất cả các lĩnh vực phân tích và đối với sự tiến bộ của khoa học nói chung. Sắc ký khác với các phương pháp tách khác ở chỗ có thể sử dụng nhiều loại vật liệu, thiết bị và kỹ thuật. Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc khái niệm, cách phân loại và ứng dụng của sắc ký.
1 Giới thiệu
1.1 Khái niệm
Sắc ký là một thuật ngữ chung cho nhiều kỹ thuật phân tách dựa trên sự phân bố (distribution) hoặc phân chia (partitioning) của mẫu (chất tan) giữa pha chuyển động được gọi là pha động và pha cố định được gọi là pha tĩnh. Sắc ký có thể được xem như một loạt các cân bằng giữa pha động và pha tĩnh. Tương tác tương đối của chất tan với hai pha này được mô tả bằng hằng số phân bố (K) hoặc hệ số dung lượng (k’) (tỷ số giữa nồng độ hoặc khối lượng của chất tan trong pha tĩnh và nồng độ hoặc khối lượng của chất tan trong pha động). Pha động có thể là chất khí, chất lỏng hoặc chất lỏng siêu tới hạn. Pha tĩnh chủ yếu là chất rắn, cũng có thể là chất lỏng. Sắc ký có thể được chia nhỏ theo các kỹ thuật khác nhau (Hình 1.1) hoặc theo các nguyên tắc hóa lý liên quan đến quá trình phân tách. Vì bản chất của tương tác giữa các phân tử chất tan và pha động hoặc pha tĩnh là khác nhau, các phương pháp này có khả năng phân tách các loại phân tử khác nhau.
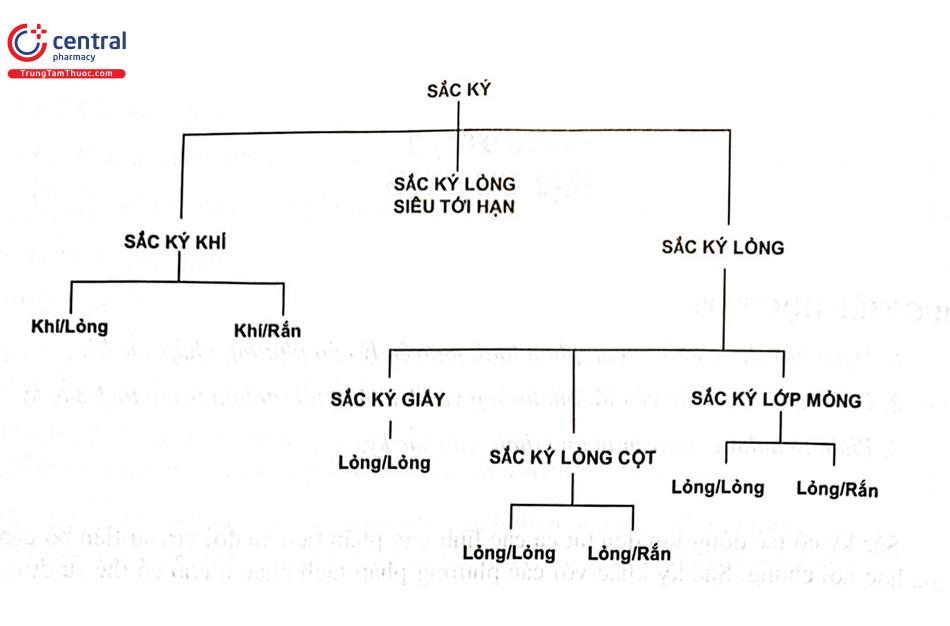
1.2 Phân loại sắc ký
1.2.1 Sắc ký khí
Sắc ký khí là kỹ thuật sắc ký cột, trong đó pha động là khí và pha tĩnh là chất lỏng cố định hoặc chất rắn được nhồi trong cột. Sắc ký khí được sử dụng để tách các thành phần dễ bay hơi, ổn định nhiệt của hỗn hợp. Sắc ký khí, cụ thể là sắc ký khí - lỏng, bao gồm các giai đoạn làm bay hơi mẫu thử và bơm nó lên đầu cột, dưới điều kiện nhiệt độ được kiểm soát, mẫu được di chuyển qua cột theo dòng của pha động khí trơ. Các chất bay hơi sau đó được tách ra dựa trên một số đặc tính như điểm sôi, kích thước phân tử và độ phân cực.
1.2.2 Sắc ký lỏng
Sắc ký lỏng chia ra làm hai loại là sắc ký phẳng gồm sắc ký giấy và sắc ký lớp mỏng; sắc ký cột là sắc ký cột cổ điển và sắc ký lỏng cao áp hay sắc ký lỏng hiệu năng cao. Tất cả các loại đều liên quan đến pha động lỏng và một chất rắn hoặc một pha tĩnh lỏng. Tuy nhiên, dạng vật lý của pha tĩnh là khá khác nhau trong mỗi trường hợp.
1.2.2.1 Sắc ký giấy
Pha tĩnh trong sắc ký giấy là nước có sẵn trong sợi celulose của giấy, hoặc thành phần thân nước từ hỗn hợp dung môi của pha động được hút chọn lọc vào giấy. Pha động là một hệ dung môi thích hợp cho sự tách đối tượng phân tích. Sự tách các chất bằng phương pháp sắc ký giấy dựa chủ yếu trên sự khác nhau về hệ số phân bố của chúng giữa hai pha lỏng. Phương pháp này được ứng dụng nhiều trong phân tích thực phẩm.
1.2.2.2 Sắc ký lớp mỏng
Pha tĩnh trong sắc ký lớp mỏng là chất hấp phụ được chọn phù hợp theo từng yêu cầu phân tích, được trải thành lớp mỏng đồng nhất và được cố định trên phiến kính hoặc phiến kim loại. Pha động là một hệ dung môi đơn hoặc đa thành phần được trộn với nhau theo tỷ lệ qui định. Trong quá trình di chuyển qua lớp hấp phụ, các cấu tử trong hỗn hợp mẫu thử được di chuyển trên lớp mỏng, theo hướng pha động, với những tốc độ khác nhau. Kết quả thu được là một sắc ký đồ trên lớp mỏng. Cơ chế của sự tách có thể là cơ chế hấp phụ, phân bố, trao đổi ion, sàng lọc phân tử hay sự phối hợp đồng thời của nhiều cơ chế tùy thuộc vào tính chất của chất làm pha tĩnh và dung môi làm pha động. Phương pháp này được ứng dụng rộng rãi trong nhiều lĩnh vực và có vai trò quan trọng trong phân tích thuốc.
1.2.2.3 Sắc ký cột
Sắc ký cột là phương pháp hữu ích nhất để tách các chất trong hỗn hợp. Sự phân đoạn các chất hòa tan xảy ra do sự di chuyển khác biệt qua một ống kín chứa pha tĩnh và các chất phân tích có thể được theo dõi trong khi quá trình phân tách đang diễn ra. Trong sắc ký lỏng cột, pha động là chất lỏng và pha tĩnh có thể là chất rắn hoặc chất lỏng được bao trên chất rắn trơ. Hệ thống sắc ký lỏng cột áp suất thấp ở áp suất khí quyển còn gọi là sắc ký cột cổ điển, còn sắc ký lỏng trên cột áp suất cao còn được gọi là sắc ký lỏng hiệu năng cao.
