Hội chứng Meckel-Gruber: bệnh học, biểu hiện lâm sàng và cách chẩn đoán
Trungtamthuoc.com - Hội chứng Meckel-Gruber là hội chứng di truyền lặn với rối loạn chức năng lông chuyển trong quá trình tạo phôi, dẫn đến: loạn sản thận dạng nang, thoát vị não, thừa ngón sau trục, dị dạng tấm ống gan. Đây là hội chứng nguy hiểm có thể khiến thai chết lưu hoặc chết trong vòng vài giờ.
Biên dịch: Bác sĩ Vũ Tài
Tải PDF bản dịch TẠI ĐÂY
1 CÁC ĐIỂM CHÍNH
1.1 THUẬT NGỮ
- Hội chứng bao gồm 3 dấu hiệu trên siêu âm
• Loạn sản thận dạng nang (Renal cystic dysplasia) (98-100%)
• Thoát vị não hoặc các bất thường hệ thần kinh trung ương khác (60-90%)
• Thừa ngón sau trục (70-87%)
- Phải có ít nhất 2/3 đặc điểm cổ điển
1.2 HÌNH ẢNH
- Loạn sản thận dạng nang gần như luôn có
• Hình ảnh thận có thể thay đổi
+ Phổ biến nhất là thận to và tăng âm nhưng có thể chứa đầy các nang lớn
• Kích thước thận thường to, chu vi bụng lớn
- Thoát vị não vùng chẩm là dấu hiệu hệ thần kinh trung ương cổ điển nhưng có thể có các dị tật khác
• Dị tật Dandy-Walker, tật đầu nhỏ, holoprosencephaly, anencephaly
- Có thể chẩn đoán trong quý 1
- Thiểu ối nặng hoặc vô ối vào quý thứ 2
• Làm cho việc đánh giá các bất thường kín đáo trở nên khó khăn hơn
- Xơ hóa gan (Hepatic fibrosis) khó phát hiện được trước sinh
2 CHẨN ĐOÁN PHÂN BIỆT HÀNG ĐẦU
- Trisomy 13
• Có sự trùng lặp đáng kể về đặc điểm hình ảnh
• Xét nghiệm chẩn đoán để đánh giá NST/đơn gen
- Cần tư vấn về nguy cơ tái phát khác nhau
3 BỆNH HỌC
• Bệnh lông chuyển (ciliopathy) gây chết, không đồng nhất về mặt di truyền
• Di truyền lặn trên NST thường với nguy cơ tái phát 25%
4 CÁC VẤN ĐỀ LÂM SÀNG
• Thiểu ối dẫn đến thiểu sản phổi
• Hầu hết thai chết lưu hoặc chết trong vòng vài giờ
• Cần tư vấn di truyền cho các lần mang thai sau
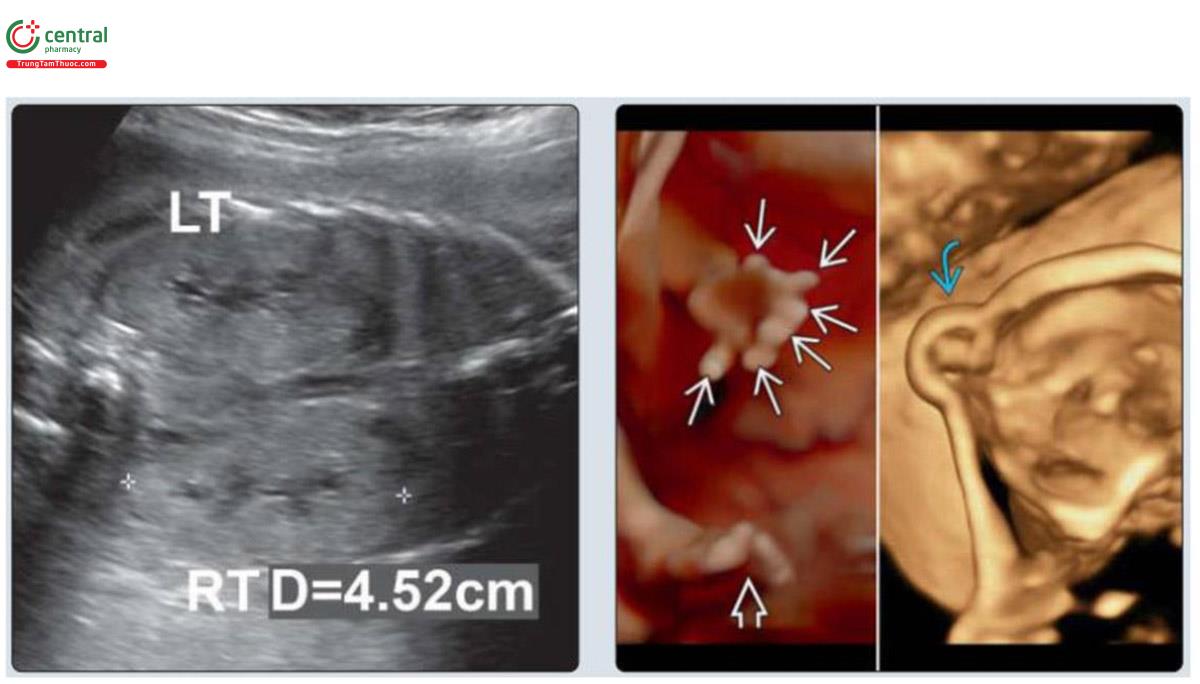
