Sắc ký khí là gì? 14 loại detector sử dụng trong sắc ký khí

Sắc ký khí (Gas Chromatography: GC) là kỹ thuật tách các hợp chất ở trạng thái khí, được ứng dụng trong phân tích những hợp chất dễ bay hơi và bền nhiệt. Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc thông tin về kỹ thuật sắc ký khí.
1 Sắc ký khí là gì?
1.1 Khái niệm
Sắc ký khí (Gas Chromatography: GC) là kỹ thuật tách các hợp chất ở trạng thái khí, được ứng dụng trong phân tích những hợp chất dễ bay hơi và bền nhiệt. Pha động trong GC là chất khí. Pha tĩnh có thể là chất rắn hoặc chất lỏng phủ trên một chất mang rắn trơ tạo nên hai dạng sắc ký khí tương ứng là sắc ký khí rắn (GSC) và sắc ký khí lỏng (GLC). Chất phân tích được pha động đưa vào cột dưới dạng hơi. Trong cột, xảy ra quá trình tương tác giữa chất phân tích trong pha động (hơi) với pha tĩnh. Thành phần hòa tan nhiều hơn trong pha tĩnh di chuyển chậm hơn trong cột, ngược lại thành phần ít hòa tan hơn trong pha tĩnh di chuyển nhanh hơn. Do đó, các thành phần có trong hỗn hợp mẫu được phân tách theo hệ số phân chia của chúng giữa thành phần trong mẫu và pha tĩnh. Do các chất phân tích khác nhau về tính chất hóa lý, chúng được tách ra khỏi nhau.
1.2 Khí mang (Carrier gas)
Khí mang đóng vai trò là pha động trong GC. Các loại khí mang thường được sử dụng gồm có nitơ, argon, heli, hydro. Khí mang cần phải có độ tinh khiết cao, phải trơ về mặt hóa học, phù hợp với detector được sử dụng và không được gây cháy nổ. Trong các khí mang nói trên, nitơ và heli là hai loại khí mang được sử dụng phổ biến hơn cả.
Khí mang trong GC cần được kiểm soát về áp suất và tốc độ dòng khí. Áp suất khí mang được kiểm soát thông qua van điều áp. Van điều áp thường có hai cấp, một dùng để kiểm soát áp suất trong bình khí và một dùng để điều chỉnh áp suất đầu ra. Ngoài ra, trước khi vào cột sắc ký khí, khí mang phải qua bộ phận điều chỉnh tốc độ. Độ chính xác của tốc độ dòng là yếu tố quan trọng để đảm bảo độ lặp lại của kết quả.
Khí mang được sử dụng trong sắc ký khí có độ tinh khiết cao, tuy nhiên vẫn có lẫn các khí tạp khác như hơi nước, hydrocarbon, oxy,... Những khí này nếu lượng nhiều đi vào trong hệ thống cột sẽ gây giảm tuổi thọ của cột, làm giảm độ nhạy của các detector và có thể ảnh hưởng đến quá trình phân tích. Do đó, trước khi vào cột, thường có các bộ phận giữ các khí trên lại, còn gọi là bẫy khí.
Yêu cầu đối với khí mang trong GC là phải trơ về hóa học, có khả năng giảm thiểu sự khuếch tán khí và tinh khiết. Độ nhớt của khí mang là thông số cần lưu tâm, đặc biệt đối với cột dài vì nó ảnh hưởng tới độ chênh lệch áp suất đầu và cuối cột tách (∆p).
∆p = (n/Bo) L x u
Trong đó:
- Bo là độ thẩm thấu riêng của khí mang (cm2)
- n là độ nhớt của khí mang
- L chiều dài cột
- u là tốc độ tuyến tính của pha động (cm/s)
Bảng 4.1. Một số đặc điểm của khí mang dùng trong sắc ký khí
| Khí | He | H2 | N2 | Ar |
| M (g/mol) | 4,003 | 2,016 | 28,01 | 39,95 |
| Độ nhơt n ở 0 độ C (uPa.s) | 18,69 | 8,362 | 16,84 | 21,35 |
| Hệ số tự khuếch tán Dg ở 1 atm và 0 độ C (cm2/s) | 1,26 | 1,12 | 0,162 | 0,144 |
1.3 Cột sắc ký và pha tĩnh
Cột sắc ký đóng vai trò rất quan trọng, quá trình tách các chất xảy ra toàn bộ trên cột sắc ký. Trong GC, có hai loại cột chủ yếu được sử dụng là cột nhồi và cột mao quản. Trong mao quản là loại cột được sử dụng phổ biến hiện nay, trong đó loại cột có pha tĩnh là chất lỏng bao lên thành mao quản (Wall Coated Open Tubular WCOT) sử dụng nhiều nhất. Sau đây sẽ trình bày một số loại pha tĩnh thông dụng loại WCOT.
1.3.1 Poly (dimethylsiloxan)
Pha tĩnh quan trọng nhất là poly (dimethylsiloxan) - PDMS, còn gọi là dimethylpolysiloxan, methylsilicon hoặc vật liệu loại 1. Nó có cấu trúc như sau:
(CH3)3Si-O- [Si(CH3)2 - O - ]nSi(CH3)3 hay MDnM
PDMS dùng chế tạo pha tĩnh trong GC, số lượng đơn vị dimethylsiloxan (n) hoặc đơn vị D thường nằm trong khoảng từ 80 (loại silicone oil) đến 40.000 (loại silicone gums); khối lượng phân tử tương ứng từ khoảng 6.000 đến 2.500.000. Đơn vị lặp lại của polyme mạch thẳng này là dimethylsiloxan (silicon).
Độ bền nhiệt của methylsilicon phụ thuộc vào độ tinh khiết của chúng. Poly (dimethylsiloxan) (Hình 4.1) tinh khiết cao, bền nhiệt đến 400°C, nhưng nhóm silanol cuối cùng cũng như các tạp chất kiềm hoặc acid (chất xúc tác từ quá trình tổng hợp) các vết oxy trong khí mang làm tăng tốc độ phân hủy nhiệt sẽ làm giảm hiệu lực cột.
Nếu các nhóm methyl được thay thế bằng các nhóm phân cực thì thu được pha tĩnh silicon phân cực có đặc tính solvat hóa tốt hơn đối với chất phân tích phân cực. Đồng thời, độ hòa tan của chất phân tích không phân cực giảm khi độ phân cực của pha silicon tăng lên. Vì lý do ổn định, chỉ một số nhóm như phenyl, cyanopropyl và trifluoropropyl được sử dụng. Chúng được gắn vào khung silica với tỷ lệ phần trăm và kết hợp thay đổi dẫn đến một loạt các cột với độ phân cực và độ chọn lọc khác nhau.
1.3.2 Poly (methylphenylsiloxan)
Poly (methylphenylsiloxan) (Hình 4.1) có hàm lượng phenyl nằm trong khoảng từ 4 đến 75 mol % (chủ yếu là 5, 20, 35, 50, hoặc 65%). Ví dụ, 5% phenylmethylpolysiloxan có nghĩa là 5% đơn vị monome có nhóm phenyl gắn với nguyên tử silicon trong mạch và 95% là nhóm methyl. Pha tĩnh này phù hợp để phân tích nhiều nhóm chất có đặc tính khác nhau tùy thuộc tỷ lệ phenyl.
1.3.3 Cyanopropylphenyl Polysiloxan
Cyanoalkylsiloxan (Hình 4.1) là pha tĩnh lỏng có độ phân cực cao và độ ổn định nhiệt hợp lý. Cột này có thể ổn định nhiệt lên đến 300°C. Hàm lượng cyanopropyl dao động từ 3 đến 100%. Tùy thuộc vào hàm lượng cyanopropyl, các silicon nitril thuộc pha tĩnh lỏng có độ phân cực từ cao đến trung bình.
Pha tĩnh cyanopropylphenyl polysiloxan được ứng dụng phân tích các chất phân tích mang electron π, các cặp electron đơn, rượu, ceton, ester,...
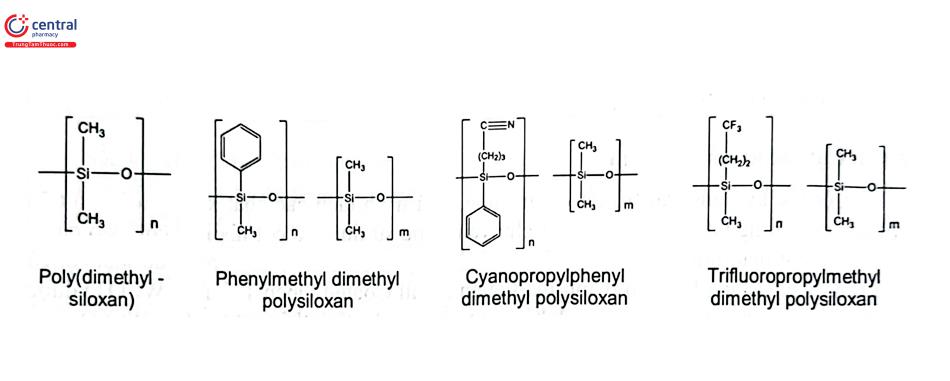
1.3.4 Trifluoropropylmethyl Polysiloxan
Trifluoropropyl được thế vào polysiloxan là poly (organosiloxan) chứa halogen duy nhất đã được sử dụng rộng rãi làm pha tĩnh lỏng trong GC. Chúng được gọi là poly (trifluoropropylmethylsiloxan) hoặc trifluoropropylmethyl polysiloxan. Hàm lượng trifluoropylmethylsiloxan (Hình 4.1) thường là 35%, 50% hoặc 100%. Độ bền nhiệt của loại pha tĩnh này là từ 260°C đến 320°C.
Poly (trifluoropylmethylsiloxan) là pha tĩnh có độ phân cực trung bình. Chúng có thể tương tác với các cặp điện tử đơn và điện tử x, do đó có khả năng lưu giữ các hợp chất giàu điện tử như hợp chất không bão hòa và thơm, hợp chất halogen, ceton, aldehyd, hợp chất nitro...
1.3.5 Polyetylen glycol (PEG)
Polyetylen glycol (PEG) và polyetylen oxyd là các pha tĩnh có tính phân cực cao và các đặc tính phân tách độc đáo. Polyethylen glycol có cấu tạo như sau:
R-O-[CH2-CH2-O]n-R
R: H, CH3, alkyl
Ứng dụng của pha tĩnh PEG là tách các hợp chất có chứa oxy, nitơ, Lưu Huỳnh hoặc halogen như rượu, phenol, amin bậc 1 và bậc 2,... Nhược điểm của PEG là giới hạn nhiệt độ làm việc thấp (từ 225 đến 280 độ C) rất nhạy với oxy và nước ở nhiệt độ cao. Do đó, phải loại vết của oxy và nước khỏi khí mang bằng cách sử dụng các bộ lọc thích hợp.
Pha tĩnh PEG được sử dụng nhiều nhất là Carbowax 20M, một chất rắn dạng sáp có trọng lượng phân tử từ 16.000 đến 20.000 Da. Các vật liệu cải tiến với trọng lượng phân tử cao hơn, phân bố trọng lượng phân tử hẹp hơn và ít tạp chất hơn được bán trên thị trường với tên thương mại Superox, Pluoronic hoặc Stabilwax.
1.3.6 Squalan
Squalan là một hydrocacbon mạch thẳng phân nhánh (2,6,10,15,19,23 - hexamethyltetracosane - C3H62)
Squalan là pha tĩnh không phân cực có tính ổn định nhiệt thấp (nhiệt độ cột tối đa 150°C). Được ứng dụng phân tích các alcol mạch thẳng.