Quá trình pha động di chuyển qua cột được gọi là rửa giải và phần xuất hiện ở đầu ra của cột được gọi là dịch rửa giải. Rửa giải có thể là đẳng dòng hay isocratic (thành phần pha động không đổi) hoặc gradient (thay đổi thành phần, tỷ lệ, tốc độ pha động) trong quá trình rửa giải để tăng độ phân giải và giảm thời gian phân tích. Khi tiến hành rửa giải, các thành phần của mẫu được lưu giữ chọn lọc bởi pha tĩnh dựa trên cường độ tương tác với pha tĩnh, do đó chúng được rửa giải ở các thời điểm khác nhau. Dịch rửa giải ra khỏi cột có thể được đưa đến máy đo (detector) thu tín hiệu đáp ứng dùng để phân tích định tính, định lượng hoặc lấy vào bộ thu mẫu phân đoạn được đặt theo chương trình để thu dịch rửa giải vào các khoảng thời gian xác định hoặc sau khi đã thu được một thể tích nhất định.
1.2.3 Sắc ký lỏng siêu tới hạn
Sắc ký lỏng siêu tới hạn dùng để chỉ sắc ký được thực hiện trên áp suất tới hạn và nhiệt độ tới hạn của pha động. Chất lỏng siêu tới hạn không phải là chất lỏng cũng không phải là khí điển hình. Sự kết hợp của áp suất tới hạn và nhiệt độ tới hạn được gọi là điểm tới hạn. Carbon dioxid thường được sử dụng làm pha động trong sắc ký lỏng siêu tới hạn; tuy nhiên nó không phải là dung môi tốt cho các hợp chất phân cực và phân tử lượng cao. Có thể thêm một lượng nhỏ dung môi hữu cơ, phân cực như methanol vào chất lỏng siêu tới hạn không phân cực để tăng cường khả năng hòa tan của chất tan, cải thiện hình dạng píc và thay đổi độ chọn lọc.
Chất lỏng siêu tới hạn có các đặc tính sắc ký trung gian giữa sắc ký lỏng và sắc ký khí. Độ khuếch tán cao và độ nhớt thấp của chất lỏng siêu tới hạn có khả năng giảm thời gian phân tích và cải thiện độ phân giải so với sắc ký lỏng. Sắc ký lỏng siêu tới hạn cung cấp một loạt các điều chỉnh độ chọn lọc bằng những thay đổi về áp suất và nhiệt độ cũng như những thay đổi về thành phần pha động và pha tĩnh. Ngoài ra, sắc ký lỏng siêu tới hạn có thể phân tách các hợp chất không bay hơi, không bền nhiệt, không thích ứng với sắc ký khí. Nó được sử dụng chủ yếu cho các hợp chất không phân cực như chất béo, dầu, ...
1.3 Nguyên tắc hóa lý của quá trình tách sắc ký
Một số nguyên tắc hóa lý (Hình 1.2) liên quan đến cơ chế sắc ký được sử dụng để tách các hợp chất khác nhau.
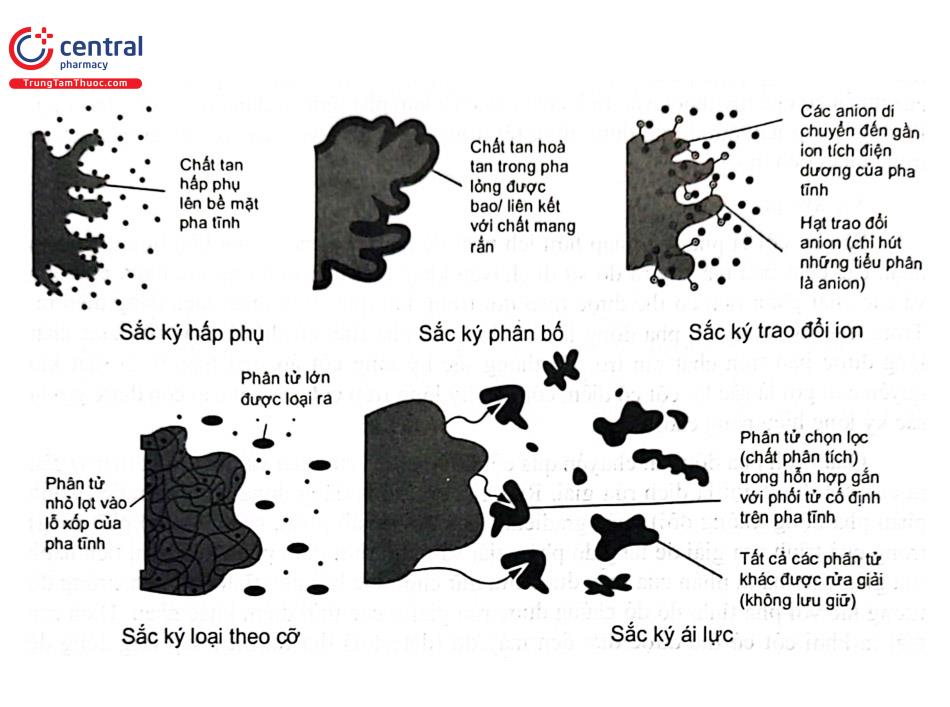
1.3.1 Sắc ký hấp phụ (Adsorption Chromatography)
Trong sắc ký hấp phụ các phân tử chất tan được liên kết trực tiếp với bề mặt của pha tĩnh. Nói một cách đơn giản, sắc ký hấp phụ có thể được giải thích là chất khí hoặc chất lỏng được hấp phụ lên bề mặt chất rắn. Pha tĩnh có nhiều vị trí hấp phụ, các vị trí hấp phụ này khác nhau về độ bền liên quan đến các phân tử mà chúng liên kết với sự đa dạng tương đối của chúng. Sắc ký hấp phụ sử dụng pha động ở trạng thái lỏng hoặc khí và pha tĩnh ở trạng thái rắn. Mỗi chất tan có sự cân bằng giữa sự hấp phụ với bề mặt chất rắn và độ hòa tan trong dung môi. Do đó, dung môi sẽ di chuyển lên theo pha động và tại điểm đạt đến trạng thái cân bằng, dung môi sẽ được hấp phụ vào pha tĩnh. Các lực liên phân tử có vai trò chính cho sự hấp phụ sắc ký bao gồm lực Van der Waals, lực tĩnh điện, liên kết hydro và tương tác kỵ nước.
1.3.2 Sắc ký phân bố (Partition Chromatography)
Trong sắc ký phân bố, pha tĩnh là chất lỏng được bao lên chất mang hoặc liên kết hóa học với chất mang rắn, chủ yếu là silica; pha động là chất lỏng có bản chất khác nhau theo qui định. Quá trình phân bố được điều khiển bằng cách thay đổi bản chất của hai pha lỏng, thường là bằng cách thay đổi thành phần dung môi hoặc điều chỉnh pH của hệ đệm. Trường hợp pha tĩnh phân cực được giữ cố định trên chất mang trơ, dung môi ít phân cực hơn được sử dụng để rửa giải các thành phần mẫu sẽ gọi là sắc ký pha thường hay pha thuận. Ngược lại, nếu sử dụng pha tĩnh không phân cực và pha động phân cực được gọi là sắc ký pha đảo.
Trong thực tế, pha tĩnh liên kết hóa học được sử dụng nhiều trong phân tích với các loại mẫu rất đa dạng. Các loại pha tĩnh thường dùng là silica được liên kết với các nhóm C8 hoặc C18 và dung môi phân cực như nước, acetonitril hoặc methanol. Điều quan trọng cần lưu ý là các cơ chế khác có thể tham gia vào việc phân tách do ảnh hưởng của các nhóm hoạt động của chất mang.
