Hội chứng Marfan là đột biến gì? Nguyên nhân, triệu chứng và cách điều trị
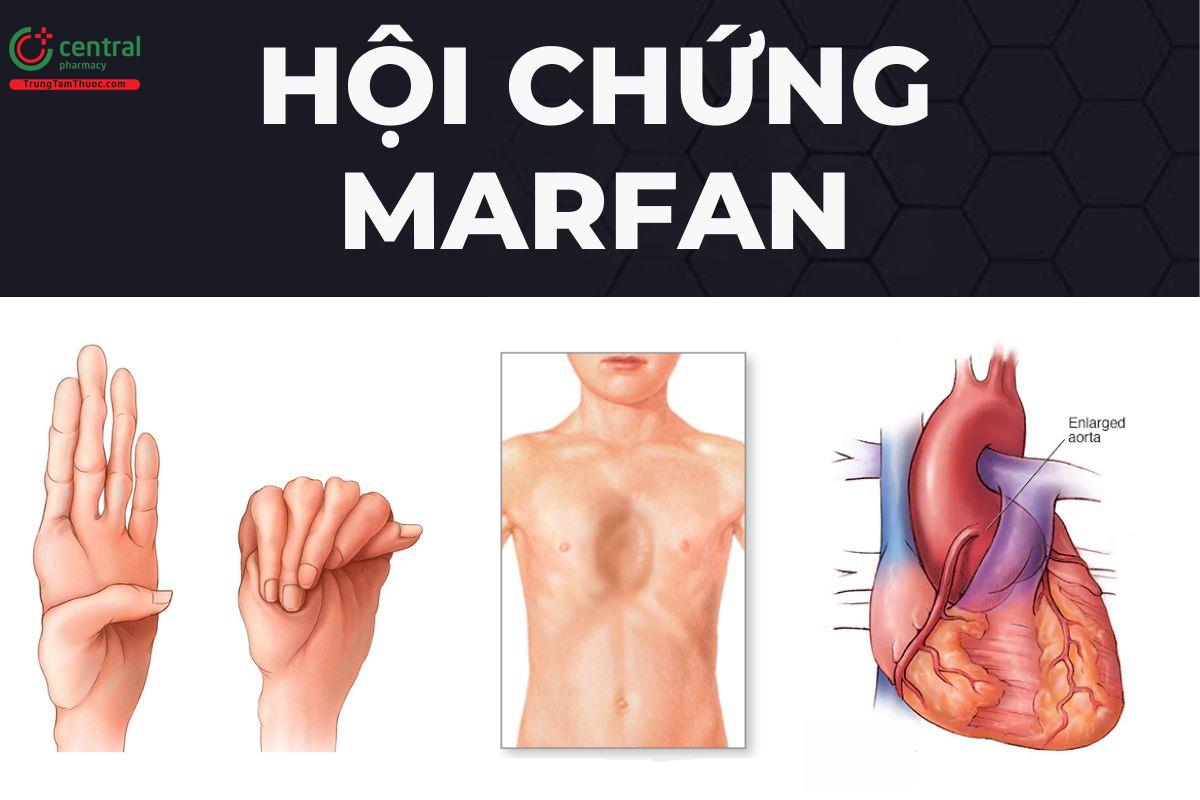
Trungtamthuoc.com- Hội chứng Marfan là một rối loạn di truyền liên quan đến các mô liên kết của cơ thể, gây ra những tổn thương đến tim, mạch máu, xương và các cơ quan nội tạng khác. Hãy cùng Trung Tâm Thuốc Central Pharmacy cùng tìm hiểu trong bài viết dưới đây.
1 Hội chứng Marfan do đột biến gen trội trên NST số mấy?
Hội chứng Marfan do đột biến gen trội trên NST 15 khá hiếm gặp, cụ thể là rối loạn di truyền do đột biến trong gen FBN1 (fibrillin-1) mã hoá của mô liên kết. Mô liên kết đóng vai trò quan trọng trong xây dựng cấu trúc xương, mạch máu, da cũng như các cơ quan cơ thể.