(Trái) Hình ảnh siêu âm ở mặt cắt vành của thai nhi 25 tuần tuổi cho thấy thận to tăng âm hai bên. Đây là dấu hiệu hay gặp nhất trên siêu âm trong hội chứng Meckel-Gruber (MKS). (Phải) Hình ảnh 3D của các chi và xương sọ cho thấy thừa ngón tay (mũi tên trắng nhỏ); cũng lưu ý, bàn chân có vẻ rộng (mũi tên trắng to) và thừa ngón cũng được xác nhận. Thoát vị não nhỏ (mũi tên xanh). Khoảng 60% trường hợp có tất cả 3 đặc điểm cổ điển này. Khi tình trạng thiểu ối tiến triển, có thể khó thấy các dấu hiệu kín đáo.
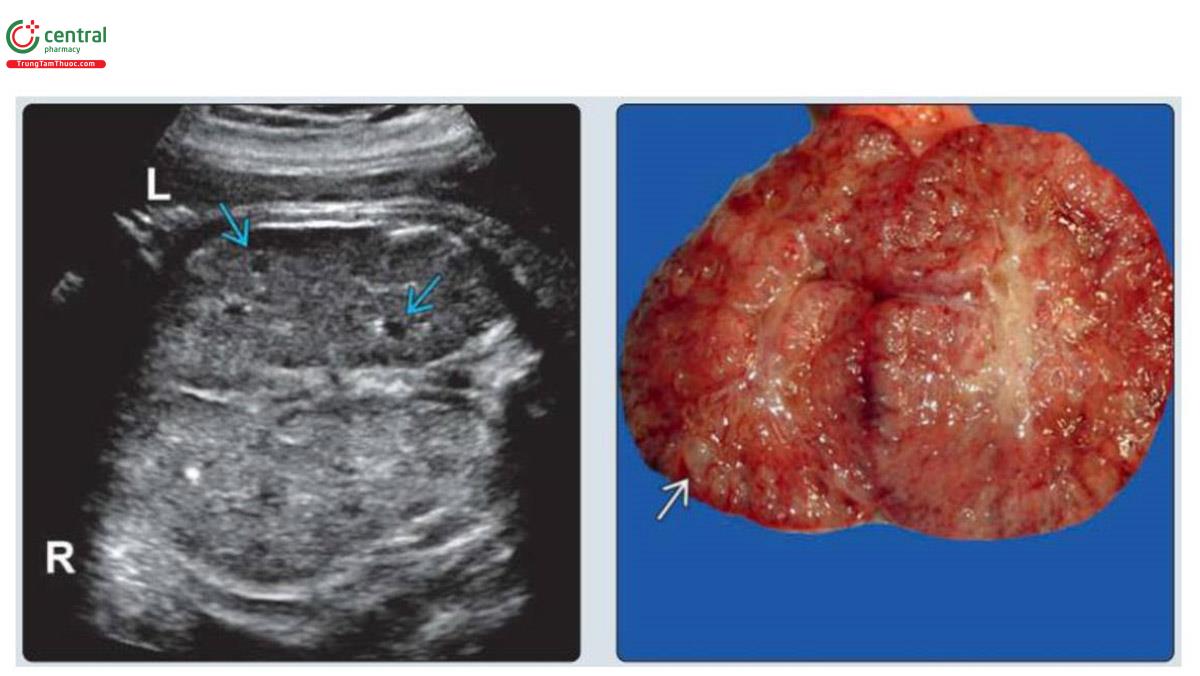
(Trái) Hình ảnh siêu âm ở mặt cắt chếch (Oblique) của thai nhi bị MKS cho thấy thận rất to gần như chạm vào đường giữa. Mất biệt hóa vỏ-tuỷ bình thường với các nang lớn nhỏ rải rác (mũi tên xanh). (Phải) Khi khám nghiệm tử thi, cắt đôi thận cho thấy vô số nang nhỏ (mũi tên trắng) và mất hoàn toàn biệt hóa vỏ-tủy. Loạn sản dạng nang có thể khiến thận rất to và thường là dấu hiệu dễ thấy nhất ở thai nhi.
5 THUẬT NGỮ
Từ đồng nghĩa
• Hội chứng Meckel
Định nghĩa
• Được Johann Meckel mô tả vào năm 1822 ở một cặp anh/chị/em ruột
• Năm 1934, Georg Gruber đã mô tả 6 thai nhi tương tự bị “dysencephalia splanchnognostica"
• Hội chứng di truyền lặn với rối loạn chức năng lông chuyển trong quá trình tạo phôi, dẫn đến
- Loạn sản thận dạng nang (98-100%)
- Thoát vị não hoặc các bất thường hệ thần kinh trung ương khác (60-90%)
- Thừa ngón sau trục (70-87%)
- Dị dạng tấm ống gan (Hepatic ductal plate malformation) → xơ hóa (> 95%)
6 HÌNH ẢNH
Đặc điểm chung
- Manh mối chẩn đoán tốt nhất
• Thai nhi có NST bình thường có ít nhất 2 đặc điểm trên siêu âm; ~ 60% có cả 3
Dấu hiệu trên siêu âm
• Đường niệu dục
- Loạn sản thận dạng nang (~98-100%)
- Hình ảnh thận trên siêu âm thay đổi
- Thận rất to, tăng âm, là phổ biến nhất
- Gấp 10-20 lần kích thước thận bình thường
- Có thể có các nang lớn (2-10 mm)
- Chu vi bụng có thể tăng đáng kể
- Bàng quang có thể nhỏ hoặc không có