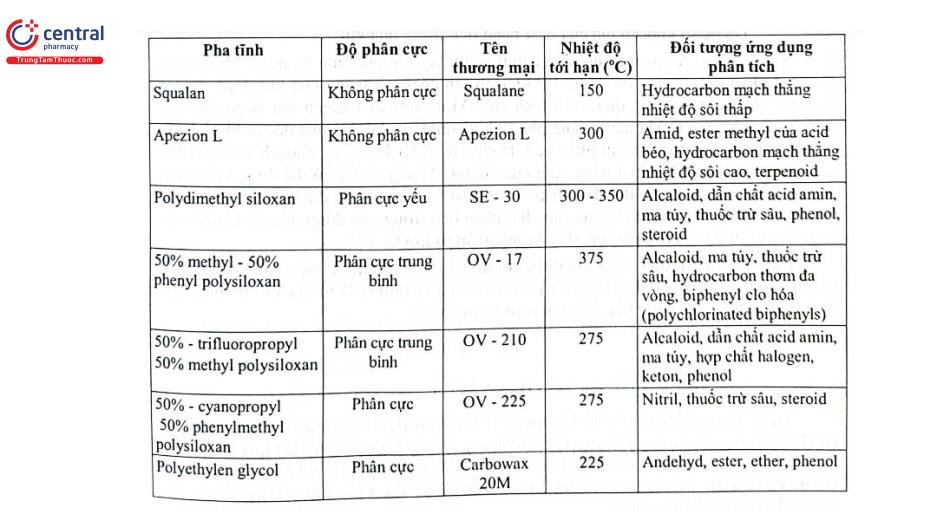
Mỗi loại pha tĩnh sẽ phù hợp cho những đối tượng cụ thể. Bảng 4.2 trình bày đặc điểm và ứng dụng phân tích của một số loại cột sắc ký thông dụng
1.4 Tối ưu hóa tốc độ khí mang
Để phân tích hiệu quả, điều kiện sắc ký cần phải được tối ưu hóa. Phương trình do Golay phát triển cho cột mao quản (A = 0) có pha tĩnh lỏng dựa trên phương trình Van Deemter như sau:
H=B/ū+ (Cs +CM)ū
Thông số CM đặc trưng cho sự mở rộng dải gây ra bởi sự khuếch tán trong pha khí, thông số này không có trong dạng ban đầu của phương trình Van Deemter. Do đó, phương trình Golay chứa hai số hạng C là Cs và CM, dùng để mô tả sự chuyển khối pha tĩnh và pha động. Các thông số B, Cs và Cm được xác định theo công thức sau:
B = 2. DG
Cs = (2.k.df2) / [3(1 + k)2.DL
CM = (1 + 6k + 11k2).dc2 / [96(1 + k)2.DG]
Trong đó:
k hệ số lưu giữ
df độ dày lớp pha tĩnh
DL hệ số khuếch tán của chất phân tích trong pha tĩnh lỏng
dc đường kính trong cột mao quản
DG hệ số khuếch tán của chất phân tích trong pha khí
Như vậy yếu tố quan trọng nhất chính là chính là độ dày lớp pha tĩnh và hệ số khuếch tán của chất phân tích trong pha động. Sự khuếch tán chất tan trong pha khí đóng vai trò tích cực và tiêu cực trong quá trình tách GC. Một mặt, sự khuếch tán là cơ chế vận chuyển các chất tan từ khí mang sang pha tĩnh và quay trở lại. Về mặt đó, sự khuếch tán là điều cần thiết cho quá trình phân tách trong GC. Mặt khác, sự khuếch tán mở rộng các píc, do đó làm giảm khả năng tách của chúng. Thông số Cs có thể được kiểm soát với lớp pha tĩnh mỏng và độ nhớt thấp. Thông số Cm phụ thuộc đường kính trong của mao quản và hệ số khuếch tán của các chất phân tích trong pha động, tốc độ khuếch tán sẽ giảm khi sử dụng khí mang có khối lượng phân tử lớn hơn.
Hơn nữa, lưu lượng dòng khí được tối ưu (ūopt) để thu được hiệu lực cột cao nhất (chiều cao đĩa lý thuyết H là tối thiểu) do đó số đĩa lý thuyết là tối đa và cho hiệu quả tách với độ phân giải cao nhất. Với cột mao quản, ta có:
ūopt = √(B/C)
Hmin = 2√(B.C)
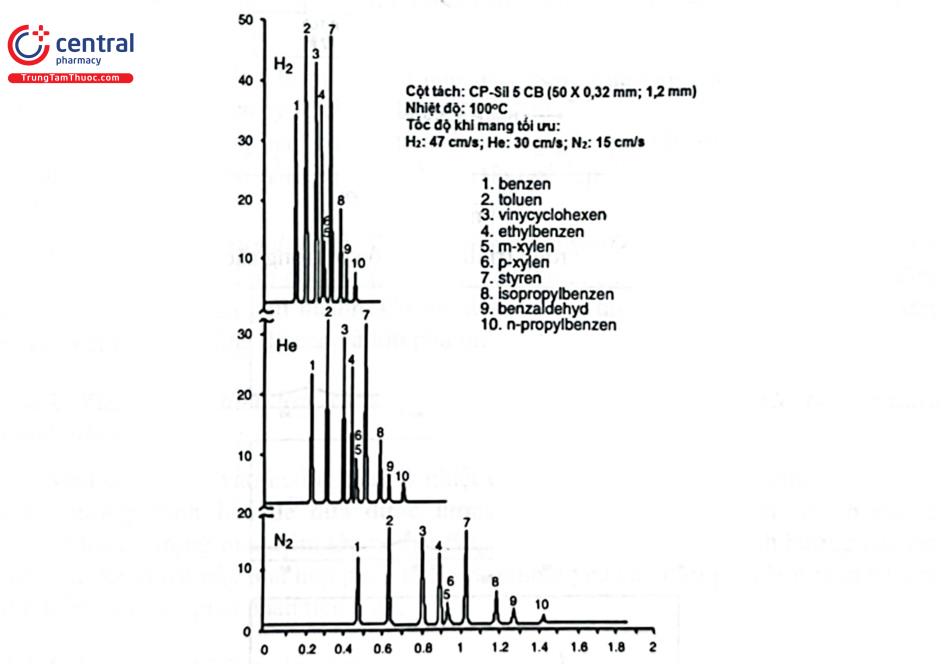
Trong thực nghiệm, tốc độ khí tuyến tính tối ưu (Optimal practical gas velocity: OPGV) thường nằm trong khoảng 1,5 - 2 lần ūopt. Bảng 4.3 trình bày kết quả tối ưu hóa đối với các cột mao quản có đường kính trong và độ dày lớp pha tĩnh khác nhau sử dụng khí mang heli. Hình 4.2 trình bày sắc ký đồ tách các chất tại lưu lượng dòng tối ưu của một số khí mang thông dụng.

1.5 Kiểm soát nhiệt độ cột
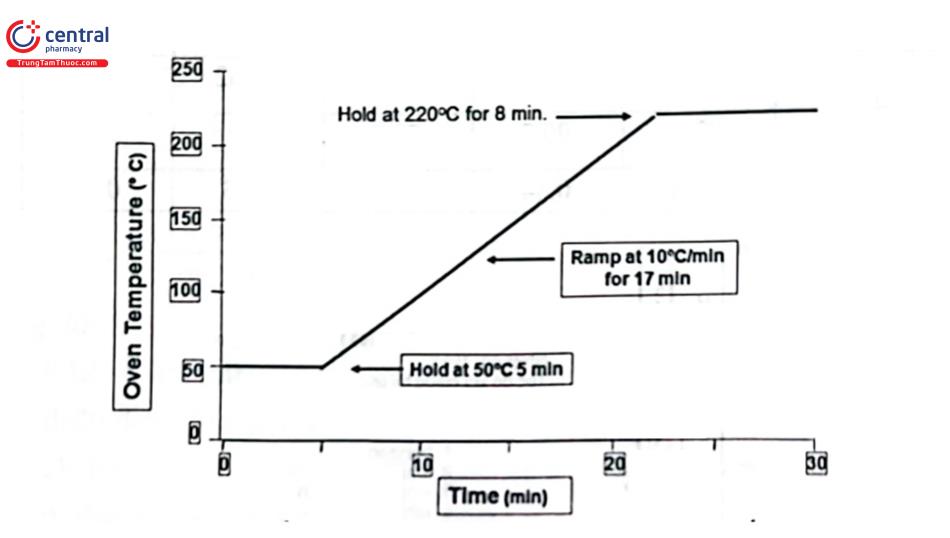
Trong sắc ký khí, cần phải kiểm soát nhiệt độ cột vì nó là một trong các yếu tố quan trọng ảnh hưởng đến khả năng tách và tốc độ rửa giải chất phân tích. Người ta đã chứng minh, khi nhiệt độ cột tăng 20°C hệ số phân bố giảm khoảng 2 lần và thời gian lưu cũng giảm tương ứng. Nhiệt độ cột có thể đặt ở chế độ đẳng nhiệt hoặc chương trình hóa nhiệt độ (gradient nhiệt độ). Phân tích ở chế độ đẳng nhiệt có nhược điểm là nếu đặt ở nhiệt độ cao thì những chất khó bay hơi tách tốt nhưng các chất dễ bay hơi rửa giải nhanh hoặc có thể không đạt độ phân giải; còn nếu ở nhiệt độ thấp thì những chất dễ bay hơi tách tốt, còn các chất khó bay hơi rửa giải chậm, píc tù dẫn đến độ nhạy giảm. Đối với chương trình nhiệt độ sẽ tăng nhiệt độ cột từ từ trong quá trình sắc ký nên sẽ tăng độ phân giải cho hỗn hợp có nhiều thành phần có điểm sôi khác nhau và giảm thời gian phân tích, nhiệt độ ban đầu thấp nên có thể tách các chất dễ bay hơi, sau đó tăng nhiệt độ tới nhiệt độ cao để rửa giải các chất có nhiệt độ sôi cao. Tuy nhiên, cần chú ý tới nhiệt độ làm việc của cột tách để đặt nhiệt độ phân tích phù hợp, tránh làm bay hơi đồ so pha tĩnh. Hình 4.3 minh họa một chương trình nhiệt độ lò cột, Hình 4.4 sắc bà sánh tách hỗn hợp 9 chất với các điều kiện sắc ký giống nhau, nhưng cột ở chế độ đẳng nhiệt và chương trình nhiệt độ.
1.6 Tiêm mẫu
Tùy thuộc vào đặc tính chất phân tích, mục đích phân tích, loại cột sử dụng mà sử dụng kỹ thuật tiêm mẫu khác nhau. Mẫu thử có thể tiêm tay bằng syring tiêm mẫu hoặc tiêm mẫu tự động, trong đó tiêm mẫu tự động cho độ lặp lại tốt hơn và dễ dàng tự động hóa.
1.6.1 Tiêm mẫu trực tiếp
Đối với một số hợp chất dễ bị phân hủy ở nhiệt độ cao, thường sử dụng kỹ thuật tiêm mẫu trực tiếp vào cột. Mẫu được đưa vào đầu cột ở trạng thái lỏng, thường với thể tích thấp (khoảng 0,3 ul) sau đó tăng dần nhiệt độ cột để tách các thành phần mẫu. Loại tiêm mẫu này thường áp dụng với cột nhồi hoặc cột mao quản có đường kính lớn. Phương pháp này phù hợp đối với phân tích vết. Hình dạng píc thu được có thể xấu, píc doãng.