Hội chứng Marfan được đặt tên theo bác sĩ người Pháp đã tìm ra nó vào năm 1986 tên là Antoine Marfan. Bệnh di truyền trội trên nhiễm sắc thể thường, có nghĩa là khả năng di truyền lại cho đời con khoảng 50% ở người mắc bệnh. Tuy nhiên người bệnh có thể sống hoàn toàn bình thường nếu mức độ biểu hiện của gen lên cơ thể thấp.

Hội chứng Marfan ở người có chân tay dài ngón tay dài là dấu hiệu nhận biết đặc trưng nhất, ngoài ra cũng gặp các biểu hiện trên mắt và tim mạch nhưng xuất hiện muộn hơn.
2 Hội chứng Marfan là bệnh di truyền do nguyên nhân gì?
Hội chứng Marfan gây ra bởi một khiếm khuyết trong gen fibrillin-1 (FBN1), đây là một protein thiết yếu của mô liên kết, tạo sự đàn hồi cho cơ thể. Do đó khi đột biến này xảy ra chức năng liên kết của các mô bị ảnh hưởng trực tiếp, suy yếu cấu trúc.[1]
Đa số bệnh nhân chịu sự di truyền từ gen bất thường của bố mẹ, phần nhỏ khoảng 25% trường hợp bệnh xảy ra do đột biến mới. Bệnh di truyền gen trội trên nhiễm sắc thể, nên chỉ cần 1 bố hoặc mẹ mắc đột biến truyền lại đủ để gây bệnh. Nếu cả 2 bố mẹ không có gen trội này thì nguy cơ con cái họ bị bệnh là 1/10000. Bệnh có tỷ lệ mắc ngang nhau ở cả nam và nữ, tương tự với các chủng tộc và khu vực trên thế giới.