- Thiểu ối/vô ối trong quý 2
- Trong quý 1, nước ối vẫn bình thường, trước khi thận đóng vai trò chính trong việc sản xuất nước ối
- Bệnh lý tiết niệu do tắc nghẽn; hiếm gặp, bất sản thận
- tinh hoàn ẩn (Cryptorchidism); rối loạn biệt hóa giới tính
• Hệ thần kinh trung ương (60-90%)
- Thoát vị não vùng chẩm là hay gặp nhất
- Dị tật Dandy-Walker
- Đầu nhỏ, giãn não thất
- Bất sản thể chai
- Holoprosencephaly
- Khiếm khuyết xương sọ (Cranial rachischisis)
- Bất sản thùy nhộng dưới, thiểu sản tiểu não
• Các chi
* Thừa ngón sau trục (70-87%)
– Ngón thừa có thể nhỏ hoặc gập góc
- Thường ảnh hưởng cả 4 chỉ như nhau, biểu hiện đa dạng nhất trong tam chứng cổ điển
• Ít gặp hơn: Bàn chân khoèo, xương dài ngắn, cong
• Gan
• Dị dạng tấm ống gan → xơ hóa gan
– Khó đánh giá chính xác trên siêu âm; có thể xác nhận khi khám nghiệm tử thi
– Tìm dấu hiệu gan to và lưu lượng máu trong gan kém nếu biểu hiện trong quý 3
• Dị tật trên khuôn mặt
- Sứt môi/hở hàm ếch, cằm nhỏ
- Mắt nhỏ; lưỡi to
- Trán dốc
• Tim
- Khiếm khuyết vách ngăn
- Hẹp eo động mạch chủ
- Kênh nhĩ thất không cân bằng
Khuyến nghị về hình ảnh
- Có thể chẩn đoán được trong quý 1
• Dấu hiệu đầu tiên có thể là độ mờ da gáy dày
• Sử dụng siêu âm qua ngả âm đạo để đánh giá thoát vị não, bệnh nang thận nếu tiền sử gia đình dương tính
+ Siêu âm sớm bình thường không loại trừ hoàn toàn hội chứng Meckel-Gruber (MKS)
- Siêu âm lại lúc 18-20 tuần tuổi nếu tiền sử gia đình dương tính
- MR rất hữu ích nếu thiểu ối làm hạn chế khả năng quan sát
7 CHẨN ĐOÁN PHÂN BIỆT
Trisomy 13
• Có sự trùng lặp đáng kể về các đặc điểm hình ảnh
• Bất thường thận ở 50%
- Loạn sản dạng nang
+ Thận tăng âm kèm nang rải rác
+ Thận có thể lớn nhưng thường nhỏ hơn trong MKS
- Thận ứ nước
• CNS
- Holoprosencephaly ở 40%
- Thoát vị não đã được báo cáo, nhưng ít gặp hơn
• Các chi
- Thừa ngón sau trục ở 75%