1.6.2 Tiêm mẫu chia dòng/ không chia dòng
Sử dụng bộ tiêm mẫu chia dòng/ không chia dòng (Split/Splitless: SSL) được thiết kể để có thể tiêm mẫu theo hai kiểu chia dòng và không chia dòng.
Chia dòng (split): Chỉ một phần nhỏ mẫu được đưa vào cột, phần còn lại được dẫn vào đường thải theo tỷ lệ chia đặt trước. Tỷ lệ chia dòng có thể dao động từ 1:10 đến 1:500. Phương pháp này có ưu điểm là không gây quá tải cột, chất lượng píc tốt, phù hợp đối với mẫu có nồng độ cao và pha tĩnh có lớp phim mỏng; nhưng không phù hợp phân tích vết.
Không chia dòng (splitless): Toàn bộ mẫu tiêm vào buồng tiêm được đưa vào cột. Phương pháp này phù hợp đối với phân tích vết nhưng có thể xảy ra hiện tượng chồng píc với những chất rửa giải nhanh. Có thể dẫn tới quá tải cột, hình dạng píc thu được xấu đối với mẫu có nồng độ cao và lớp pha tĩnh mỏng.
1.6.3 Tiêm mẫu hóa hơi chương trình nhiệt độ (Programmable temperature vaporization: PTV)
Mẫu được đưa vào buồng tiêm ở nhiệt độ thấp, sau đó nhiệt độ của buồng tiêm được chương trình hoá để đưa được lượng mẫu về trạng thái hơi nhanh chóng (450°C/phút). Lượng mẫu tiêm lớn có thể tăng đến 5 ul mà không bị ảnh hưởng của pic dung môi. Kỹ thuật này phù hợp phân tích vết, nhưng yêu cầu thời gian làm lạnh và làm nóng mẫu nên thời gian phân tích tăng
1.6.4 Tiêm mẫu không gian hơi
Mẫu thử được chuẩn bị trong dung môi thích hợp và đựng trong lọ kín. Ở nhiệt độ không đổi, trạng thái cân bằng được thiết lập giữa các thành phần dễ bay hơi của mẫu thử (chất lỏng rắn) và pha khí phía trên nó (không gian hơi). Sau thời gian cân bằng (~15 phút), một phần của không gian hơi - thể tích phía trên nền mẫu trong đó chứa các chất dễ bay hơi dạng khí - được đưa qua vách ngăn cao su bằng syring kín khí và tiêm vào cột sắc ký. Phương pháp tiêm mẫu không gian hơi tiến hành đối với chất phân tích bay hơi ở nhiệt độ dưới 290°C, mẫu thử không thể tiêm trực tiếp vào hệ thống sắc ký do mẫu thử khó tan trong dung môi pha mẫu thành dung dịch, các mẫu có nền mẫu dạng rắn, lỏng, bột nhão.
Khi thực hiện chế độ tiêm mẫu không gian hơi cần lưu ý: Không gia nhiệt lên quá nhiệt độ sôi của dung môi pha mẫu; thể tích lọ mẫu và thể tích mẫu phải phù hợp (thường thể tích mẫu lỏng từ 1/2 - 2/3 thể tích lọ mẫu); với mẫu có độ nhớt lớn cần phải để thời gian cân bằng đủ lâu; các bộ phận như kim, syringe, dây nối chuyển (transfer line) cũng cần được gia nhiệt để tránh ngưng tụ mẫu.
So với cách tiêm mẫu thông thường, tiêm mẫu không gian hơi có ưu điểm là chuẩn bị mẫu đơn giản hơn; có thể phân tích trực tiếp một loạt các mẫu thử như dạng lỏng, dạng rắn và bột nhão; cột bền hơn do mẫu thử dạng khí (không gian hơi phía trên mẫu thử) sạch hơn; độ chính xác cao; píc dung môi nhỏ hơn hoặc không xuất hiện. Tuy nhiên, nhược điểm của nó là chỉ phân tích được những cấu tử nhẹ và yêu cầu nền mẫu phải bay hơi kém hơn chất phân tích.
2 Dẫn chất hóa phân tích
Hạn chế của phương pháp GC là chỉ áp dụng được cho các chất có thể chuyển thành dạng khí mà không bị phân hủy bởi nhiệt. Các chất có đặc tính này có thể phân tích trực tiếp, như tinh dầu, xăng dầu, chất hữu cơ dễ bay hơi, ... Đối với các chất bay hơi kém hoặc không bay hơi thì sử dụng biện pháp dẫn xuất hóa tạo sản phẩm có khả năng hóa hơi, tăng độ bền nhiệt nên sẽ mở rộng phạm vi áp dụng của GC.
Mỗi chất phân tích phải có áp suất hơi phù hợp để chuyển sang thể khí mà không bị phân hủy nhiệt. Nhưng áp suất hơi giảm làm tăng khối lượng phân tử, tăng độ phân cực, nên không thể giảm áp suất đến khi bay hơi mà không phân hủy. Nếu khả năng hóa hơi kém do có tương tác mạnh giữa các phân tử, chẳng hạn như có liên kết hydro, thì bước dẫn xuất có thể che khuất các nhóm phân cực, làm tăng đáng kể khả năng hóa hơi. Sau dẫn xuất hóa, chất phân tích bị biến đổi hóa học trở thành một chất khác phù hợp cho phân tích GC. Dẫn xuất không chỉ nhằm mục đích tăng khả năng hóa hơi, tăng độ bền nhiệt của chất phân tích mà còn cải thiện khả năng đáp ứng với sắc ký khí của chất phân tích do nó đã biến đổi thành dẫn xuất nhạy hơn hoặc chọn lọc hơn với hệ thống phát hiện. Ngoài ra, các dạng dẫn xuất hóa của một chất phân tích có thể tách tốt hơn khỏi các chất khác vì nó rửa giải ở vị trí khác trên sắc ký đồ với ít các hợp chất đồng rửa giải hơn. Dẫn xuất hóa cũng có thể hỗ trợ trong định tính các chất chưa biết. Sự thay đổi của píc trên sắc ký đồ sau khi tiến hành phản ứng dẫn xuất hóa nhóm chức đặc hiệu của chất phân tích giúp định tính nhóm chức, kết hợp với khối phổ sẽ cho biết số lượng nhóm chức dựa vào sự thay đổi khối lượng. Dẫn xuất hóa cũng có thể tạo ra nhiều phổ khối đặc hiệu hơn, ví du, tạo ra các mảnh khối đặc hiệu, giúp định tính các chất chưa biết. Hơn nữa, dẫn xuất hóa với thuốc thử chọn lọc đồng phân có thể chuyển dạng đồng phận trong cặp đối quang và chúng có thể tách ra khỏi nhau mà không cần sử dụng cột tách đồng phân.
Một phản ứng dẫn xuất lý tưởng phải đáp ứng các yêu cầu sau: Phản ứng xảy ra nhanh, lặp lại tốt, lý tưởng nhất chỉ tạo ra một dẫn xuất duy nhất. Sản phẩm dẫn chất tạo thành phải bền nhiệt và có đáp ứng tốt với sắc ký, hiệu suất phản ứng cao và ổn định. Thuốc thử và sản phẩm phụ của quá trình dẫn xuất phải là tối thiểu và không được ảnh hưởng đến quá trình phân tích; thuốc thử cũng không được phá hủy cột; nếu có phải loại bỏ trước khi phân tích. Các phản ứng phải dễ dàng và an toàn khi thực hiện. Các phản ứng dẫn xuất thường được sử dụng trong GC là silyl hóa, alkyl hóa, acyl hóa, oxi hóa khử, và tạo vòng.
2.1 Silyl hóa
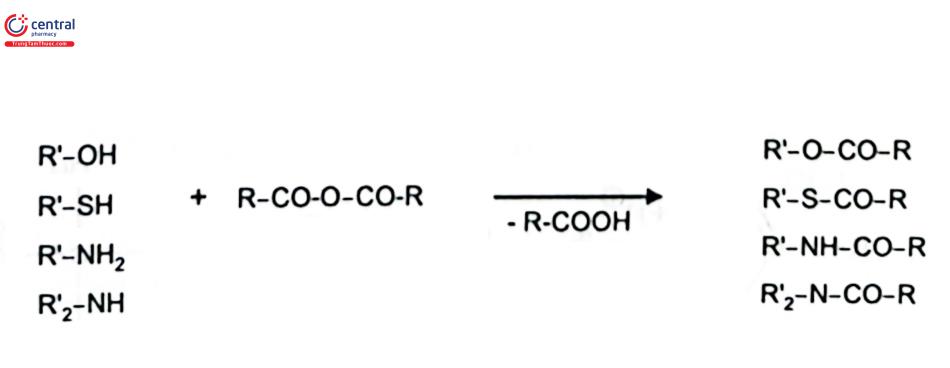
Silyl hóa là một phản ứng dẫn xuất hóa được sử dụng rất rộng rãi. Phản ứng thay thế các proton acid trong các nhóm chức bằng một nhóm trialkylsilyl. Trong đó, trimetylsilyl là chất dẫn xuất hóa được sử dụng nhiều nhất. Phản ứng có thể tiến hành với alcol, acid carboxylic, thiol, amin, amid và các dạng enol của các hợp chất carbonyl. Khả năng phản ứng của các nhóm chức với thuốc thử silyl giảm theo thứ tự alcol > phenol > carboxyl > amin > amid. Sơ đồ phản ứng chung cho quá trình dẫn xuất hóa với thuốc thử trimetylsilyl (TMS) này được thể hiện trong sơ đồ phản ứng 1.
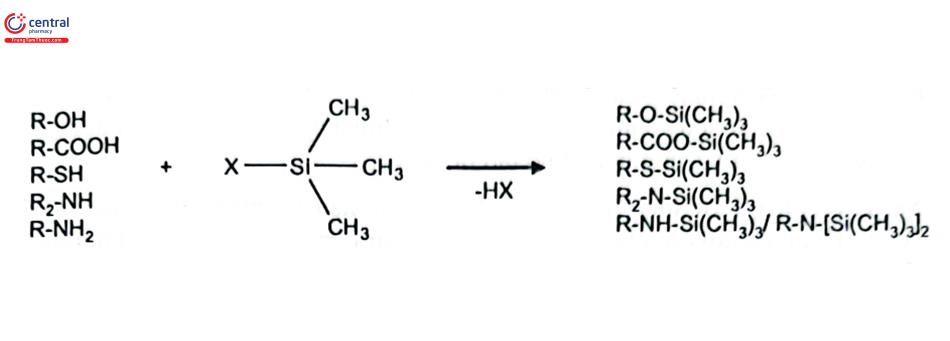
Các phản ứng bị ảnh hưởng do cản trở không gian, khả năng phản ứng với alcol giảm dần theo thứ tự sau: Bậc 1 > Bậc 2 > bậc 4; và với các amin: bậc 1 > bậc 2.