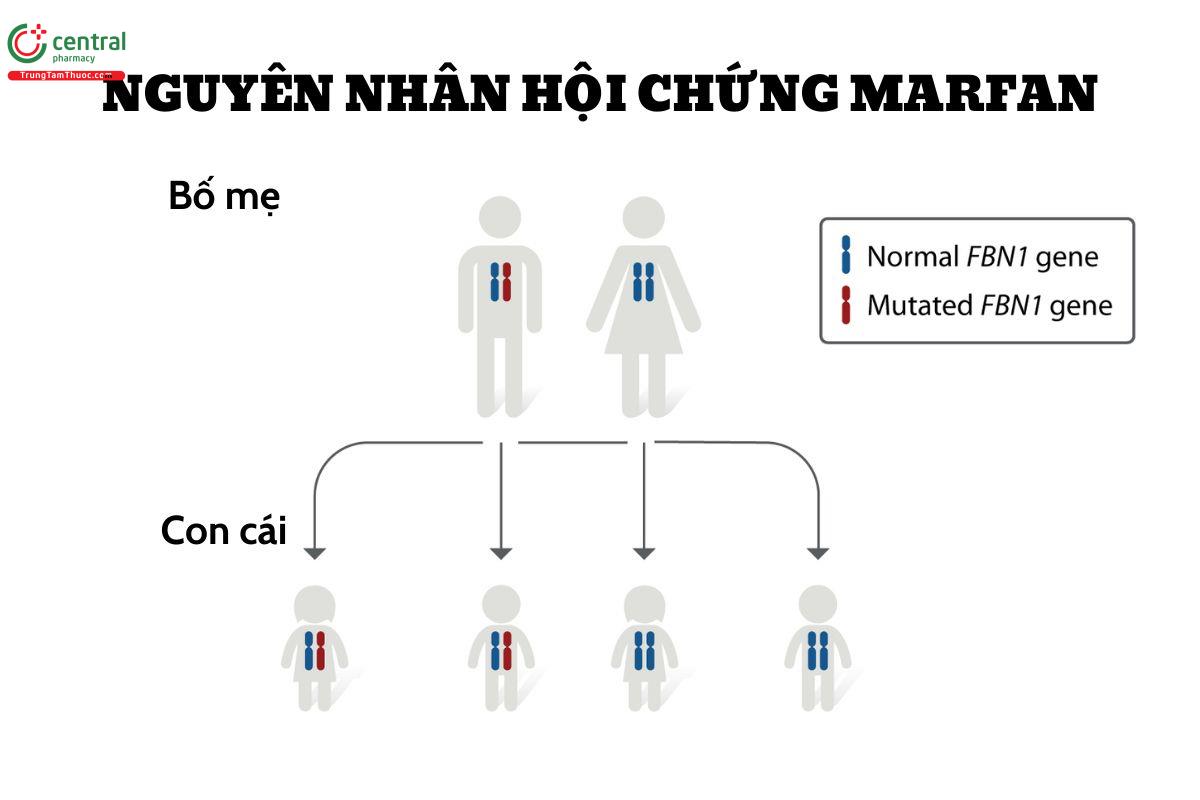
3 Tại sao hội chứng Marfan lại biểu hiện muộn?
Hội chứng Marfan liên quan đến mô liên kết nên các biểu hiện của bệnh tiến triển theo thời gian phát triển của cơ thể. Bên cạnh đó do mức độ ảnh hưởng của đột biến FBN1 có sự khác nhau về vị trí và loại đột biến, cụ thể:
Trong quá trình tăng trưởng, đặc biệt là giai đoạn dậy thì các mô liên kết sẽ phát triển nhanh hơn nên chịu áp lực lớn khiến chân tay dài nhanh chóng, vẹo cột sống. Ngoài ra biểu hiện trên mạch máu như phình động mạch chủ sẽ xảy ra nhiều hơn ở người lớn giai đoạn trưởng thành khi mạch máu bị tăng áp lực từ tim cao hơn.
Vị trí và loại đột biến gen FBN1 có thể khiến bệnh biểu hiện muộn hoặc triệu chứng nhẹ, tuy nhiên các triệu chứng nguy hiểm trên mạch máu như phình, bóc tách động mạch chủ thường chỉ phát hiện được khi xảy ra biến chứng.
====> Xem thêm bài viết: Bệnh Wilson (rối loạn chuyển hoá đồng) có nguy hiểm không? Triệu chứng và thuốc điều trị
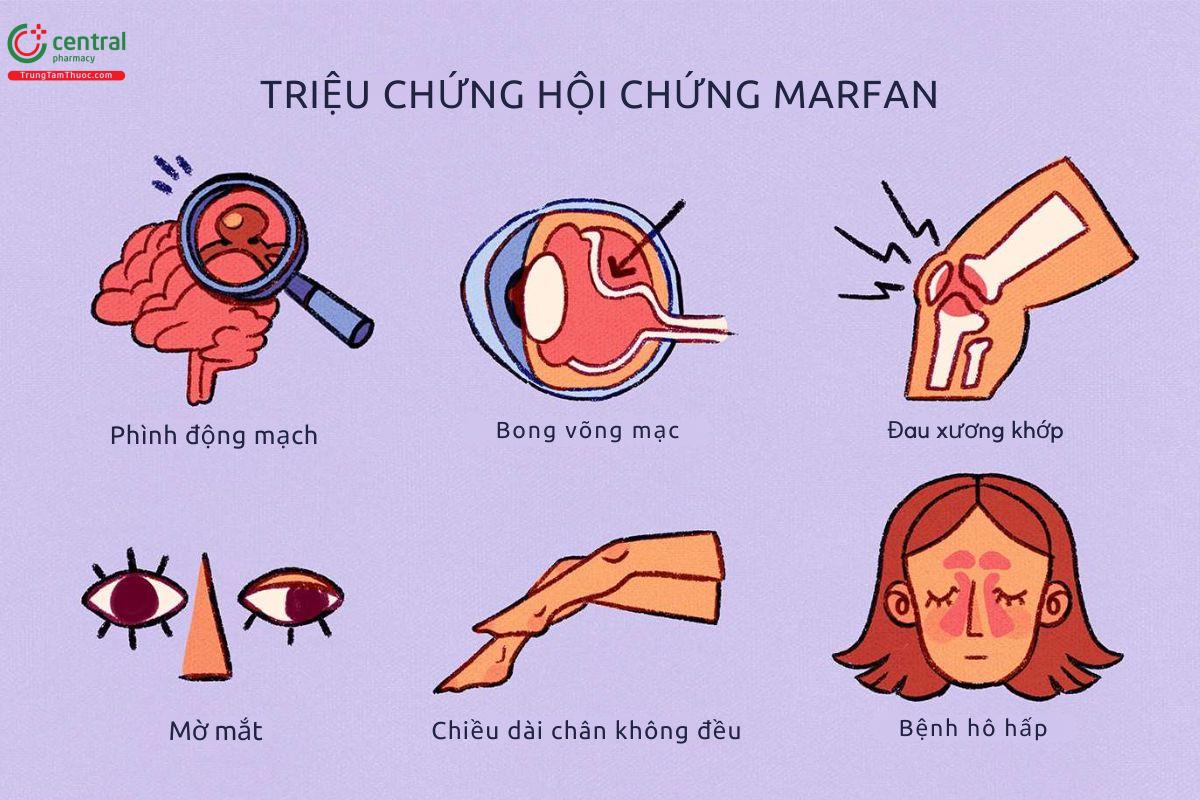
4 Triệu chứng hội chứng Marfan
Triệu chứng của hội chứng Marfan có thể rất khác nhau ở nhiều mức độ biểu hiện từ nhẹ đến trung bình, bao gồm:[2]
4.1 Hệ tim mạch
Các đặc điểm tim mạch của hội chứng Marfan bao gồm phình gốc động mạch chủ, tách gốc động mạch chủ, sa van hai lá, vôi hóa sớm vòng van hai lá và giãn động mạch phổi.