- Bàn chân đu lắc (Rocker-bottom foot)
• Thiểu ối ít gặp hơn
- Có thể đa ối
• Dị tật tim 80%
- khiếm khuyết vách ngăn
- Thiểu sản tim trái
- Hẹp van động mạch chủ/van hai lá
• Thai giới hạn tăng trưởng
• Thoát vị rốn
Hội chứng Smith-Lemli-Opitz (DHCR7)
• CNS
- Đầu nhỏ, holoprosencephaly, não úng thủy, bất sản thể chai
• Dị tật tim
- Kênh nhĩ thất, thông liên thất, thiểu sản tim trái
• Đường niêu-dục
- Rối loạn biệt hóa giới tính, bệnh nang thận
• Thừa ngón sau trục
• Các dạng nặng có thể trùng lặp với MKS
• Đặc điểm khuôn mặt riêng biệt
- Hai hốc mắt xa nhau, mũi ngắn hếch, tai đóng thấp, cằm nhỏ, trán cao rộng, nếp mí mắt rẻ quạt (epicanthal folds)
Bệnh thận đa nang di truyền lặn trên NST thường (PKHD1)
• Thận to, tăng âm
• Không có thoát vị não hoặc thừa ngón
• Thiểu ối/hiếm khi vô ối
Thận loạn sản nhiều nang hai bên (Bilateral Multicystic Dysplastic Kidneys)
• Không có đặc điểm nào khác của MKS
Hội chứng Hydrolethalus (HYLS1, KIF7)
• Thừa ngón (thường gấp đôi ngón chân cái), não úng thủy, bất thường tim
• Không có nang thận
Hội chứng Bardet-Biedl (các gen BBS)
• Thừa ngón, loạn dưỡng thận tiến triển, bất thường gan, dị tật tim
• Không bị thoát vị não; loạn dưỡng võng mạc, béo phì
Hội chứng Joubert (> 10 gen)
• Dị tật tiểu não: thiểu sản thùy nhộng (dấu hiệu răng hàm (molar tooth sign))
• Thoát vị não vùng chẩm, thừa ngón, bệnh thận: Ít gặp hơn
• Giảm trương lực cơ, khuyết tật nhận thức
Hội chứng COACH (TMEM67)
• Thiểu sản/bất sản thùy nhộng tiểu não (Cerebellar vermis hypo-/aplasia), thiểu năng trí tuệ (oligophrenia), thất điều bẩm sinh (congenital ataxia), khuyết mô mắt (coloboma), xơ hóa gan (hepatic fibrosis)
8 BỆNH HỌC
8.1 Đặc điểm chung
• Di truyền học
- Bệnh lông chuyển (ciliopathy) làm hạn chế cuộc sống
+ Các allen ở cùng locus (Allelism) với phổ bệnh lông chuyển (spectrum of ciliopathies) (Joubert, Bardet-Biedl, Oro-facio-digital, v.v.)