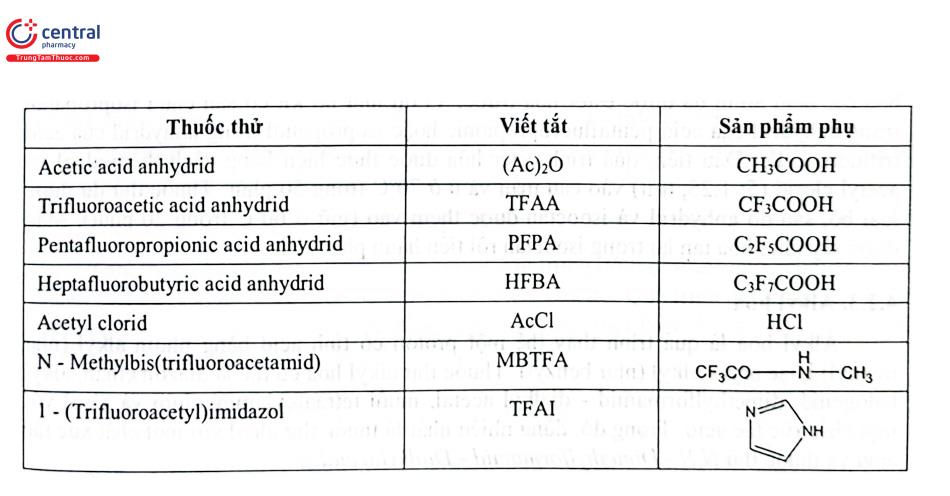
Có nhiều thuốc thử silyl hóa trên thị trường với hoạt tính và ứng dụng khác nhau. Các thuốc thử phản ứng phổ biến nhất được liệt kê trong Bảng 4.4 theo thứ tự tăng dần khả năng phản ứng của nhóm silyl.
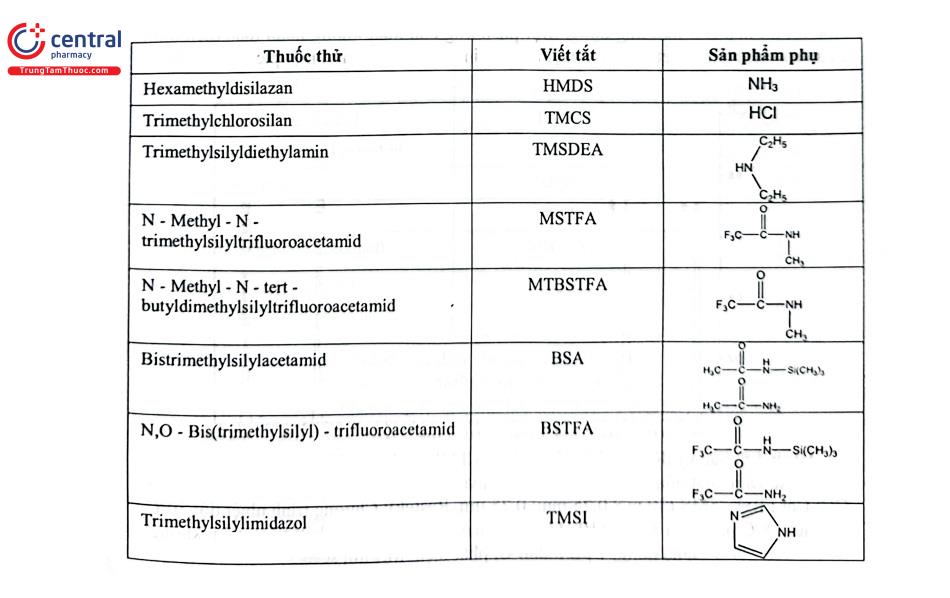
TMCS ít khi được sử dụng đơn lẻ, mà thường được kết hợp với thuốc thử khác để tăng khả năng phản ứng, lượng TMCS thường được thêm khoảng 1%. Các thuốc thử thường sử dụng và có hoạt tính tương tự nhau là BSA, BSTFA, và MSTFA. BSTFA và các sản phẩm phụ của phản ứng là trifluoroacetamid và N- trimethyl silyltrifluoroacetamid rất dễ bay hơi, ít gây ảnh hưởng tới các píc rửa giải sớm trong trong phân tích GC. MSTFA và sản phẩm phụ chủ yếu là N - methyltrifluoroacetamid dễ bay hơi nhất nên được lựa chọn trong phân tích các chất có dẫn xuất silyl rửa giải sớm. BSA, BSTFA, và MSTFA là thuốc thử ở dạng lỏng và cũng được sử dụng như là dung môi cho các phản ứng. Nếu có vấn đề về độ tan hoặc mẫu thử phải được pha loãng trước khi tiêm, phải sử dụng dung môi không có proton acid. Pyridin là một dung môi lý tưởng trong các phản ứng silyl hóa vì nó tạo ra base.
Silyl hóa phải được thực hiện trong điều kiện khan vì nước phản ứng với thuốc thử và cũng làm biến đổi các chất dẫn xuất. Có thể sử dụng một lượng dư lớn thuốc thử silyl hóa nếu không thể loại được nước.
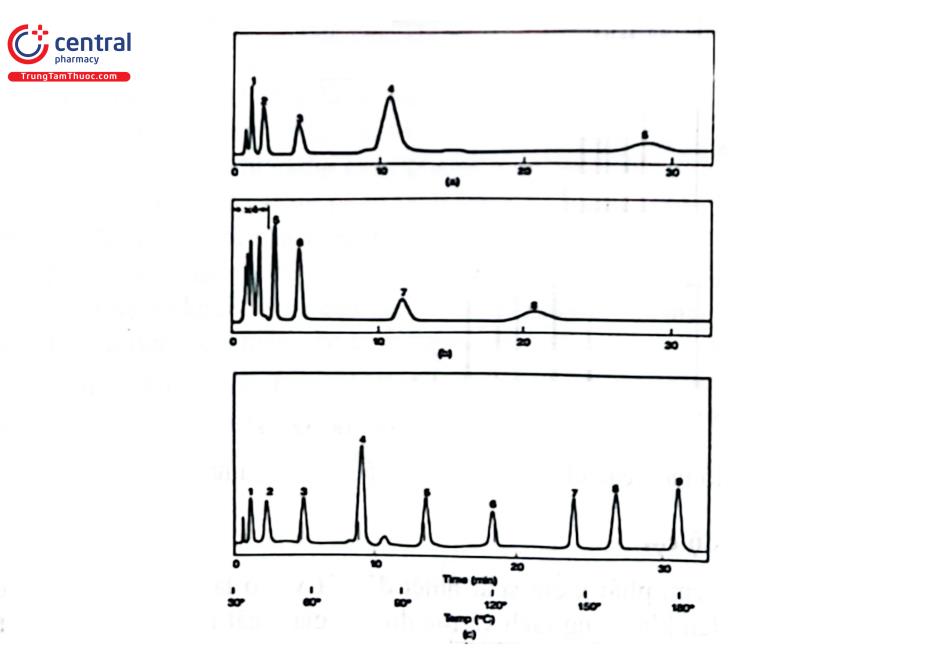
Quá trình dẫn xuất hóa phụ thuộc vào chất cần phân tích. Silyl hóa nhanh các chất phân tích trong buồng tiêm có nhiệt độ cao giúp phản ứng xảy ra dễ dàng, như với alcol hoặc acid carboxylic. Chất phân tích và thuốc thử dẫn xuất được tiêm đồng thời bằng cách sử dụng kỹ thuật là thuốc thử và mẫu thử được tiêm trực tiếp, xen kẽ nhau để thực hiện dẫn xuất hóa trên cột. Với chất phân tích có khả năng phản ứng kém hơn, phải giữ chất phân tích và thuốc thử ở nhiệt độ cao trước khi tiêm. Thông thường phản ứng thực hiện ở 60 độ C trong 1 giờ. Nói chung, thời gian và nhiệt độ dẫn xuất phải được tối ưu hóa cho mỗi ứng dụng. Một phản ứng dẫn xuất không hoàn toàn điển hình là của các acid amin, kết quả là từ một acid amin sẽ cho nhiều hơn một píc (Hình 4.5).
Khi dẫn xuất hóa, chủ yếu sử dụng thuốc thử trimetylsilyl, nhưng thuốc thử là đồng đẳng alkyl lớn hơn cũng có thể được sử dụng. Một trong những chất phổ biến nhất là N-methyl - N - tertbutyl dimethyl silyltrifluoroacetamide (MTBSTFA), đây là nhóm tert - butyl - dimethylsilyl. Dẫn xuất này ít nhạy với độ ẩm và có thể chịu được hàm lượng nước lên đến 2%. Một số thuốc thử bán sẵn như SILMAS chứa trimethylsilyl, triethylsilyl, và nhóm tert-butyl dimethylsilyl (Fluka).
Một vấn đề thường gặp khi silyl hóa trong phân tích bằng detector ion hóa ngọn lửa là detector bị bẩn do lắng đọng silica trong phản ứng đốt và sẽ rõ rệt hơn nếu sử dụng lượng dư lớn thuốc thử. Do đó, phải thường xuyên làm sạch detector.
Phân tích mẫu silyl hóa phải đảm bảo rằng các dung môi có chứa proton, như nước hay methanol, được loại bỏ hoàn toàn khỏi mẫu. Nếu không, sẽ làm bất hoạt các thuốc thử và các dẫn xuất silyl bị phân hủy bởi các dung môi có proton. Không phân tích các mẫu silyl hóa bằng pha tĩnh polyethylen glycol hoặc các pha tĩnh có chứa proton acid vì thuốc thử có thể phản ứng với pha tĩnh.
2.2 Acyl hóa
Acyl hóa là quá trình hình thành nhóm acyl ( - CO - R) ở chất phân tích có chứa các nhóm chức OH -, SH -, NH2 - hoặc NH. Sự thay thế các proton có tính acid của các chất phân tích bằng một nhóm acyl sẽ cải thiện đáng kể khả năng sắc ký. Các thuốc thử acyl hóa phổ biến nhất được tóm tắt trong Bảng 4.5.
Thuốc thử dẫn xuất thường sử dụng là các anhydrid acetic acid, anhydrid trifluoroacetic acid, pentafluoropropionic anhydrid acid hoặc anhydrid heptafluorobutyric acid. Phản ứng tổng quát được thể hiện trong sơ đồ phản ứng 2.
Các phản ứng thường được thực hiện với lượng dư thuốc thử. Acid tạo thành trong phản ứng phải được loại bỏ trước khi phân tích GC để bảo vệ cột sắc ký. Điều này cũng áp dụng cho các phản ứng với halogen acyl. Thông thường, các dung môi còn được sử dụng để tạo môi trường base cho phản ứng, ví dụ như pyridin. Về điều kiện phản ứng, như thời gian và nhiệt độ phản ứng tùy thuộc vào chất phân tích và thuốc thử, thường nằm trong khoảng từ 15 phút đến 1 giờ, nhiệt độ từ nhiệt độ phòng đến điểm sôi. Môi trường phản ứng có tính acid trong quá trình dẫn xuất hóa với anhydrid acid và acyl halogen có thể ảnh hưởng tới độ ổn định của chất sau dẫn xuất. Các thuốc thử acyl hóa nhẹ với môi trường không có tính acid là các amid hoạt động, như N - methyl - bis (trifluoroacetamid) (MBTFA) và imdiazol acyl như trifluoroacetyl imidazol. MBTFA được sử dụng cho các phản ứng trifluoroacetyl hóa của các amin bậc một và bậc 2, nhóm hydroxyl và nhóm thiol trong điều kiện không có acid. Sản phẩm phụ tạo thành của phản ứng là N - Methyltrifluoroacetamid. Trifluoroacetylimidazol được sử dụng cho các phản ứng trifluoroacetyl hóa với các amin bậc 1, bậc 2 cũng như các nhóm hydroxyl. Sản phẩm phụ tạo ra là Imidazol. Thuốc thử halogen acyl hóa tạo thuận lợi để phát hiện bằng detector cộng kết điện tử.