1.3.3 Sắc kí trao đổi ion (lon - Exchange Chromatography: IEC)
Sắc ký trao đổi ion dựa vào lực hút của ion chất tan và vị trí mang điện tích trên pha tĩnh. Chất trao đổi anion có nhóm mang điện tích dương trên pha tĩnh hút anion chất tan. Chất trao đổi cation có nhóm mang điện tích âm sẽ hút cation chất tan. Chất trao đổi anion và chất trao đổi cation là polymer không tan trong nước mang các nhóm trao đổi ion được gọi là chất trao đổi ion (ion exchangers) hoặc nhựa trao đổi ion (ion - exchange resins). Sự phân tách sắc ký bằng trao đổi ion dựa trên sự khác biệt về ái lực của các chất trao đổi đối với các ion (hoặc các loại tích điện) được tách ra. Các yếu tố chi phối tính chọn lọc của chất trao đổi đối với một ion cụ thể bao gồm hóa trị, bán kính và nồng độ của ion; bản chất của chất trao đổi ion; thành phần và pH của pha động.
1.3.4 Sắc ký loại theo cỡ (Size - Exclusion Chromatography: SEC)
Sắc ký loại theo cỡ, còn được gọi là loại trừ phân tử, thẩm thấu qua gel (Gel Permeation Chromatography: GPC) và sắc ký lọc gel (Gel - Filtration Chromatography: GFC), đây là phương thức sắc ký dễ thực hiện và dễ hiểu nhất. Nó được sử dụng rộng rãi trong nghiên cứu sinh học để tách các đại phân tử như protein và carbohydrat, nó cũng được sử dụng để phân đoạn và xác định đặc tính của các polyme tổng hợp.
Trong hệ thống SEC lý tưởng, các phân tử được phân tách chỉ dựa trên cơ sở kích thước của chúng; không xảy ra tương tác giữa các chất tan và pha tĩnh. Pha tĩnh trong SEC là cột nhồi vật liệu có các lỗ xốp kích thước tương đương với các phân tử được phân đoạn. Các chất tan quá lớn (kích thước lớn hơn kích thước cực đại của lỗ xốp của pha tĩnh) sẽ di chuyển theo pha động trong khoảng kẽ (giữa các hạt) bên ngoài lỗ xốp và được rửa giải. Do đó, các phân tử lớn nhất được rửa giải đầu tiên từ cột SEC. Các phân tử có kích thước nhỏ hơn kích thước nhỏ nhất của lỗ xốp sẽ khuếch tán hoàn toàn qua pha tĩnh. Các phân tử có kích thước trung gian được tách dần theo mức độ khuếch tán qua pha tĩnh theo thứ tự phân tử lớn hơn sẽ được rửa giải ra trước, phân tử nhỏ hơn sẽ ra sau.
1.3.5 Sắc ký ái lực (Affinity Chromatography)
Sắc ký ái lực là duy nhất ở chỗ sự phân tách dựa trên sự tương tác cụ thể, thuận nghịch giữa phân tử chất tan và phối tử được cố định trên pha tĩnh. Các phối tử này có thể là kháng thể, chất ức chế enzym, lectin hoặc các phân tử khác có khả năng liên kết chọn lọc và thuận nghịch với các phân tử chất phân tích trong mẫu. Phối tử được chọn dựa trên tính đặc hiệu và cường độ tương tác của nó với phần tử cần tách (chất phân tích) và được cố định trên chất mang thích hợp.
Sự phân tách khai thác sự khóa và mở của hệ thống sinh học nên sắc ký ái lực có thể được xem như là phần mở rộng của sắc ký hấp phụ được biết đến với tên gọi là hấp phụ đặc hiệu sinh học. Khi mẫu được đưa qua cột, các phân tử chất cần phân tích được hấp phụ vào phối tử cố định trên pha tĩnh, trong khi các thành phần khác được rửa giải. Chất phân tích liên kết sau đó được rửa giải thông qua sự thay đổi thành phần pha động. Phương pháp rửa giải trong sắc ký ái lực có thể được chia thành phương pháp không đặc hiệu và đặc hiệu sinh học. Rửa giải không đặc hiệu liên quan đến việc phá vỡ liên kết của phối tử bằng cách thay đổi pH của pha động, cường độ ion, hằng số điện môi hoặc nhiệt độ. Nếu muốn đạt được tính chọn lọc trong quá trình rửa giải, ví dụ, trong trường hợp pha tĩnh là các phối tử chung thì sử dụng kỹ thuật rửa giải đặc hiệu sinh học. Phối tử tự do, giống hoặc khác với phối tử của pha tĩnh, được thêm vào pha động. Phối tử tự do này cạnh tranh các vị trí liên kết trên chất phân tích. Ví dụ, glycoprotein liên kết với cột concanavalin A (lectin) có thể được rửa giải bằng cách sử dụng đệm chứa lượng lectin dư. Nói chung, phối tử rửa giải phải có ái lực lớn hơn với chất cần phân tích so với phối tử cố định của pha tĩnh.
2 Độ phân giải và tối ưu quá trình tách sắc ký
2.1 Độ phân giải
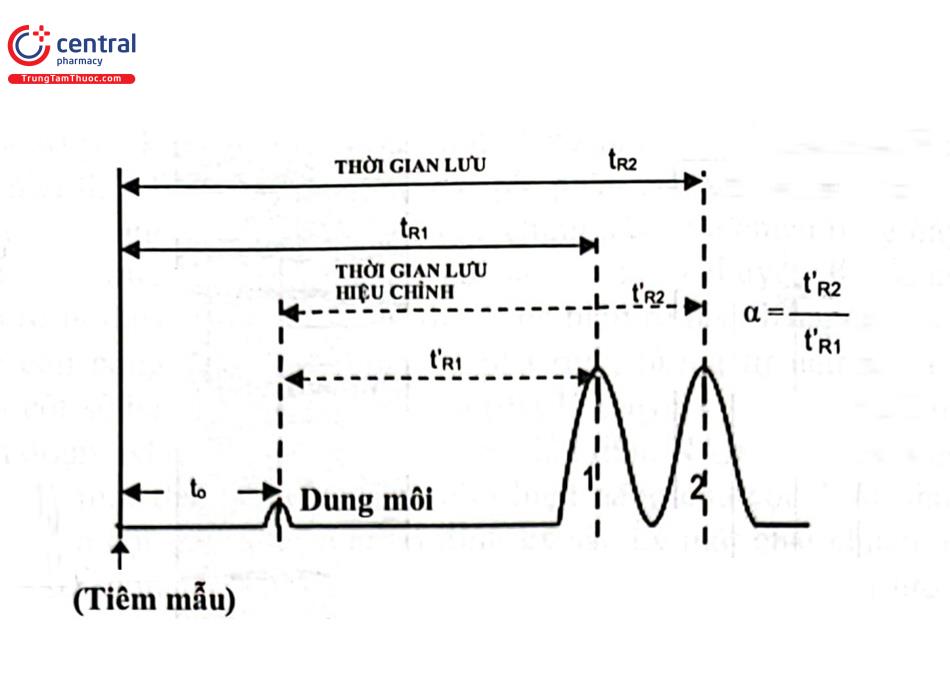
Mục tiêu chính của sắc ký là tách các thành phần của mẫu thành các dải hoặc píc riêng biệt khi chúng di chuyển qua cột. Trong sắc ký cột, một píc sắc ký được xác định bởi một số thông số bao gồm thời gian lưu, chiều rộng píc và chiều cao píc. Thể tích của pha động cần thiết để rửa giải một hợp chất từ cột sắc ký được gọi là thể tích lưu giữ, VR.
Thời gian từ khi tiêm mẫu đến khi phát hiện chất phân tích ở nồng độ cực đại là thời gian lưu, tr. Sự thay đổi thời gian lưu và chiều rộng píc ảnh hưởng lớn đến độ phân giải sắc ký. Sự khác biệt về kích thước cột, lượng mẫu, nhiệt độ, tốc độ dòng pha động, thể tích chết của hệ thống và loại detector có thể dẫn đến sự khác biệt về thời gian lưu. Bằng cách trừ đi thời gian cần thiết để pha động hoặc chất tan không lưu giữ (tm hoặc to) di chuyển qua cột đến detector được thời gian lưu hiệu chỉnh, ta như được mô tả trong Hình 1.3. Thời gian lưu hiệu chỉnh có thể được coi là thời gian mẫu được hấp phụ vào pha tĩnh.