- Di truyền lặn trên NST thường; cực kỳ không đồng nhất về mặt di truyền
+ Nguy cơ tái phát 25%
- Ít nhất 15 gen gây bệnh MKS
+ MKS1, TMEM216 (MKS2), TMEM67 (MKS3), CEP290 (MKS4), RPGRIPIL (MKS5), CC2D2A (MKS6), NPHP3 (MKS7), TCTN2 (MKS8), B9D1 (MKS9), B9D2 (MKS10), TMEM231 (MKS11), KIF14 (MKS12), TMEM107 (MKS13), TXNDC15, TCTN1
TMEM67 (16%) và MKS1 (7%) là 2 gen phổ biến nhất
+ Biến thể gây bệnh ở các gen này: Chỉ chiếm 50-60% các trường hợp MKS lâm sàng
8.2 Đặc điểm vi thể
• Thận
- Loạn sản dạng nang
- Nephron bị thiếu trầm trọng; tủy: nang nhỏ
- Biệt hóa vỏ tủy kém/không có
- Có thể lớn gấp 10-20 lần kích thước bình thường
• Các tế bào nguyên bào xơ-cơ ở gan và thận
• Xơ hóa gan (dị tật tấm ống)
- Ngừng phát triển hệ thống đường mật trong gan
+ Tăng sinh ống mật phản ứng
+ Giãn ống mật
- Xơ hóa quanh cửa
+ Dẫn đến tắc mạch cửa
9 CÁC VẤN ĐỀ LÂM SÀNG
9.1 Biểu hiện kiểu hình
• Các dấu hiệu/triệu chứng hay gặp nhất
- Hầu hết phát hiện được trên siêu âm quý 1
+ 90% được chẩn đoán lúc 14,3 ± 2,6 tuần tuổi
- Thiểu ối trong quý 2 và 3
• Các dấu hiệu/triệu chứng khác
- Có thể có tiền sử đứa trẻ bị ảnh hưởng trước đó
- Tăng a-fetoprotein huyết thanh mẹ (do thoát vị não)
+ Có thể bình thường nếu được bao phủ bởi màng
- Đa dạng về kiểu hình
+ Các bất thường kèm theo khác nhau đáng kể giữa các trường hợp
9.2 Dịch tễ học
• 1 trên 135.000 ca sinh trên toàn thế giới; khác nhau giữa các khu vực; M =F
- Dân số Bỉ: 1:3.000
- Dân số Phần Lan: 1:9.000
- Tỷ lệ tăng khi kết hôn cận huyết (ví dụ: Kuwati Bedouin)
- Tỷ lệ tăng: người Ấn Độ Gujarati, người Tatar và Hutterite
• 5% thai nhi bị thoát vị não có MKS
9.3 Diễn tiến tự nhiên & Tiên lượng
• Hạn chế cuộc sống