Tuy nhiên, dẫn xuất của cloroacetyl và bromoacetyl có thời gian lưu lâu hơn so với perfluoroacyl và có đáp ứng sắc ký kém hơn nên thường sử dụng pentafluoropropionic và heptafluorobutyric anhydrid acid do sản phẩm có khả năng bay hơi và đáp ứng sắc ký tốt.
Acyl hóa bằng anhydrid acid thường là phương pháp được lựa chọn để dẫn xuất hóa các acid amin đã được ester hóa trước. Ví dụ như bộ kít có sẵn chứa isopropanol trong anhydrid của acid pentafluoropropionic hoặc isopropanol trong anhydrid của acid trifluoroacetic. Đầu tiên, quá trình ester hóa được thực hiện bằng cách thêm alcol và acetyl clorid : 1,25, tt/tt) vào cặn mẫu và ủ ở 70°C trong 50 phút. Thuốc thử dư được loại bỏ, sau đó anhydrid và isooctan được thêm vào (giữ ở 60°C trong 20 phút). Mẫu được sấy khô, hòa tan lại trong isooctan rồi tiến hành phân tích.
2.3 Alkyl hóa
Alkyl hóa là quá trình thay thế một proton có tính acid bằng nhóm alkyl (như methyl) hoặc aryl - alkyl (như benzyl). Thuốc thử alkyl hóa có thể là diazomethan, alkyl halogenid, dimethylformamid - dialkyl acetal, muối tetraalkylammonium và alcol với một chất xúc tác acid. Trong đó, dùng nhiều nhất là thuốc thử alcol với một chất xúc tác acid và thuốc thử N,N - Dimethylformamid - Diallylacetal
2.3.1 Acid Methanolic
Phản ứng ester hóa với acid methanolic cùng chất xúc tác acid theo sơ đồ phản ứng 3.

Ester hóa là một phản ứng thuận nghịch và theo chiều thuận nếu nước tạo thành được loại khỏi hỗn hợp phản ứng và alcol dư. Có thể sử dụng 2,2 - di methoxy propan làm chất hút nước.
H2O + (CH3)2C(OCH3)2 → CH3 - CO-CH3 + 2 CH3OH
Chất xúc tác acid phổ biến nhất là acid hydrocloric và acid sulfuric. Các chế phẩm bán sẵn như H2SO4 10% trong methanol (tt/tt) hoặc methanolic HCl 0,5 N và 3 N. Acid hydrocloric có ưu điểm là có thể bốc hơi cùng với alcol sau khi phản ứng kết thúc. Nếu sử dụng acid sulfuric, phải chiết các ester vào một dung môi không phân cực, ngoài ra còn có nguy cơ xảy ra phản ứng oxy hóa. Dẫn xuất với methanolic HCl để chuẩn bị mẫu có thể thực hiện bằng cách sử dụng 100 LL thuốc thử và hỗn hợp phản ứng, giữ ở 70°C trong 30 phút, tiếp theo là bay hơi alcol, còn lại ester.
Thay vì bốc hơi, nước có thể được thêm vào hỗn hợp phản ứng và các ester được tách ra nhiều lần bằng một dung môi không cực (n - hexan), sau đó có thể được làm khô bằng natri sulfat khan.
Methanolic HCl có thể được tạo ra trong Dung dịch bằng cách sử dụng acetyl clorid và một lượng dư methanol. Một sản phẩm phụ là methyl acetat.
Các thuốc thử này đã được sử dụng cho phân tích các dẫn xuất của acid carboxylic và acid béo trong lipid.
2.3.2 Chuyển dạng ester
Quá trình chuyển dạng ester mô tả phản ứng trong đó một ester được chuyển dạng thành một ester khác bằng cách thay thế phần alcol của nó. Quá trình này được gọi là sự rượu phân. Nó có thể được xúc tác bằng một acid hoặc một base. Phản ứng xúc tác bởi acid được chỉ ra ở sơ đồ phản ứng 4.
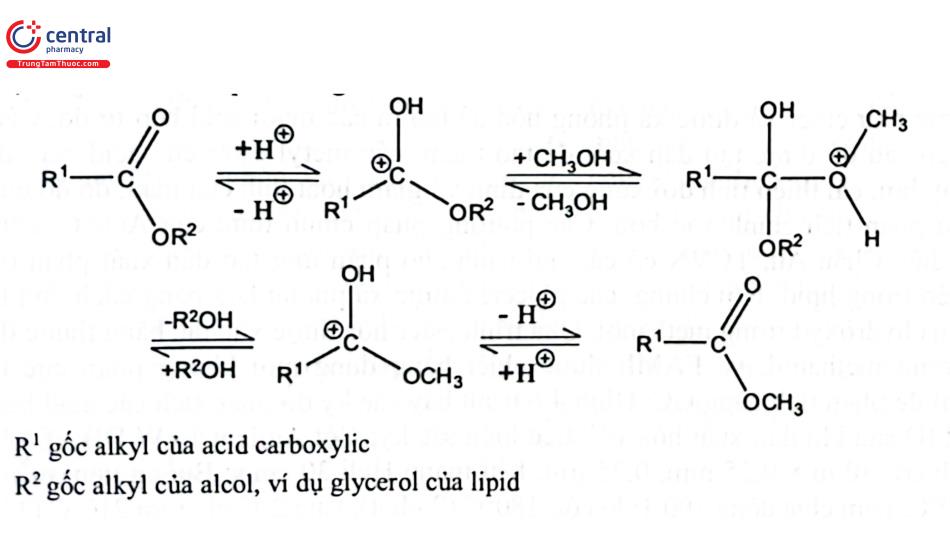
Ester hóa được sử dụng trong phân tích lipid để chuyển dạng liên kết acid béo trong lipid (ví dụ như trong triglycerid) thành dạng ester giữa methanol và acid béo (Fatty Acid Methyl Ester: FAME). Phản ứng này thường sử dụng Methanolic HCl.
Kiềm hóa ester có thể được tạo ra bằng cách sử dụng methanolic natri methoxid. Ưu điểm của phương pháp này là phản ứng diễn ra nhanh ở nhiệt độ phòng, trong khi phản ứng xúc tác bằng acid yêu cầu phải đun nóng.
2.3.2.1 Xúc tác ester hóa bằng boron triflourid (BF3)
Acid Lewis BF3 là một xúc tác thường được sử dụng để tạo ester alkyl. Hay sử dụng nhất là dạng BF3 trong methanol. Các tác nhân sẵn có trên thị trường là loại dung dịch 10% hoặc 14%. Ngoài ra, alcol bậc cao hơn cũng có thể sử dụng thay thế methanol. Tác nhân phổ biến là loại BF3 trong propanol và BF trong butanol.
Nói chung, ester hóa có xúc tác BF3 nhanh; hầu hết các phản ứng hoàn tất chỉ trong một vài phút dưới điều kiện đun nóng ở 100°C. Các dẫn chất sau đó có thể thu được bằng cách chiết vào dung môi heptan. BF3 trong methanol thường được dùng làm tác nhân của quá trình dẫn xuất hóa của các acid béo và trong chuyển dạng ester của các lipid. Quá trình ester hóa của các acid béo tự do được hoàn tất trong vòng 2 phút ở 100°C và quá trình chuyển dạng ester của các lipid diễn ra trong khoảng 90 phút.
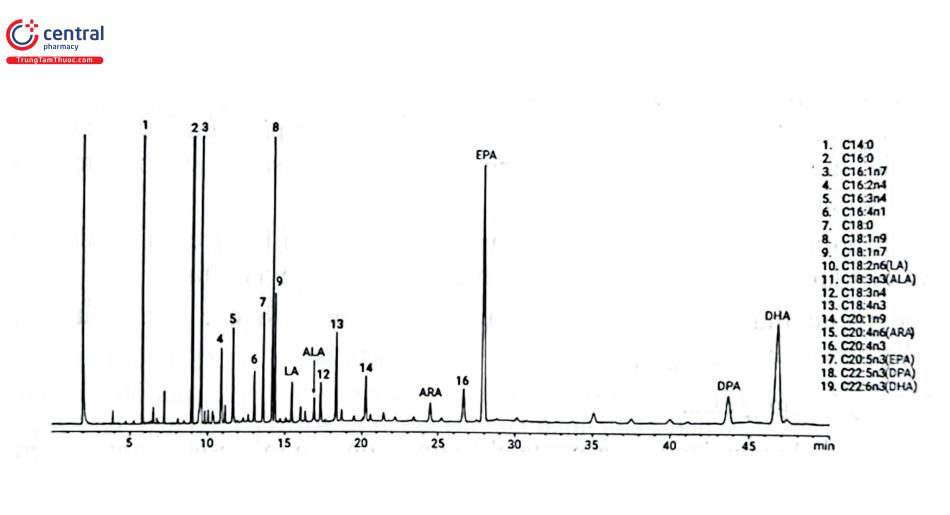
Để tạo FAME, lipid được chiết từ mẫu nền bằng cách sử dụng dung môi không phân cực như ether và được xà phòng hóa để tạo ra các muối acid béo tự do. Các muối acid béo sau đó được tạo dẫn xuất để tạo thành các metyl ester của acid béo, để tăng tính bay hơi, cải thiện tính đối xứng của đỉnh và giảm hoạt tính của mẫu, do đó cung cấp dữ liệu phân tích chính xác hơn. Các phương pháp chính thức của AOAC, cũng như Dược điển Châu u, TCVN có các qui trình cho phản ứng tạo dẫn xuất phân tích các acid béo trong lipid. Nói chung, các glycerid được xà phòng hóa bằng cách đun hồi lưu với natri hydroxyd trong methanol. Quá trình ester hóa được xúc tác bằng thuốc thử như BF3 trong methanol, và FAME được chiết bằng dung môi không phẫn cực (ví dụ, heptan) để phân tích bằng GC. Hình 4.6 trình bày sắc ký đồ phân tích các acid béo bằng GC - FID sau khi dẫn xuất hóa với điều kiện sắc ký: Cột Agilent J&W DB - FATWAX Ultra Inert, 30 m × 0,25 mm, 0,25 um; Khí mang Heli 30 cm/s; Buồng tiêm mẫu: nhiệt độ 250°C, tiêm chia dòng 100:1; lò cột: 180°C (2 phút), tăng 2°C/phút tới 210°C (35 phút); Detector FID: 280°C, hydro: 40 mL/phút, không khí: 400 mL/phút, khí phụ trợ: 25 mL/phút; Thể tích tiêm mẫu: 1 uL.
Ngoài ra, dẫn chất hóa trong phân tích GC còn có thể sử dụng các tác nhân khác như:
- Oxim hóa và tạo ra các hydrazon để phân tích các hợp chất carbonyl phân tử lượng thấp.
- Đóng vòng: Ứng dụng với các chất phân tích có ít nhất hai nhóm chức ở gần nhau như diol, keto acid,...
- Dẫn xuất trong dung dịch nước: Phân tích các hợp chất phân cực, hòa tan trong nước như acid amin. Thực hiện tạo damn và hóa môi trường nước tại chỗ (in situ) với thuốc thử alkyl cloroformat, thuốc thử alkyl hóa, acyl hóa,...