Độ phân giải (Rs) của hai píc liền kề có liên quan đến hệ số tách, a. Giá trị của a (Hình 1.3) phụ thuộc vào nhiệt độ, pha tĩnh và pha động sử dụng. Độ phân giải Rs được tính theo công thức sau:
Rs = 2∆t / (w2 + w1) (CT 1.1)
Trong đó
- ∆t khoảng cách giữa 2 píc 1 và 2 (tR2 - tR1)
- w1 và w2 là độ rộng đáy píc 2 và píc 1 đo ở đường nền
- Thời gian lưu và độ rộng đáy píc phải được biểu thị bằng cùng một đơn vị
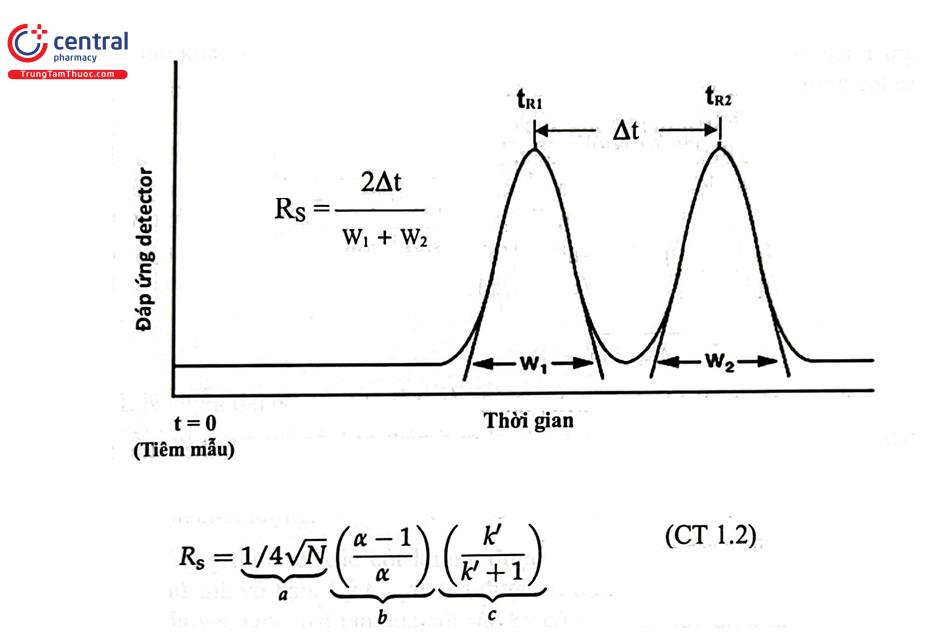
Độ phân giải sắc ký là một hàm của hiệu năng cột, độ chọn lọc và hệ số dung lượng. Về mặt toán học, mối tương quan này được biểu thị theo CT 1.2
Trong đó:
- a là thông số biểu thị cho hiệu năng của cột;
- b là thông số biểu thị cho độ chọn lọc của cột;
- c thông số biểu thị cho dung lượng cột.
Trong sắc ký phẳng, đại lượng đặc trưng cho mức độ di chuyển của chất phân tích là hệ số lưu giữ Rf (Retardation factor) hay còn gọi là thừa số chậm. Đó là tỷ số giữa khoảng cách từ điểm chấm mẫu đến tấm của vết sắc ký và khoảng cách di chuyển của dung môi tính từ điểm chấm mẫu (Hình 1.5).
Rf = b/a (CT 1.3)
Trong đó:
- b là quãng đường di chuyển của chất phân tích;
- a là quãng đường di chuyển của tuyến dung môi và
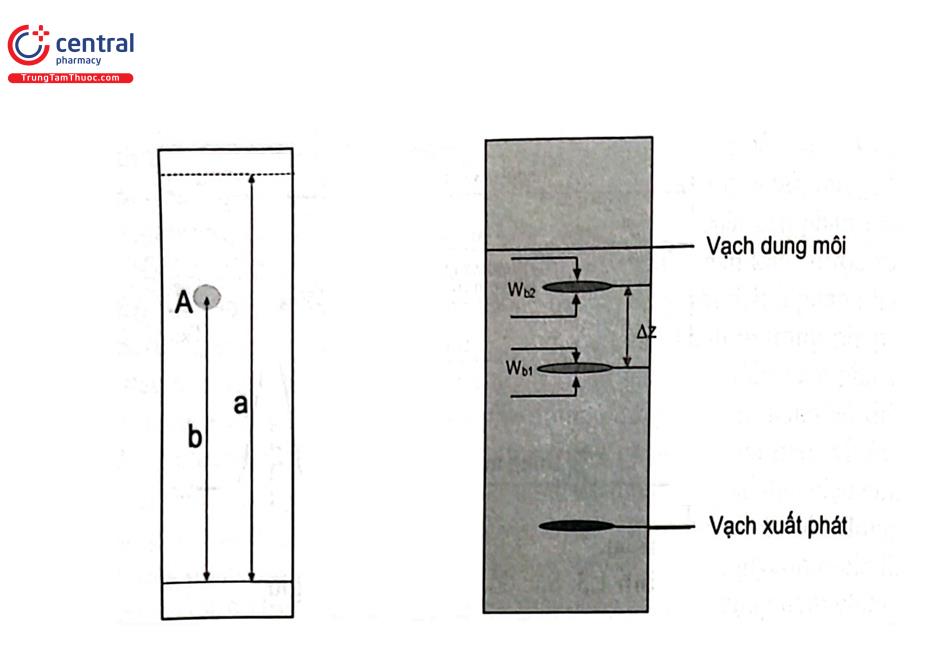
Khi sắc ký liên tục không xác định được tuyến dung môi, vị trí vết chất thử trên sắc đồ có thể xác định bằng hệ số dịch chuyển tương đối R. và được xác định bằng tỷ số giữa khoảng cách dịch chuyển của vết chất thử và khoảng cách dịch chuyển của vết chất chuẩn đối chiếu được sắc ký trong cùng điều kiện và trên cùng bản mỏng với mẫu thử.
Thông số đánh giá khả năng tách rời của 2 vết liền kề đảm bảo yêu cầu định tính, định lượng là độ phân giải Rs và được tính theo công thức sau:
Rs = 2∆z / (Wb1 + Wb2) (CT 1.4)
Trong đó:
- ∆z là khoảng cách giữa 2 vết chất phân tích;
- Wb1 và Wb2 là đường kính của vết chất phân tích theo đường di chuyển của dung môi.
- Thông thường, yêu cầu Rs ≥ 1,5; trong phép tách khó có thể giá trị Rs thấp hơn vẫn được chấp nhận miễn là phân biệt được rõ ràng chất phân tích.
2.2 Các yếu tố ảnh hưởng tới độ phân giải
2.2.1 Hiệu năng cột
Nếu gặp phải vấn đề trong quá trình tách sắc ký cần phải cải thiện độ phân giải, đầu tiên người phân tích phải kiểm tra hiệu quả của cột tách. Một cột có hiệu năng cao sẽ tách các píc dạng dải hẹp. Hiệu năng cột được biểu thị bằng số đĩa lý thuyết N có thể được tính theo công thức sau:
N = (tR/ꝍ)2 = 16(tR/w)2 = 5,54(tR/w1/2)2 (CT 1.5)
Trong đó
- tR là thời gian lưu
- ꝍ là độ lệch chuẩn cho một píc dạng phân bố chuẩn Gaussian;
- w là chiều rộng đáy píc (w = 4ꝍ);
- w1/2 là chiều rộng píc đo ở nửa chiều cao píc.