- Thiểu ối dẫn đến thiểu sản phổi
- Hầu hết thai chết lưu hoặc chết trong vòng vài giờ; đã có trường hợp sống được vài tháng năm (5-28 tháng)
9.4 Điều trị
• Microarray (aCGH)/NST đồ để loại trừ trisomy 13
• Đề nghị đình chỉ thai nghén hoặc chăm sóc giảm nhẹ
• Cố gắng hạn chế các can thiệp làm tăng tỷ lệ bệnh tật; theo dõi thai nhi/mổ lấy thai
• Chu vi bụng lớn có thể gây đẻ khó do bụng (abdominal dystocia)
• Lấy máu cuống rốn để xét nghiệm NST/đơn gen
• Khám/khám nghiệm tử thi bởi chuyên gia bệnh học/di truyền học có kinh nghiệm để xác nhận chẩn đoán
• Tư vấn di truyền cho các lần mang thai sau
- Nguy cơ tái phát 25%
10 BẢNG KIỂM CHẨN ĐOÁN
Cân nhắc
• MR khi thiểu ối làm hạn chế khả năng đánh giá hình thái thai nhi
Các điểm cần lưu ý
• Có sự trùng lặp đáng kể về các đặc điểm hình ảnh với trisomy 13
- Xét nghiệm chẩn đoán để đánh giá NST, xét nghiệm đơn gen để xác định nguyên nhân, cho biết nguy cơ tái phát
+ nguy cơ tái phát ~ 1% đối với trisomy 13 so với 25% đối với MKS
• Hình ảnh thận rất đa dạng, từ thận to tăng âm cho đến thận bị thay thế hoàn toàn bằng những nang lớn
- Kích thước thận thường rất to khiến chu vi bụng lớn
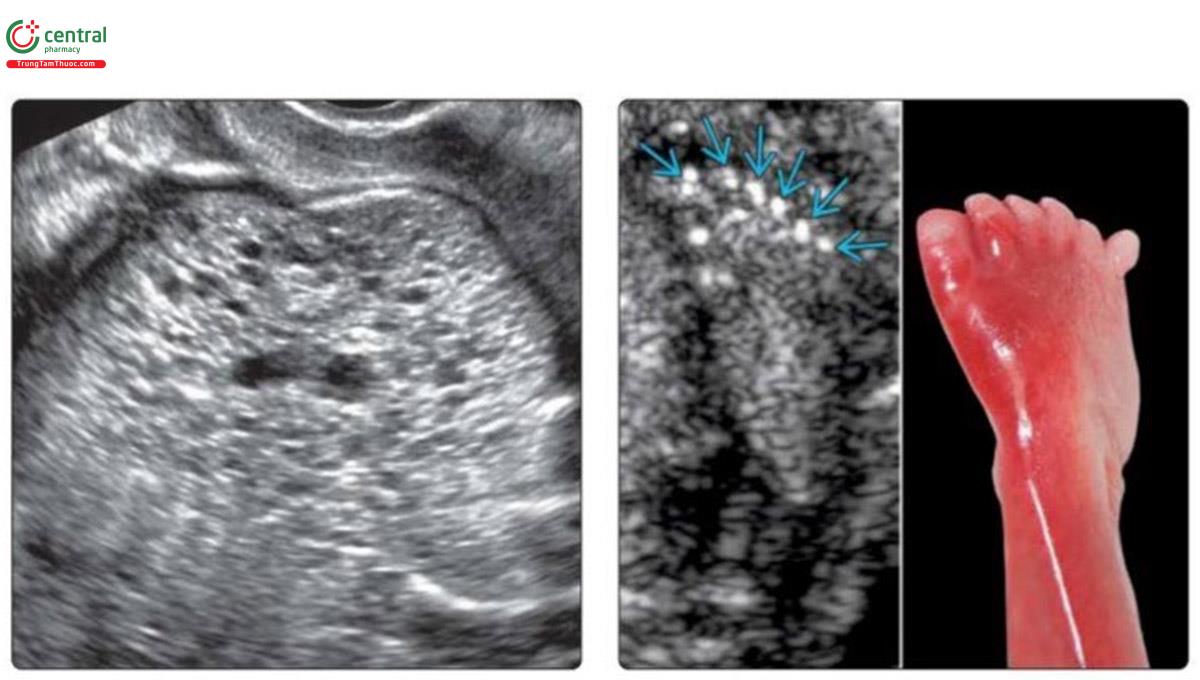
(Trái) Thai nhi bị MKS này ở tư thế nằm ngang, cột sống hướng xuống dưới nên được siêu âm qua ngả âm đạo để đánh giá thận tốt hơn. Hình ảnh chi tiết thận phải cho thấy nó bị thay thế bằng nhiềnang nhỏ và hầu như không còn nhu mô bình thường. (Phải) Siêu âm bàn chân của cùng một thai nhi cho thấy thừa ngón (mũi tên xanh). Thừa ngón là dấu hiệu ít gặp nhất trong MKS và có thể dễ bị bỏ sót thứ phát do thiểu ối. Thừa ngón sau trục được xác nhận trên hình ảnh khám nghiệm tử thi.