Mặc dù phương pháp dẫn xuất hóa có tiềm năng rất lớn nhưng các nhà phân tích ngần ngại sử dụng nó, bởi vì làm tăng các bước trong quá trình chuẩn bị mẫu, điều này có thể rất tẻ nhạt, tốn thời gian và có thể tạo ra sai số trong cả định tính và định lượng nếu không được thẩm định chặt chẽ. Hơn nữa, nhiều thuốc thử dẫn chất hóa nguy hiểm, do thường yêu cầu phải có hoạt tính mạnh.
3 Detector
Detector là bộ phận thu nhận, xử lý các tín hiệu của chất phân tích, chuyển thành các tín hiệu điện có thể đo lường được. Có hai loại detector được sử dụng trong GC là detector phụ thuộc khối lượng và detector phụ thuộc nồng độ.
Detector phụ thuộc khối lượng là detector đáp ứng với khối lượng trên đơn vị thời gian vào detector tức là đáp ứng tỷ lệ với mỗi nanogam chất phân phân tích đi vào detector, nó hầu như độc lập với lưu lượng dòng khí. Diện tích píc hầu như không đổi cho dù chiều rộng píc có thay đổi hoặc có pha loãng do khí phụ trợ được thêm vào ở đầu ra của cột. Điển hình cho loại này là detector ion hóa ngọn lửa và detector quang học ngọn lửa. Các detector phụ thuộc khối lượng có tính phá hủy và các hợp chất rửa giải từ cột được biến đổi hoặc phá hủy về mặt hóa học trong detector.
Detector phụ thuộc nồng độ không phá hủy chất phân tích nên có thể được sử dụng nối tiếp với các detector khác. Các detector phụ thuộc nồng độ điển hình là detector dẫn nhiệt, quang hóa, bắt điện tử và ion hóa heli phóng điện xung. Diện tích và chiều cao của píc sẽ phụ thuộc vào tốc độ khí mang qua cột cũng như sự pha loãng do thêm khí phụ trợ. Nếu thời gian bất kỳ píc nào bị thay đổi, như khi thay đổi chương trình nhiệt độ thì cần phải chuẩn hóa lại tất cả các thành phần. Tín hiệu phát hiện phụ thuộc nồng độ chất phân tích và một số đặc tính vật lý như độ dẫn điện.
Một số tiêu chí quan trọng phải được xem xét khi lựa chọn detector để đáp ứng yêu cầu phân tích. Khoảng tuyến tính hoặc khoảng động học tuyến tính cần xem xét khi tiến hành định lượng. Độ nhạy, độ tin cậy, tính ổn định, tính dễ vận hành cũng như chi phí vận hành cũng cần lưu ý.
Có nhiều loại detector được sử dụng trong GC bao gồm:
3.1 Detector ion hóa heli phóng điện xung (Pulsed discharge helium ionisation detector: PDHID)
Khí mang heli đi vào detector được ion hóa dưới tác động của tia điện phóng dạng xung ở điện áp cao và heli dạng ion hóa là nguồn năng lượng chính để ion hóa chất phân tích rửa giải khỏi cột. Vì heli có thể ion hóa là 17,7 eV, nên bất kỳ hợp chất nào có năng lượng ion hóa thấp hơn sẽ bị ion hóa. Chất phân tích duy nhất có năng lượng ion hóa cao hơn là neon sẽ không cho phản ứng. Do đó, detector này có thể phân tích được đa số chất phân tích ở thể khí. Tuy nhiên, detector này ít được ứng dụng trong thực tế do yêu cầu khí heli dạng siêu tinh khiết và thường yêu cầu tinh chế 'theo yêu cầu tạo ra heli rất khô với tổng tạp chất ít hơn 1 ppm.
3.2 Detector dẫn nhiệt (Thermal Conductivity Detector: TCD)
Nguyên tắc phát hiện dựa vào sự khác biệt về độ dẫn nhiệt của khí mang và hỗn hợp khí mang với tích. Detector dẫn nhiệt thường sử dụng để xác định chất khí, đặc biệt là khí trơ mà các detector khác không xác định được.
3.3 Detector ion hóa ngọn lửa (Flame Ionization Detector: FID)
Ngọn lửa không khí - hydro đốt và phân hủy các chất hữu cơ (chứa C) thành ion. Ion di chuyển về điện cực tạo thành dòng điện. Dòng điện được khuếch đại và phát hiện. Detector này chọn lọc với các hợp chất hữu cơ chứa C, cho tín hiệu nhạy nhất với hydrocarbon.
FID đáp ứng với khối lượng trên một đơn vị thời gian đi vào detector, do đó độ nhạy gần như không phụ thuộc vào lưu lượng khí.
Trừ methan, bất kỳ hợp carbon sẽ cho phản ứng kém với FID, như CO, acid formic, HCN và hết các hợp chất cháy trong không khí sẽ cho đáp ứng tốt trên FID. Ngược lại, hợp chất không carbon mà đáp ứng của nó giảm khi độ phân cực tăng lên. Ví dụ, một hỗn hợp chuẩn có chứa 1000 ppm (tt/tt) methan, ethan, propan và butan sẽ cho diện tích píc theo tỷ lệ 1: 2: 3: 4. Tỷ lệ diện tích píc của các anken như ethylen, propen và buten cũng sẽ là 2: 3: 4 nhưng giá trị diện tích píc sẽ khác một chút so với ankan. Điều này cũng đúng đối với chất thơm và chất thơm đa vòng. Tuy nhiên, khi hydrocarbon có oxy, tỷ lệ đáp ứng đối với số lượng carbon sẽ giảm đáng kể. Ví dụ, Ethanol (C2H6O) sẽ có diện tích píc bằng khoảng một nửa của ethan (C2H6) mặc dù đáp ứng của nó vẫn sẽ gấp đôi so với methanol. Tương tự như vậy sẽ có các bộ giá trị tương tự cho ceton, aldehyd, ether và acid.
Khi phân tích bằng FID có thể sử dụng bất kỳ khí mang nào không có hydrocarbon. Khí mang phải được lọc để loại bỏ các tạp chất hydrocarbon để giảm tín hiệu nền. Khí phụ trợ cũng phải không có hydrocarbon. Nếu FID được sử dụng để xác định hydrocarbon vết thì độ tinh khiết của không khí cũng rất quan trọng và không khí cũng phải được lọc với các bộ lọc trong dòng thích hợp.
Detector này có khoảng động tuyến tính khoảng 107. Để cải thiện độ nhạy và để ổn định ngọn lửa, thông thường phải có khí phụ trợ để có hiệu suất tối ưu. Tốc độ dòng khí điển hình đối với FID là 1 mL/ phút heli qua cột, 30 - 40 mL/ phút khí phụ trợ nitơ hoặc 40 mL/ phút hydro và 400 mL/ phút không khí. Trong thực tế, hầu hết các nhà sản xuất pha trộn hydro và khí phụ trợ trong mô - đun điều khiển lưu lượng và cấp hỗn hợp này qua một ống duy nhất vào detector.
FID ít thay đổi hiệu năng theo nhiệt độ nhưng vẫn cần kiểm soát do liên quan tới các bộ phận khác. Detector không được đặt cao hơn nhiệt độ làm việc tối đa cho phép của vật liệu chế tạo cột sử dụng. Thông thường nhiệt độ detector được đặt cao hơn nhiệt độ tối đa của lò cột khoảng 20°C. Do nước có thể hình thành ở nhiệt độ thấp, do đó FID không được hoạt động dưới 120°C. Hầu hết các FID đều có khả năng hoạt động ở nhiệt độ tối đa là 450°C.
3.4 Detector methan hóa ngọn lửa (FID methaniser)
FID methan hóa là một FID với một buồng phản ứng sau cột được gắn giữa đầu ra của cột sắc ký và FID. Phản ứng sử dụng chất xúc tác niken - zirconi với hydro dư để chuyển CO và CO2 thành methan để xác định chúng ở dạng vết.
3.5 Detector quang học ngọn lửa (Flame Photometric Detector: FPD)
FPD tương tự như FID với điểm khác biệt chính là sự phát quang của ngọn lửa được điều khiển bởi một quang kế với bộ lọc được gắn theo chiều ngang. Để cải thiện khả năng phát xạ quang học, ngọn lửa sử dụng dòng hydro lưu lượng cao hơn, nhiệt độ cao hơn và ngọn lửa lớn hơn so với FID. Ngọn lửa kích thích sự phát xạ của các tia đặc trưng cho S (392 nm), P (526 nm) và Sn (610 nm). Detector này nhạy và chọn lọc với S, P và Sn
3.6 Detector quang học ngọn lửa xung (Pulsed Flame Photometric Detector: PFPD)
Detector này có lợi thế khác biệt so với FPD, có thể phát hiện 28 nguyên tố bao gồm lưu huỳnh, phospho, nitơ, arsen và nhiều kim loại đất hiếm, nhưng chủ yếu ứng dụng phân tích lưu huỳnh và nitơ. PFPD khác với FPD ở chỗ sau khi phân tử mẫu bị phân hủy tạo thành các gốc, đầu ngọn lửa chạm đến đáy buồng đốt, nó sẽ tự bị dập tắt vì thiếu nhiên liệu, kèm theo tín hiệu nhận biết được (âm thanh “bốp”) và trình tự tương tự được lặp lại. Tốc độ phát xung tối ưu là 3 - 4 lần mỗi giây.
3.7 Detector quang hóa lưu huỳnh (Sulphur chemiluminescence detector: SCD)
Detector đặc hiệu đối với lưu huỳnh, rất ít bị ảnh hưởng của vấn đề nền mẫu nên sử dụng phân tích lưu huỳnh trong các hỗn hợp phức tạp. Về nguyên tắc, chất phân tích rửa giải từ cột được đốt cháy ở nhiệt độ rất cao, thường cao hơn 1800°C để tạo thành lưu huỳnh monoxyd (SO). Chất này phản ứng với ozon (O3) và tín hiệu của phản ứng phát quang hóa học này tạo ra tín hiệu đo. Tuy nhiên, detector này ít sử dụng trong thực tế do yêu cầu khí mang và ozon phải có độ tinh khiết cao và có thể cần bảo trì nhiều hơn vì nó hoạt động ở áp suất giảm.
3.8 Detector quang hóa nitơ (Nitrogen chemiluminescence detector: NCD)
Sử dụng phân tích các hợp chất chứa nitơ trừ nitơ. NCD có tính đặc hiệu cao nên được sử dụng rộng rãi trong phân tích nitơ trong các hỗn hợp phức tạp. Tương tự như SCD, dịch rửa giải từ cột đi vào lò đốt plasma, tại đó các hợp chất chứa nitơ chuyển thành oxyd nitric. Khi phản ứng với ozon, oxyd nitric trở nên bị kích thích điện tử và phát ra ánh sáng hồng ngoại trong vùng 600 - 3200 nm khi nó trở về trạng thái cơ bản tạo ra tín hiệu đo.
3.9 Detector cộng kết điện tử (Electron Capture Detector: ECD)
Nguồn 63Ni tạo nên electron có động năng lớn. Nếu chất phân tích có khả năng cộng kết điện tử (do cấu tạo thiếu electron) đi vào detector sẽ bắt electron từ khí mang đã được ion hoá. Để duy trì dòng electron yêu cầu, tần suất của xung sẽ tăng lên để bu đắp và tạo ra tín hiệu.