Mặc dù một số píc không thực sự có hình dạng phân bố chuẩn Gaussian, nhưng thực tế vẫn xử lý như thế. Trong trường hợp các píc phân giải không hoàn toàn hoặc hơi không đối xứng, chiều rộng píc ở nửa chiều cao chính xác hơn chiều rộng đáy píc. Giá trị N tính được từ phương trình trên được gọi là số đĩa lý thuyết. Khái niệm địa lý thuyết, bắt nguồn từ lý thuyết chưng cất có thể được hiểu rõ nhất bằng cách xem sắc kỷ là một chuỗi các cân bằng giữa pha động và pha tĩnh, tương tự như phân bố ngược dòng. Do đó, một cột sẽ bao gồm N phân đoạn (đĩa lý thuyết) với một sự cân bằng xảy ra trong mỗi phân đoạn. Như một giá trị gần đúng đầu tiên, N không phụ thuộc vào thời gian lưu và do đó là một thước đo hữu ích cho hiệu năng của cột. Một phương pháp theo dõi hiệu năng của cột theo thời gian là định kỳ sắc ký một chất chuẩn, trong điều kiện không đổi, và so sánh các giá trị N thu được. Điều quan trọng cần lưu ý là cùng một cột tách nhưng nó có số đĩa khác nhau đối với các chất tan khác nhau trong hỗn khác nhau có nhau và do đó có chuỗi cân bằng riêng biệt giữa pha động và pha tĩnh. Việc mở rộng dải do sự suy giảm chất lượng cột sẽ dẫn đến giảm N đối với một chất tan cụ thể. Sự mở rộng dải là kết quả của một thời gian kéo dài để chất tan đạt đến trạng thái cân bằng giữa pha động và pha tĩnh.
Số lượng đĩa lý thuyết nói chung tỷ lệ thuận với chiều dài cột. Vì các cột có sẵn với nhiều độ dài khác nhau, rất hữu ích khi có một thước đo hiệu quả của cột mà không phụ thuộc vào độ dài của cột. Thông số chiều cao tương đương với đĩa lý thuyết (Height Equivalent to a Theoretical Plate: HETP ) được sử dụng và được tính như sau:
HETP = L/N (CT 1.6)
Ở đây, L là chiều dài cột
HETP đôi khi được mô tả đơn giản hơn là chiều cao đĩa lý thuyết (H). Nếu một cột bao gồm các đoạn rời rạc, HETP sẽ là chiều cao của mỗi đoạn tưởng tượng. chiều cao đĩa lý thuyết nhỏ (số đĩa lý thuyết lớn) cho thấy hiệu quả tách tốt. Ngược lại, số đĩa giảm đi dẫn đến sự phân tách kém do thời gian cân bằng kéo dài trong một cột bị suy giảm chất lượng.
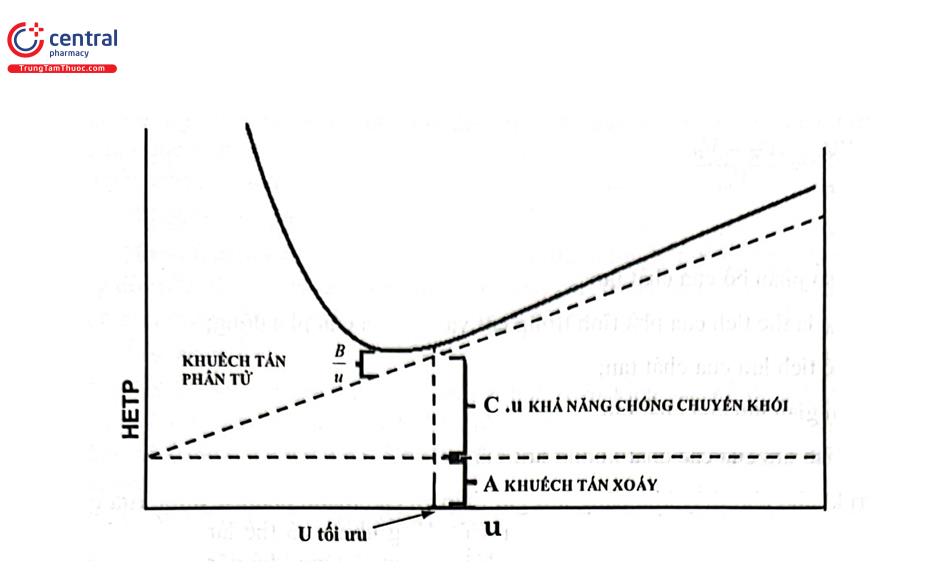
Trong thực tế, các cột không được chia thành các đoạn rời rạc và sự cân bằng không nhanh vô hạn. Lý thuyết đĩa được sử dụng để đơn giản hóa khái niệm cân bằng. Sự di chuyển của chất tan qua cột sắc ký có tính đến tốc độ hữu hạn mà tại đó chất tan có thể tự cân bằng giữa pha tĩnh và pha động. Do đó, hình dạng dải phụ thuộc vào tốc độ rửa giải và bị ảnh hưởng bởi sự khuếch tán chất tan. Bất kỳ cơ chế nào khiến dải chất tan mở rộng sẽ làm tăng HETP và giảm hiệu năng cột. Các yếu tố khác nhau ảnh hưởng tới chiều cao đĩa lý thuyết biểu thị bằng phương trình Van Deemter:
HETP = A + B/u + Cu (CT 1.7)
Trong đó:
- A, B, C là các hằng số
- u là vận tốc tuyến tính dòng pha động
u chính là tốc độ pha động di chuyển dọc theo cột và được tính theo công thức sau:
u = 66,67.F / (dc2πεT) (CT 1.8)
Trong đó:
- F lưu lượng dòng pha động
- dc là đường kính trong của cột (mm)
- εT là độ xốp tổng của các hạt nhồi trong cột (cột chứa hạt pha tĩnh hình cầu, xốp, giá trị εT ~ 0,7).
Các hằng số A, B và C đặc trưng cho cột, pha động và nhiệt độ nhất định. Còn u có thể tính đơn giản u = L/to.
Hằng số A đại diện cho sự khuếch tán xoáy hoặc nhiều đường dẫn dòng pha động. Khuếch tán xoáy đề cập đến các dòng chảy vi mô khác nhau mà pha động có thể di chuyển giữa các hạt nhồi trong cột (tương tự như dòng xoáy xung quanh hòn đá trong một con suối). Do đó, các phân tử mẫu cũng có thể đi theo các con đường khác nhau, tùy thuộc vào dòng pha động mà chúng đi theo. Kết quả là, các phân tử chất tan lan truyền từ một vùng ban đầu hẹp đến một vùng rộng hơn trong cột. Sự khuếch tán xoáy có thể được giảm thiểu bằng các kỹ thuật nhồi cột tốt và sử dụng các hạt có đường kính nhỏ, đồng nhất.
Hằng số B của phương trình Van Deemter, biểu thị cho sự khuếch tán dọc, nó xuất hiện bởi vì tất cả các chất tan đều có khả năng khuếch tán từ vùng có nồng độ cao (trung tâm của dải sắc ký) đến vùng có nồng độ thấp (cạnh đầu hoặc cạnh sau của một dải sắc ký). Trong sắc ký lỏng, ảnh hưởng của B đối với HETP là nhỏ, trừ khi tốc độ dòng pha động thấp. Với tốc độ dòng pha động thấp chất tan sẽ lưu giữ trên cột lâu hơn, do đó độ khuếch tán của nó sẽ lớn hơn.