- Giãn gốc động mạch chủ thường tăng theo tuổi và thường đi kèm với trào ngược động mạch chủ. Bệnh lý động mạch chủ có thể dẫn đến hình thành phình động mạch; giãn; và cuối cùng là tách động mạch, nguyên nhân chính gây tử vong ở bệnh nhân.
- Sa van hai lá với các lá van dài cũng thường xảy ra ở những bệnh nhân mắc hội chứng Marfan, mặc dù đây được coi là một phát hiện không đặc hiệu, tuy nhiên sa van hai lá đôi khi dẫn đến suy tim ở trẻ em gây biến chứng nghiêm trọng.
- Ít phổ biến hơn, bệnh nhân có thể bị bệnh cơ tim không liên quan đến bệnh van tim như rối loạn nhịp tim.
4.2 Triệu chứng ở mắt
Tổn thương mắt được thấy ở phần lớn bệnh nhân mắc hội chứng Marfan.
- Lồi thủy tinh thể được thấy ở 50–80 % bệnh nhân; tình trạng này tiến triển, có thể làm suy giảm thị lực và thường cần can thiệp phẫu thuật.
- Các bất thường về nhãn khoa khác bao gồm nhược thị, lác cận thị, tăng chiều dài nhãn cầu và phẳng giác mạc. Nhãn cầu dài góp phần gây cận thị nặng và nguy cơ bong võng mạc, có thể dẫn đến suy giảm thị lực hoặc mù vĩnh viễn.
- Sự hình thành đục thủy tinh thể sớm từ khi còn trẻ, đây là tình trạng phần trong suốt thuỷ tinh thể bị mờ đục.
- Nguy cơ phát triển bệnh tăng nhãn áp, một biến chứng đã biết của hội chứng Marfan, tăng lên đáng kể sau khi phẫu thuật sửa chữa bong võng mạc.