Đây là detector nhạy nhất trong GC (trừ MS/MS), có tính chọn lọc đối với các hợp chất âm điện, đặc biệt là các halogen. Độ nhạy đối với một số hợp chất này có thể tới phạm vi phần nghìn tỷ (ppt). Nguồn phóng xạ Niken 63 (nguồn phát xạ B) 5 - 10 mCi trong detector được đựng trong một hộp kín. Do đó, việc mua, sở hữu và tiêu hủy nó được qui định bởi các cơ quan có thẩm quyền xử lý các nguyên tố phóng xạ. Chu kỳ bán rã của 63Ni là khoảng 90 nằm ở nhiệt độ môi trường nên rất ít khả năng nguồn cần phải được thay thế trong suốt thời gian hoạt động của hệ thống GC trừ khi nó bị nhiễm bẩn nghiêm trọng bởi các mẫu không bay hơi hoặc bị phá hủy do sử dụng nhiệt độ quá cao.
ECD là detector phụ thuộc nồng độ, do đó, để có độ nhạy tối đa, lượng khí phụ trợ phải được giữ ở mức thấp. Khoảng động tuyến tính của ECD khá hạn chế (nhỏ hơn 104) nên phải chú ý để đảm bảo tất cả mẫu thử có nồng độ nằm trong khoảng tuyến tính. ECD rất chọn lọc đối với các khí chứa halogen, nhưng cũng sử dụng phân tích peroxyd, nhóm nitro và các hợp chất chứa oxy, nhưng không có cùng độ nhạy. Nó kém nhạy với hydrocarbon, amin và rượu. ( (Bảng 4.6).

Khí mang đối với ECD là nitơ hoặc hỗn hợp argon - methan. Vì độ nhạy cao nên yêu cầu khí mang và khí phụ trợ không những phải có độ tinh khiết cao mà còn phải được loại bỏ tất cả các tạp có tính chất ái điện tử, do đó thường dùng các bộ lọc loại hơi ẩm trên đường đi của dòng khí và phía trước bẫy oxy.
Một số pha tĩnh, chẳng hạn như cyano, không tương thích với ECD vì nó có tính chất điện phân và sẽ tạo ra tín hiệu nền rất cao do cột bị chảy. Điều này cũng sẽ phụ thuộc nhiều vào nhiệt độ và sẽ làm giảm nghiêm trọng phạm vi hoạt động tuyến tính của detector mặc dù điều này ít gây ra vấn đề hơn trong phân tích khí do nhiệt độ được kiểm soát khi sử dụng.
Nhiệt độ detector thường được đặt ở nhiệt độ cao hơn nhiệt độ làm việc tối đa của cột khoảng 50°C để đảm bảo rằng mẫu không thể ngưng tụ. Mặc dù ECD có nhiệt độ làm việc tối đa là 400°C, nhưng ở nhiệt độ này 63Ni sẽ bị mất đi nên chỉ sử dụng trong thời gian ngắn ở nhiệt độ này.
3.10 Detector ion hóa phát xạ ngọn lửa (Flame Thermionic Detector: FTD)
Tương tự detector ion hóa ngọn lửa chỉ khác là có thêm hạt muối kim loại kiềm (RbCl) đặt trên ngọn lửa. Các chất chứa N và P bị đốt trên ngọn lửa sẽ tạo thành CN- và PO2- . Các ion này liên kết với H+ (sinh ra khi Rb bị chuyển thành Rb+) và tạo thành dòng ion. Detector này nhạy với hợp chất của N và P nên còn được gọi là detector nitơ - phosphor (NPD).
3.11 Detector ion hóa quang học (PhotoIonnization Detector: PID)
Detector này có thêm một đèn UV năng lượng khoảng 8,3 - 11,7 eV (Hình 4.7). ẫu thử khi đi vào detector nếu năng lượng ion hóa của nó thấp hơn năng lượng tia UV chiếu xạ thì sẽ xảy ra quá trình ion hóa tạo ra electron. Các electron này tạo thành dòng và di chuyển đến bộ phát hiện. PID được sử dụng để phân tích các hợp chất thơm và có nối đôi, đồng thời có thể phát hiện nhiều chất vô cơ như arsin, phosphin và hydrogen sulfid.
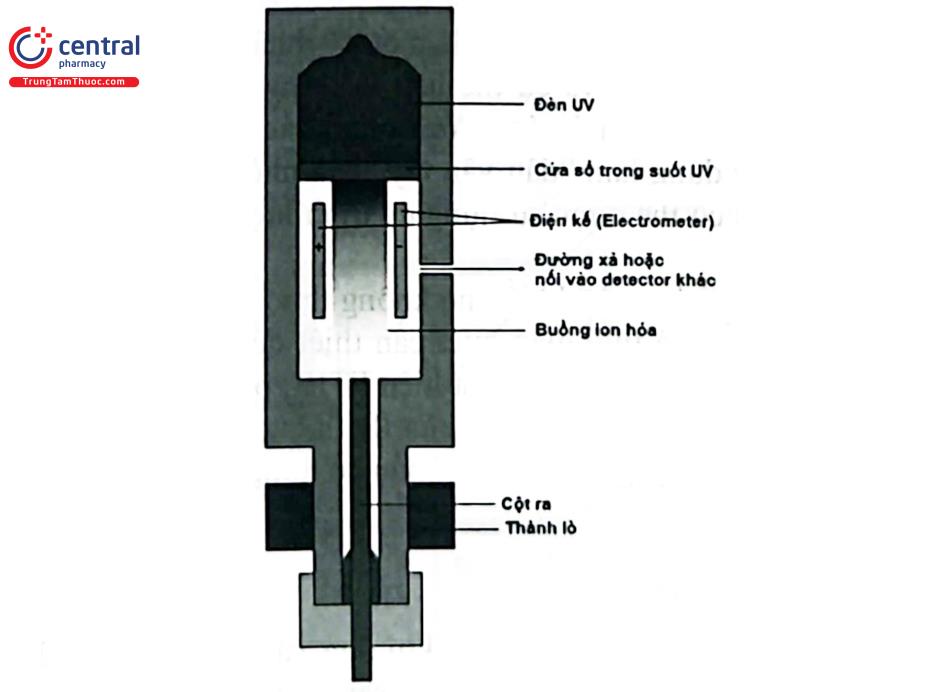
PID là detector không phá hủy và có thể lắp nối tiếp với các detector khác như FID. Nó có tính chọn lọc và độ nhạy cao, có khả năng phát hiện ở mức ppb. PID không yêu cầu khí nhiên liệu, có thể sử dụng không khí làm khí mang. Nó có khoảng tuyến tính rộng và nhiệt độ hoạt động tối đa là 270°C. Các vấn đề có thể gặp là đường nền kém, thời gian ổn định lâu, đèn UV là thiết bị tiêu hao do đó không nên bật đèn khi không sử dụng trong thời gian dài.
3.12 Detector phát xạ nguyên tử (Atomic Emission Detector: AED)
Detector phát xạ nguyên tử có thể phân tích 23 nguyên tố khác nhau bao gồm carbon, hydro, oxy, chì, Mangan, flo và silicon, thêm cả lưu huỳnh và nitơ. Về nguyên tắc, chất phân tích rửa giải từ cột GC được trộn với khí phản ứng có chứa heli và oxy có độ tinh khiết cao, được đưa vào một ống thạch anh và được đốt cháy tạo thành plasma. Các bước sóng phát xạ đặc trưng của các nguyên tố được đo trên máy quang phổ có cách tử quay cho phép phân tích đồng thời một số nguyên tố.
AED là thiết bị duy nhất được sử dụng phát hiện đồng thời một loạt các nguyên tố mặc dù có thể cần phải tiêm riêng để phát hiện một số nguyên tố do điều kiện sắc ký, đặc biệt là khí phải thay đổi để có được các điều kiện tối ưu cho việc phát hiện các nguyên tố khác nhau. Ví dụ, để phát hiện carbon, lưu huỳnh và nitơ trong một mẫu, sẽ sử dụng khí hydro và oxy, phát hiện carbon ở 179 nm và lưu huỳnh ở 181 nm; đối với nitơ cần bổ sung thêm methan và đo sự phát xạ ở bước sóng 388 nm.
Tuy nhiên, detector này ít sử dụng trong thực tế do yêu cầu tất cả các loại khí sử dụng phải có độ tinh khiết cao, có thể phải sử dụng các bộ lọc theo yêu cầu. Hơn nữa, do AED có thể phát hiện oxy nên toàn bộ hệ thống cần đảm bảo tình trạng chân không và dịch vụ bảo trì không phổ biến.
3.13 Detector hồng ngoại biến đổi Fourier (Fourier transform - infrared: FT-IR)
Quang phổ hồng ngoại biến đổi Fourier là một kỹ thuật nổi tiếng và được thiết lập vững chắc trong hóa học phân tích và cụ thể hơn là trong phân tích khí. Phổ hấp thụ cung cấp các thông tin đỉnh hấp thụ ở các bước sóng khác nhau là đặc trưng của từng hợp chất và có thể được sử dụng làm “dấu vân tay” của nhóm hợp chất. Cường độ hấp thụ ở các bước sóng cụ thể có thể được sử dụng để định lượng hàm lượng hoạt chất có trong mẫu.
Tuy nhiên, có hai yếu tố hạn chế khiến nó không thực tế cho nhiều ứng dụng đó là yêu cầu về độ dài đường dẫn và thời gian quét cần thiết để phát hiện lượng chất phân tích rửa giải từ cột GC. Để có đủ mẫu cho phát hiện FTIR, phải sử dụng cột nhồi, hạt có lỗ xốp lớn với thiết bị có chiều dài tế bào đo của IR thông thường 10 - 50 cm. Do đó, GC - FTIR được giới hạn ứng dụng đối với các mẫu có nồng độ khá cao và hữu ích cho việc phân tích bằng cột nhồi.
3.14 Detector khối phổ
Nguyên tắc chung là mẫu từ máy sắc ký khí đưa vào máy khối phổ sẽ được ion hóa trong buồng ion để tạo các phần tử mang điện, sau đó và phân tích khối để tách các ion khác nhau theo tỉ số m/z. Các ion được bộ phận phát rã chuyển đến bộ phận lọc hiện thu nhận, tín hiệu được chuyển vào máy tính có cài đặt phần mềm xử lý và lưu trữ.
Tương tự như LC - MS, detector khối phổ trong GC - MS cũng bao gồm nguồn ion hóa và bộ phân tích khối mà không cần giao diện kết nối (interface) phức tạp. Lý do là chất phân tích sau khi tách trên GC đã tồn tại ở trạng thái khí do đó đầu ra của hệ GC sẽ được kết nối trực tiếp với đầu vào của detector MS.