Hằng số C (chuyển khối) phát sinh từ thời gian hữu hạn cần thiết để chất tan cân bằng giữa pha động và pha tĩnh. Sự chuyển khối thực tế là sự phân bố của chất tan vào pha tĩnh, không xảy ra ngay lập tức và phụ thuộc vào sự phân bố của chất tan và hệ số khuếch tán. Nếu pha tĩnh là các hạt xốp, phân tử mẫu đi vào lỗ rỗng sẽ không còn được dòng dung môi vận chuyển và chỉ di chuyển bằng cách khuếch tán. Sau đó, phân tử chất tan này có thể khuếch tán trở lại dòng pha động hoặc nó có thể tương tác với pha tĩnh. Trong cả hai trường hợp, các phân tử chất tan bên trong lỗ xốp sẽ di chuyển chậm lại so với các phân tử bên ngoài lỗ xốp và xảy ra hiện tượng mở rộng vùng mẫu. Ảnh hưởng tới HETP của thông số C có thể được giảm thiểu bằng cách sử dụng các hạt xốp có đường kính nhỏ hoặc pha tĩnh nguyên khối.
Tốc độ dòng pha động, u, ảnh hưởng tới HETP theo những cách đối lập - tăng tốc độ dòng làm tăng thời gian cân bằng (Cu), nhưng giảm sự khuếch tán dọc của các tiểu phân chất tan (B /u). Biểu đồ Van Deemter (Hình 1.6) có thể được sử dụng để xác định tốc độ dòng pha động tại đó HETP là tối thiểu và hiệu năng cột sẽ là tối đa. Tốc độ dòng pha động có thể đặt trên mức tối ưu để giảm thời gian phân tích nếu vẫn đạt được độ phân giải thích hợp. Tuy nhiên, khi tốc độ pha động rất cao, sẽ có ít thời gian hơn để đạt đến trạng thái cân bằng, điều này sẽ dẫn đến việc mở rộng dải.
Ngoài tốc độ dòng, nhiệt độ có thể ảnh hưởng đến sự khuếch tán theo chiều dọc và sự chuyển khối. Nhiệt độ tăng sẽ làm tăng chuyển động của chất tan giữa pha động và pha tĩnh trong cột, do đó dẫn đến rửa giải nhanh hơn và các píc thu hẹp hơn.
2.2.2 Độ chọn lọc của cột (Hệ số tách a)
Độ phân giải sắc ký phụ thuộc vào độ chọn lọc của cột cũng như hiệu năng cột. Độ chọn lọc của cột đề cập đến khoảng cách, hoặc khoảng cách tương đối, giữa hai píc và được tính theo công thức sau:
a = ( tR2 - to) / (tR1 - to) = t'R2 / t'R1 = K2 / K1 (CT 1.9)
Trong đó:
- tR1 và tR2 là thời gian lưu của các píc (chất) 1 và 2, tương ứng;
- to (hoặc tm) thời gian lưu của các chất không bị lưu giữ;
- t'R1 và t'R2 là thời gian lưu hiệu chỉnh của các chất 1 và 2 tương ứng;
- K1 và K2 hệ số phân bố của các chất 1 và 2 tương ứng.
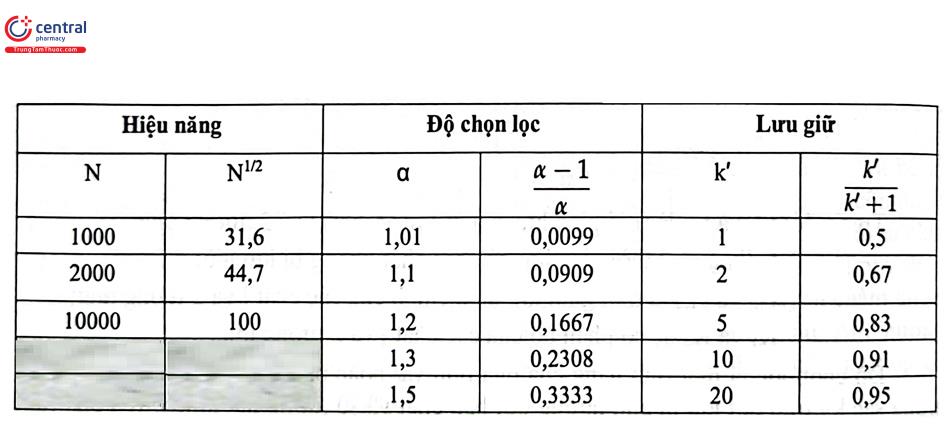
Độ chọn lọc là một tính năng của pha tĩnh và/ hoặc pha động. Ví dụ, độ chọn lọc trong sắc ký trao đổi ion bị ảnh hưởng bởi bản chất và số lượng nhóm ion trên pha tĩnh nhưng cũng có thể được điều chỉnh thông qua pH và cường độ ion của pha động. Tính chọn lọc tốt có lẽ quan trọng hơn đối với một sự phân tách nhất định hơn là hiệu năng cao, vì độ phân giải liên quan trực tiếp đến độ chọn lọc nhưng liên quan bậc hai với hiệu năng; do đó cần tăng N gấp bốn lần để tăng gấp đôi Rs.
2.2.3 Hệ số dung lượng cột
Khả năng lưu giữ hoặc hệ số dung lượng, ký, là thước đo khoảng thời gian mà một chất tan ở trong/ trên pha tĩnh so với pha động. Mối quan hệ giữa các yếu tố dung lượng và khả năng lưu giữ sắc ký (có thể được biểu thị bằng đơn vị thể tích hoặc thời gian) được trình bày dưới đây:
k' = KVs/Vm = (VR-Vm)/ Vm = (tR - to)/to (CT 1.10)
Trong đó:
- K là hệ số phân bố của chất tan
- Vs và Vm là thể tích của pha tĩnh trong cột và thể tích của pha động;
- VR là thể tích lưu của chất tan;
- tR là thời gian lưu của chất tan
- to thời gian lưu của các chất không lưu giữ.
Giá trị k' nhỏ cho thấy khả năng lưu giữ thấp và các thành phần sẽ được rửa giải gần với dung môi, dẫn đến sự phân tách kém. Cột dùng nhiều có thể làm mất một số nhóm hoạt động, do đó dẫn đến giá trị kí nhỏ. Nếu k′ lớn sẽ tăng khả năng tách nhưng có thể gây ra sự mở rộng dải và thời gian phân tích dài. Trên cơ sở thực tế, các giá trị k' yêu cầu nằm trong khoảng từ 1 - 10.
2.3 Tối ưu hóa độ phân giải
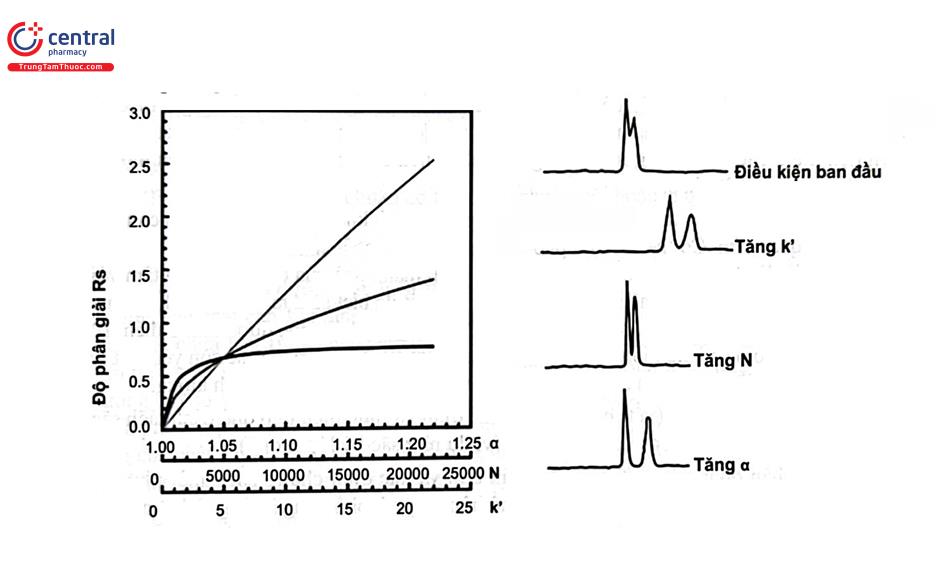
Như đã đề cập ở trên, độ phân giải chịu ảnh hưởng của 3 yếu tố chính là hiệu năng cột (N), độ chọn lọc (a) và hệ số dung lượng (k). Để thu được độ phân giải đạt yêu cầu của phép phân tích cần tác động vào các thông số này. Bảng 1.1 trình bày ví dụ đánh giá khả năng thay đổi độ phân giải khi tác động vào các yếu tố ảnh hưởng.