- Dịch chuyển hoặc trật khớp thấu kính do sự suy yếu của hệ thống dây chằng neo giữ thấu kính, xảy ra khá phổ biến ở hơn 1 nửa người mắc hội chứng này.
4.3 Hệ thần kinh
Các biểu hiện thần kinh thường bao gồm giãn màng cứng, tình trạng mở rộng túi màng cứng xung quanh tủy sống và nang màng nhện tủy sống hoặc túi thừa. Giãn màng cứng nghiêm trọng và thoát vị màng não có thể gây đau lưng dưới và đau rễ thần kinh và yếu chân, đặc biệt là khi đứng và đi bộ trong thời gian dài.
4.4 Hệ hô hấp
Các biểu hiện ở phổi như khó thở, biến dạng ngực có thể là triệu chứng của hội chứng Marfan.
- Các biến chứng về phổi hoặc phổi có thể là kết quả của các bất thường ở thành ngực góp phần gây ra xẹp phổi, và tổn thương mô liên kết khiến phế nang không đàn hồi.
- Sự mở rộng của các khoảng phổi có thể dẫn đến tràn khí màng phổi tự phát, từ đó có thể dẫn đến tình trạng mất ổn định tim phổi.
- Bệnh nhân có thể phát triển bệnh khí phế thũng ở đường thở khi lớn tuổi, đối tượng thường gặp nhất là ở cuối độ tuổi trưởng thành.
- Tổn thương hô hấp cũng có thể gây ra rối loạn hô hấp khi ngủ ở người lớn.
4.5 Hệ cơ xương
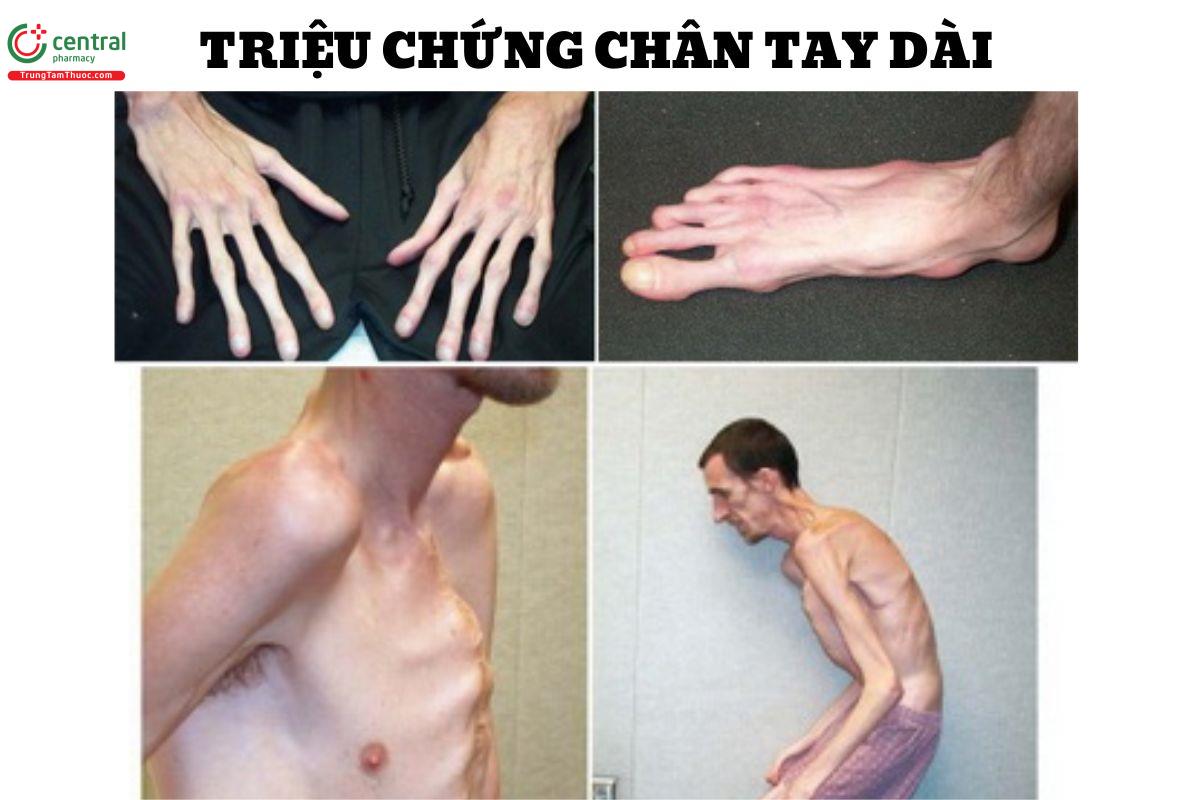
Các triệu chứng liên quan đến cơ xương khớp ở người bệnh bị hội chứng Marfan dễ nhận thấy rõ ràng nhất. Các đặc điểm cơ xương lâm sàng trong hội chứng Marfan thường bao gồm:
- Vóc dáng cao, chân tay và ngón tay dài không cân xứng, độ cong bất thường của cột sống, lõm hoặc lồi xương ức.