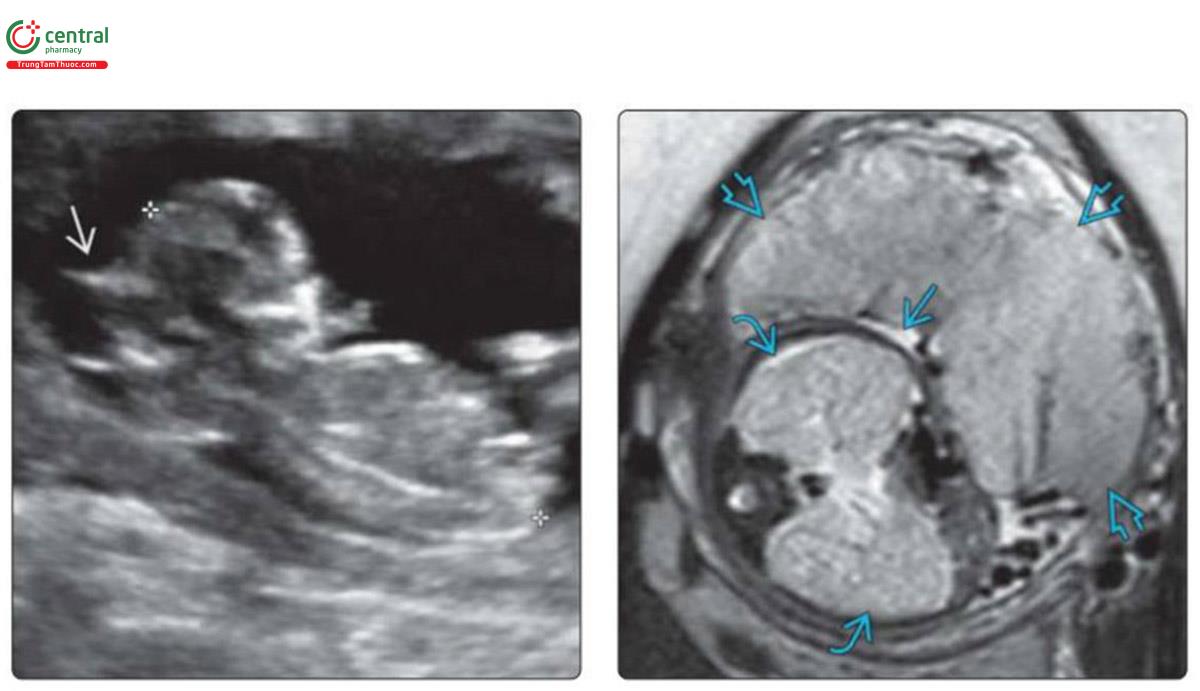
(Trái) Hình ảnh siêu âm mặt cắt dọc của thai nhi 12 tuần tuổi mắc MKS ở bệnh nhân đã có con bị bệnh trước đó. Thấy thoát vị não lớn (mũi tên trắng) với mô não lồi ra ngoài cơ thể. Xét nghiệm đơn gen phát hiện 2 biến thể gây bệnh ở gen CC2D2A (MSK6). (Phải) T2 MRI qua tử cung trong trường hợp MKS ở tuần thứ 32 cho thấy thiểu ối nặng. Thai nhi bị ép vào bánh nhau (mũi tên xanh to) chỉ có một lượng nhỏ nước ối có tín hiệu cao xung quanh (mũi tên xanh bé thẳng) Thận thai nhi (mũi tên xanh bé cong) rất to, chiếm hết ổ bụng.