Hiệu năng cột
Ta có:
N=L/H (CT 1.11)
Để tăng N có thể tăng L và/ hoặc giảm H. Có thể tăng N bằng cách dùng cột dài hơn, còn giảm H bằng cách sử dụng pha tĩnh có đường kính hạt nhỏ, đồng đều và/ hoặc giảm đường kính trong của cột.
Tuy nhiên, độ phân giải chỉ tăng theo tỷ lệ căn bậc 2 của L (Rs = L1/2). Nếu tăng chiều dài cột sẽ tăng thời gian phân tích, tăng áp suất đầu cột, píc sẽ giãn rộng hơn. Còn giảm đường kính hạt pha tĩnh, tăng độ đồng đều của hạt nhồi liên quan đến công nghệ chế tạo pha tĩnh; đồng thời giảm đường kính trong của cột cũng sẽ tăng áp suất làm việc nên yêu cầu cao hơn đối với bơm cao áp.
Độ chọn lọc (Hệ số tách)
Hệ số tách thay đổi nhỏ cũng có thể cải thiện tốt độ phân giải. Đối với sắc ký lỏng thay đổi pha tĩnh hoặc thay đổi pH, chất tạo cặp trong pha động sẽ thay đổi rõ rệt độ chọn lọc; còn đối với sắc ký khí có thể thay đổi nhiệt độ hoặc thay đổi pha tĩnh.
Khả năng lưu giữ
Trong thực nghiệm, thường điều chỉnh điều kiện sắc ký để giá trị k′ nằm trong khoảng 2 - 5. Tuy nhiên, có thể chấp nhận với k' nhỏ hơn 10 bởi vì giá trị k′ tác động không nhiều tới độ phân giải. Để tăng độ phân giải bằng cách tăng kí thì sẽ kéo dài thời gian phân tích rất nhiều.
3 Ứng dụng
Sắc ký là phương pháp phân tích được ứng dụng nhiều nhất trong các phòng phân tích hóa lý. Nó được sử dụng trong nhiều lĩnh vực như dược phẩm, thực phẩm, môi trường, sinh học,... Các phép thử trong phân tích thực hiện bằng sắc ký bao gồm định tính, định lượng, xác định tạp chất và điều chế.
3.1 Định tính
Khi điều kiện tách và độ phân giải đã được tối ưu hóa, có thể định tính các chất trong hỗn hợp bằng sắc ký. So sánh thời gian lưu của chất phân tích của mẫu thử và mẫu chuẩn được sắc ký song song, trong các điều kiện giống nhau cho phép xác định sự có mặt của một chất chưa biết. Khi cần so sánh sắc ký đồ thu được từ hai hệ thống sắc ký hoặc cột tách khác nhau, tốt hơn là so sánh thời gian lưu hiệu chỉnh.
Tuy nhiên, các chất khác nhau có thể có thời gian lưu giống nhau. Nói cách khác, ngay cả khi thời gian lưu của chất chưa biết và chất chuẩn là tương đương nhau thì hai chất đó có thể không giống nhau. Do đó, cần bổ sung thêm các kỹ thuật khác để xác nhận píc như:
- Thêm vào mẫu chưa biết một chất đã biết (thường dùng chất chuẩn) và so sánh sắc đồ của mẫu ban đầu và mẫu đã được thêm chuẩn để xem píc nào đã tăng lên. Chỉ chiều cao của píc quan tâm mới được tăng lên mà không thay đổi về thời gian lưu, chiều rộng hoặc hình dạng của píc.
- Sử dụng detector mảng diod có thể cung cấp phổ hấp thụ của các píc cần xác định. Mặc dù các phổ giống hệt nhau chưa đủ khẳng định về định tính, nhưng sự khác biệt về phổ xác nhận rằng các píc của mẫu thử và mẫu chuẩn là các chất khác nhau.
- Trong trường hợp không có thiết bị có khả năng quét phổ, có thể sử dụng các detector khác như detector đo độ hấp thụ hoặc huỳnh quang và có thể đánh giá theo tỷ lệ. Sắc ký đồ của mẫu thử và chất chuẩn được theo dõi ở hai bước sóng khác nhau. Tỷ lệ diện tích píc ở các bước sóng này phải giống nhau nếu chất phân tích trong mẫu thử và chất chuẩn giống hệt nhau.
- Sử dụng detector khối phổ có thể cung cấp các mảnh khối của ion mẹ và ion con của các píc cần quan tâm. Có thể so sánh phổ đồ thu được của chất cần quan tâm với thư viện phổ được thiết lập trong cùng điều kiện. Hoặc so sánh mảnh phổ của chất cần quan tâm với chất chuẩn làm song song. Thông thường, định tính chất đã biết sẽ dựa vào 1 mảnh ion mẹ và 2 mảnh ion con và yêu cầu phải đạt 4 điểm IP. Trong đó, 1 ion mẹ được tính 1 điểm IP (Identification Point) và mỗi ion con được tính 1,5 điểm IP. Ngoài ra, có thể còn yêu cầu tỷ lệ cường độ ion giữa mảnh ion dùng để định tính và mảnh ion dùng trong định lượng.
- Các chất quan tâm (vết) có thể được xác định và thực hiện tiếp quá trình tách sắc ký bằng cách sử dụng một chế độ tách khác (ví dụ như sắc ký lớp mỏng).
- Thu phân đoạn ứng với các píc quan tâm và xác định chúng bằng một phương pháp phân tích khác (ví dụ như khối phổ, phổ hồng ngoại, phổ cộng hưởng từ hạt nhân).
Cũng có thể định tính chất phân tích dựa vào thời gian lưu tương đối bằng cách so sánh tỷ số thời gian lưu của píc chất phân tích và thời gian lưu píc của một chất đã biết (chất chuẩn hoặc chất chuẩn nội) trên sắc ký đồ với tỷ số thời gian lưu của chất phân tích và chất chuẩn hoặc chất chuẩn nội đã xác lập được. Nếu tỷ số trùng nhau thì được coi là dương tính.
3.2 Định lượng
Sắc ký là phương pháp tách nên có thể định lượng các chất trong hỗn hợp bằng các cách sau đây:
3.2.1 Phương pháp chuẩn ngoại
Nồng độ các thành phần cần phân tích được xác định bằng cách so sánh (các) đáp ứng píc của Dung dịch thử với đáp ứng píc của chất đối chiếu làm trong cùng điều kiện. Thông thường nồng độ chất phân tích trong mẫu thử và mẫu chuẩn xấp xỉ bằng nhau.