3.14.1 Nguồn ion hóa
Nguồn ion hóa đóng vai trò chuyển các phân tử chất phân tích thành dạng ion. Các loại nguồn ion hóa phổ biến trong GC - MS bao gồm:
3.14.1.1 Ion hóa va chạm điện tử (EI - Electron impact ionization)
Chùm electron có năng lượng 70 eV tạo ra từ một sợi đốt nóng (filament heater), được tăng tốc chuyển động hướng tới anod dưới tác động của điện trường. Trên đường di chuyển tới anod, chùm electron va chạm với các phân tử khí trung hòa điện tích của mẫu phân tích có trong buồng ion làm electron của phân tử bị bắn ra và hình thành nên các gốc hoạt động mang điện tích dương hoặc các ion mang điện tích dương theo phương trình: R+e-> R*+ + 2e . Kỹ thuật EI, hầu hết chỉ hình thành các ion tích điện đơn lẻ từ một phân tử trung tính ban đầu. Trong một số trường hợp, các ion mang điện tích +2 (R+e-> R*2+ + 3e -) hoặc +3 (R+e->R*3+ + 4e-) cũng có thể hình thành cùng với các ion có điện tích +1 khi chùm electron va chạm với phân tử trung tính ban đầu tùy thuộc vào cấu tạo của phân tử. Tuy nhiên, các ion đa điện tích thường chiếm tỷ lệ rất nhỏ so với ion đơn điện tích. Các ion được hình thành sẽ đi qua một điện trường khoảng 400 - 4000V để tăng tốc đi vào bộ phận phân tích khối. Tốc độ chuyển động của các ion tỷ lệ với khối lượng và điện tích của chúng. Trong kỹ thuật EI, khí và các mẫu có áp suất hóa hơi cao được nạp trực tiếp vào nguồn ion, còn các mẫu lỏng hoặc rắn phải được làm nóng để làm tăng áp suất hóa hơi khi nạp vào buồng ion. Cấu tạo nguồn ion hóa EI của thiết bị phổ khối được trình bày ở Hình 4.8.
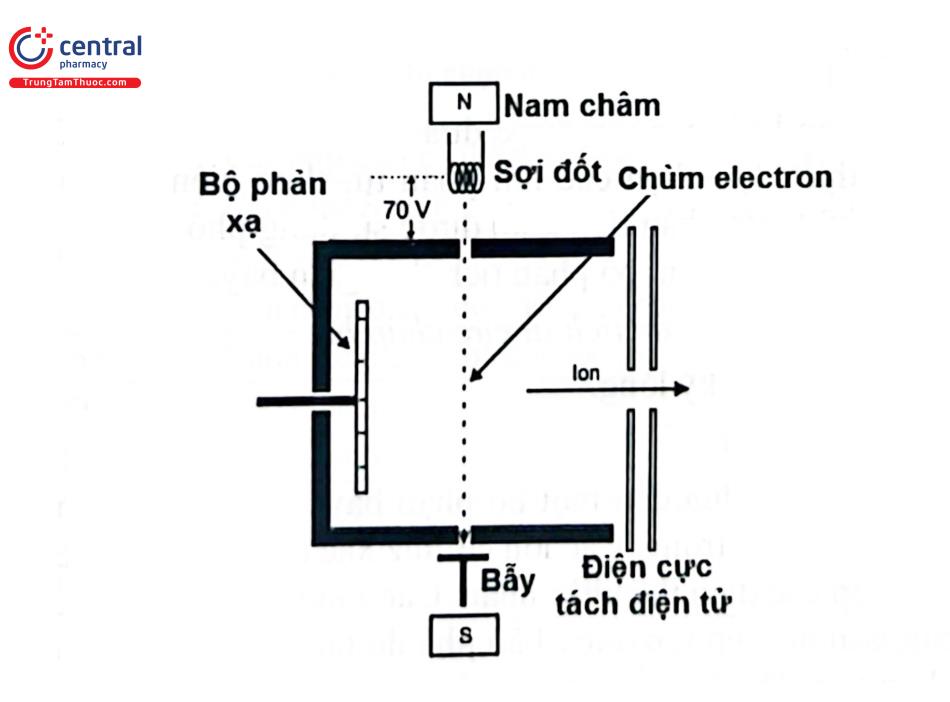
Ion hóa va chạm điện tử có ưu điểm là phổ khối tương đối dễ thu nhận và tái tạo, dễ dàng nhận dạng và xác định cấu trúc các hợp chất, dễ tra cứu thư viện phổ; nhược điểm của nó là chỉ phù hợp đối với mẫu dễ bay hơi và có tính ổn định cao.
3.14.1.2 Ion hóa hóa học (CI - Chemical ionization)
Nguồn ion hóa hóa học có cấu tạo tương tự nguồn El. Trong nguồn CI, các ion được tạo ra thông qua sự va chạm của các phân tử chất phân tích với các ion sơ cấp (thuốc thử) được cho thêm vào nguồn ion. Đầu tiên chùm electron sẽ ion hóa thuốc thử (CH4, CH3OH, NH3...) bằng cơ chế EI để tạo ra các ion thuốc thử (ký hiệu [BH]* hoặc X*); gốc mang điện tích (ký hiệu Xu+) và các electron hoạt động (thermal electrons), sau đó các ion này lại tiếp tục va chạm với nhau và va chạm với phân tử trung hoà ban đầu của chất cần phân tích (M) để tạo ra ion của chất cần phân tích bằng một trong các cách sau:
M + [BH]+ -> [M - H]+ + B (chuyển proton - proton transfer)
M+X+ -> [M+X]+ (liên kết cộng điện tử - electrophilic addition/adduct formation)
M +X+ > [M-A]+ + AX (loại gốc c anion - anion abstraction)
M+X*+ -> M*+ + X (trao đổi điện tích - charge exchange)
Tùy thuộc vào cấu trúc của phân tử trung hòa ban đầu, cấu trúc của thuốc thử mà các ion âm hoặc ion dương hoặc cả ion âm và ion dương của chất cần phân tích sẽ được tạo ra thông qua các phản ứng hóa học với các ion thuốc thử. Phần lớn các hợp chất hữu cơ cần phân tích khi ion hóa bằng kỹ thuật CI sẽ tạo ra các ion mang điện tích dương bằng một trong các cách trên, trong đó cơ chế chuyển proton là phổ biến hơn cả, đặc biệt đối với các hợp chất có nhóm amin hoặc nhóm alcol hoặc các hợp chất có tính acid hoặc base theo thuyết Bronted. Chỉ có một số ít các hợp chất hữu cơ có khả năng tạo các ion âm ổn định bền vững bằng cách cộng kết điện tử như một số hợp chất có vòng phenyl, diphenyl, đặc biệt là trong phân tử có các nguyên tố có độ âm điện cao như các halogen thế ở vòng thơm
Một trong những thuốc thử được sử dụng phổ biến nhất là khí methan (CH4), ở áp suất 1 Torr; các ion chính tạo thành là CH5+, C2H5+, C3H5+.
3.14.2 Bộ phân tích khối
Sau khi đã được ion hoá, các ion được đưa đến bộ phân tích khối nhằm loại bỏ những ion không cần thiết, lựa chọn các ion phân tử, thực hiện bắn phá thêm để thu được các ion con. Các kỹ thuật phân tích khối được sử dụng phổ biến là bộ phân tích tứ cực, ba tứ cực, bộ phân tích bẫy ion, bộ phân tích thời gian bay.
3.14.2.1 Bộ phân tích tứ cực và bộ phân tích tứ cực chập ba
Tương tự như đối với sắc ký lỏng.
3.14.2.2 Bộ phân tích khối bẫy ion
Các ion từ nguồn được đưa đến một bộ phận bẫy, có cấu tạo gồm 4 điện cực có các bề mặt hình hypebol ở bên trong. Các ion có m/z xác định được lựa chọn để đưa đến bộ phát hiện nhờ việc áp các điện thế khác nhau. Các ion này cũng có thể được lựa chọn và giữ lại trong bẫy, sau đó tiếp tục được bắn phá để tạo thành các ion con, sau đó các ion lại được lựa chọn tiếp. Do đó, về mặt nguyên tắc MS bẫy ion có thể thực hiện được MS nhiều lần nên sẽ tăng khả năng định tính.
3.14.2.3 Bộ phân tích khối thời gian bay
Dựa trên cơ sở gia tốc các ion tới detector với cùng một năng lượng. Các ion có cùng năng lượng nhưng khác nhau về khối lượng nên thời gian các ion đi tới detector sẽ khác nhau. Do có năng lượng động học hay vận tốc, các ion này sẽ bay với khoảng cách là d trong một khoảng thời gian t, trong đó t phụ thuộc vào tỉ số m/z. Hiện nay, thiết bị phân tích thời gian bay phản xạ electron giúp kéo dài quãng đường các ion đi tới detector và do đó làm tăng độ phân giải của thiết bị và tăng cường khả năng định tính với các chất chưa biết.
3.14.3 Thư viện phổ của GC - MS
Phân tích bằng GC với thiết bị MS tử cực theo chế độ ion hóa EI, năng lượng ion hóa luôn là +70 eV, do đó đã có các thư viện phổ để phục vụ tra cứu cho phép thử định tính đối với những chất chưa biết. Để định tính chất chưa biết sẽ thực hiện phân tích mẫu ở chế độ ion hóa va chạm electron ở mức năng lượng ion hóa +70 eV thu được phổ, sau đó so sánh với phổ chuẩn trong thư viện dựa vào tính năng chồng phổ của máy. Tùy thuộc vào cấu hình và tính năng của thiết bị mà độ chính xác của phép định tính là khác nhau. Đối với thiết bị có khả năng thực hiện MS nhiều lần như bộ phân tích khối tứ cực chập ba hoặc bẫy ion, năng lượng bắn phá trong quá trình ion hóa hoặc phân mảnh sẽ tùy thuộc vào vùng tối ưu của thiết bị do đó các thư viện phổ sẽ là riêng của mỗi nhà sản xuất ở điều kiện cụ thể và (hoặc) độ phân giải cao có thể định danh chính xác chất chưa biết. Hiện nay, đã có một số thư viện phổ đối với thiết bị phổ tứ cực chập các hãng như ChemStation/MassHunter của Agilent, Xcalibu của Thermo, MassLynx của Waters,...
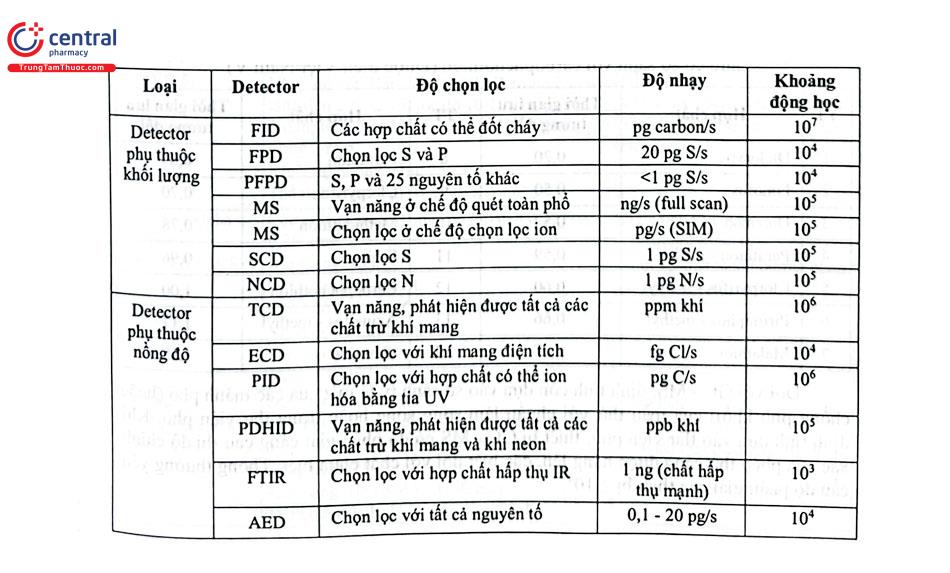
Khi tiến hành phân tích GC, căn cứ vào bản chất và nồng độ chất phân tích, nền mẫu để lựa chọn detector phù hợp. Bảng 4.7 trình bày tóm tắt về các detector của GC và một số đặc điểm của chúng.
4 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Sắc ký khí”, Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 136 - 159. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược Điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