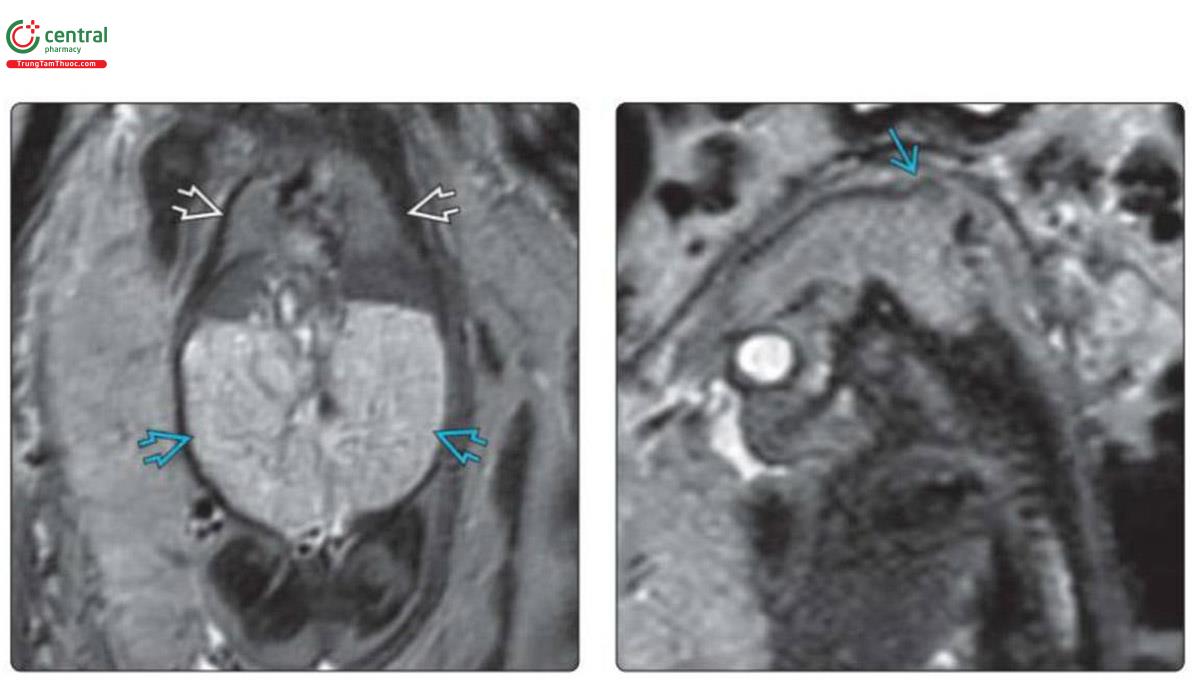
(Trái) Mặt cắt vành của cùng một thai nhi cho thấy thận rất to (mũi tên xanh to), cả 2 thận đều có kích thước > 7 cm. Thận có tín hiệu cao bất thường và không có biệt hóa vỏ tủy bình thường. Ngực (mũi tên trắng to) nhỏ và có hình chuông. (Phải) Mặt cắt dọc não của cùng một thai nhi cho thấy thoát vị não vùng chẩm nhỏ (mũi tên xanh bé). Hộp sọ nhỏ và có hình dạng không đều. Ngoài ra, còn thấy tiểu não dị dạng thoát vị xuống dưới vào cột sống cổ trên, phù hợp với Chiari 3.
11 TÀI LIỆU THAM KHẢO
1. McConnachie DJ et al: Ciliopathies and the kidney: a review. Am J Kidney Dis. 77(3):410-9, 2021
2. Radhakrishnan P et al: Meckel syndrome: clinical and mutation profile in six fetuses. Clin Genet. 96(6):560-5, 2019
3. Ridnõi K et al: A prenatally diagnosed case of Meckel-Gruber syndrome with novel compound heterozygous pathogenic variants in the TXNDC15 gene. Mol Genet Genomic Med. 7(5):e614, 2019
4. Hartill V et al: Meckel-Gruber syndrome: an update on diagnosis, clinical management, and research advances. Front Pediatr. 5:244, 2017
5. Khurana S et al: Meckel-Gruber syndrome: ultrasonographic and fetal autopsy correlation. J Ultrasound. 20(2):167-70, 2017