3.2.2 Phương pháp chuẩn nội
Chất chuẩn nội phải là một chất có thể tách khỏi chất cần định lượng, có tính chất tương tự chất phân tích. Thêm cùng lượng chuẩn nội vào dung dịch thử và dung dịch chất đối chiếu. Chất chuẩn nội không được có phản ứng với chất cần phân tích, phải bền và không chứa tạp có thời gian lưu tương tự thời gian lưu chất phân tích. Nồng độ chất phân tích được xác định dựa vào việc so sánh tỷ số diện tích píc (hay chiều cao píc) giữa chất phân tích và chuẩn nội trong dung dịch thử với tỷ số diện tích píc (hay chiều cao píc) giữa chất phân tích và chuẩn nội trong dung dịch đối chiếu.
3.2.3 Phương pháp chuẩn hoá diện tích
Hàm lượng phần trăm của một hay nhiều thành phần trong mẫu thử được tính bằng cách xác định diện tích một píc hay nhiều píc dưới dạng % so với tổng diện tích tất cả các píc trừ các píc của dung môi hay thuốc thử và các píc ở dưới mức có thể bỏ qua.
3.2.4 Phương pháp đường chuẩn
Xác lập mối quan hệ giữa đáp ứng đo được như diện tích píc hoặc tỷ lệ diện tích píc (y) và lượng (nồng độ, khối lượng) của chất phân tích (x) dựa trên chất chuẩn, rồi xây dựng phương trình đường chuẩn. Tính kết quả phân tích từ đáp ứng của chất phân tích dựa vào đường chuẩn.
Phương pháp đường chuẩn có thể dùng chuẩn nội hoặc chuẩn ngoại.
3.3 Xác định tạp chất
Xác định tạp được thực hiện trong kiểm nghiệm thuốc dạng nguyên liệu hoặc Xác định tạp được thực hiện trong kiểm nghiệm phẩm. Tạp chất cần xác định có thể là tạp chất đã biết hay còn gọi là tạp định danh hoặc tạp chất chưa biết (unknown impurities). Tạp định danh được xác định dựa vào so sánh với thời gian lưu của píc tạp chuẩn làm trong cùng điều kiện hoặc dựa vào thời gian lưu tương đối so với chất chính (thời gian lưu tương đối được thiết lập trong quá trình thẩm định hương pháp phân tích dùng tạp chuẩn). Khi tính kết quả hàm lượng tạp dựa vào đáp ứng của chất chính, cũng có thể phải dùng hệ số hiệu chỉnh do đáp ứng của chất chính và tạp khác nhau. Để định lượng tạp bằng sắc ký có thể thực hiện theo các cách như sau:
Chuẩn hóa diện tích
Hàm lượng của mỗi tạp được xác định bằng phương pháp chuẩn hóa diện tích
Nếu không có qui định riêng, khi tạp chất và hoạt chất có đáp ứng tương tự nhau với detector (± 20%) thì trong tính toán có thể chấp nhận hệ số đáp ứng bằng 1. Nếu đáp ứng khác nhau nhiều thì phải nhân thêm hệ số đáp ứng khi tính toán.
Pha loãng dung dịch thử
Thông thường, yêu cầu phép phân tích phải phát hiện được píc trong sắc ký đồ do có thể có sự khác nhau về độ nhạy của phương pháp khi tiến hành ở các phòng thí nghiệm khác nhau, của các người phân tích khác nhau. Do đó, thường so sánh píc thu được của dung dịch mẫu thử với dung dịch pha loãng của chất kiểm tra. Mẫu thử được chuẩn bị ở mức nồng độ cao, mẫu đối chiếu sẽ là pha loãng mẫu thử đến mức giới hạn (mẫu đối chiếu cũng có thể là mẫu pha từ chất chuẩn ở mức nồng độ giới hạn). Mẫu thử được phân tích ở nồng độ cao nhằm phát hiện tất cả các tạp. Mẫu đối chiếu ở nồng độ thấp nhằm đảm bảo độ tuyến tính của hoạt chất. Đáp ứng của hoạt chất có trong mẫu có nồng độ thấp tương tự tạp chất liên quan có nồng độ cao. Do đó, chỉ cần một khoảng tuyến tính hẹp để định lượng.
Sử dụng chất chuẩn
Chất chuẩn tạp chất được sử dụng như một chất đã biết để so sánh với tạp chất trong mẫu thử. Trong phép thử, nồng độ các tạp chất liên quan được xác định dựa trên đáp ứng píc (diện tích píc) tạp và đáp ứng của chuẩn tạp tương ứng. Thông thường, nồng độ dung dịch chuẩn tạp được chuẩn bị ở mức giới hạn cho phép của tạp đó.
Khi yêu cầu phép thử các tạp chất liên quan là tổng các tạp chất hay khi phải định lượng một tạp chất thì điều quan trọng là phải chọn một ngưỡng thích hợp và các điều kiện thích hợp cho việc lấy tích phân các diện tích píc. Trong các phép thử như vậy thì ngưỡng bỏ qua (nghĩa là các diện tích píc dưới ngưỡng này không được tính đến) thường là 0,05%. Như thế, ngưỡng đặt cho hệ thống thu thập dữ liệu ít nhất phải là nửa ngưỡng bỏ qua. Việc lấy tích phân các diện tích píc của các tạp không tách được hoàn toàn khỏi píc chính nên được thực hiện theo phương pháp ngoại suy từ hõm tới hõm (vạch tiếp tuyến).
3.4 Điều chế
Ngoài ứng dụng trong phân tích, sắc ký còn được ứng dụng trong điều chế để phân lập và tinh chế các cấu tử từ hỗn hợp ban đầu. Các chất thu được của sắc ký điều chế có thể sử dụng để làm chất chuẩn, làm thuốc hoặc tiếp tục thực hiện các phép phân tích khác tùy thuộc phương pháp, qui mô và mục đích của thử nghiệm. Đối với phân tích sắc ký các tiêu chí như độ nhạy, độ chính xác của phương pháp là các chỉ tiêu đánh giá hiệu năng của phương pháp thì độ tinh khiết của sản phẩm, tỷ lệ thu hồi dung môi cao, tiêu hao dung môi tối thiểu là các tiêu chí đánh giá hiệu quả của phương pháp sắc ký điều chế. Có thể thực hiện sắc ký điều chế theo phương pháp sắc ký lớp mỏng điều chế; sắc ký cột điều chế và sắc ký lỏng hiệu năng cao điều chế.
4 Kết luận
Sắc ký là một phương pháp phân tách dựa trên sự phân chia chất tan giữa pha động và pha tĩnh. Pha động có thể là chất lỏng, khí hoặc chất lỏng siêu tới hạn. Pha tĩnh có thể là chất lỏng hoặc chất rắn cố định, ở dạng phẳng hoặc dạng cột. Dựa trên các đặc điểm hóa lý của chất phân tích và sự sẵn có của thiết bị đo, một hệ thống sắc ký được chọn để tách, xác định và định lượng chất phân tích. Các chế độ sắc ký bao gồm hấp phụ, phân bố, trao đổi ion, loại theo cỡ và sắc ký ái lực. Các yếu tố cần được xem xét khi phát triển sự phân tách bao gồm các biến số của pha động (cường độ, pH, nhiệt độ và tốc độ dòng pha động) và hiệu năng cột, độ chọn lọc và dung lượng cột. Sau khi phát hiện, sắc ký đồ cung cấp cả thông tin định tính và định lượng thông qua giá trị thời gian lưu và dữ liệu diện tích hoặc chiều cao píc. Sắc ký là phương pháp được sử dụng ngày càng nhiều và đóng vai trò quan trọng trong nhiều lĩnh vực như kiểm nghiệm thuốc, kiểm nghiệm thực phẩm, phân tích môi trường, phân tích dịch sinh học,...
5 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Đại cương”, Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 11 - 27. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược Điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