- Độ linh hoạt bất thường hoặc hạn chế của các khớp.
- Bàn chân phẳng, giảm duỗi khuỷu tay và co cứng ngón tay.
- Một số người có triệu chứng rối loạn ngôn ngữ khi vòng họng cao và hàm nhỏ.
Các biểu hiện cơ xương có thể gây đau với mức đau tăng theo thời gian và biểu hiện vẹo cột sống và chứng chân nhện phổ biến hơn ở phụ nữ so với nam giới. Sự lỏng lẻo của khớp góp phần gây ra viêm khớp thoái hóa tiến triển, đặc biệt là khi uốn cong hoặc mang vác thường xuyên và có áp lực lớn.
4.6 Các hệ cơ quan khác
Các vết rạn da không rõ nguyên nhân, ngay cả khi không tăng cân hoặc mang thai ở bệnh nhân mắc hội chứng Marfan đã được báo cáo.
Các biểu hiện trên cấu trúc xương mặt hàm như trật xương hàm, hàm nhỏ dẫn đến khó nói, rối loạn ngôn ngữ.
5 Chẩn đoán hội chứng Marfan
Các phương pháp chẩn đoán hội chứng Marfan dựa trên triệu chứng lâm sàng, xét nghiệm di truyền và phương pháp hình ảnh học. Các biểu hiện của bệnh thường dễ bị nhầm lẫn với các bệnh khác và thậm chí có triệu chứng không rõ ràng ở một số trường hợp, nên để chẩn đoán chính xác nhất cần phối hợp chặt chẽ các phương pháp với nhau. Tiêu chuẩn Ghent Revised Criteria (Tiêu chuẩn Ghent sửa đổi, 2010) thường được sử dụng để chẩn đoán hội chứng này. [3]
5.1 Thăm khám lâm sàng
Bác sĩ sẽ kiểm tra các dấu hiệu điển hình trên nhiều hệ cơ quan:
- Hệ xương: Quan sát chiều cao, tầm vóc, tỷ lệ sải tay/chiều cao, cấu trúc lồng ngực, và vẹo cột sống.
- Tim mạch: Nghe tim và kiểm tra các dấu hiệu bất thường như tiếng thổi ở van tim.
- Mắt: Kiểm tra thủy tinh thể, đo thị lực.
5.2 Chẩn đoán hình ảnh
- Siêu âm tim: Đo đường kính gốc động mạch chủ, phát hiện sa van hai lá hoặc hở van tim.
- MRI hoặc CT scan: Đánh giá chi tiết động mạch chủ và các mạch máu lớn khác.
- Dùng máy sinh hiển vi để phát hiện trật thủy tinh thể ở mắt.
5.3 Xét nghiệm di truyền
Xét nghiệm đột biến gen FBN1, rất hữu ích trong trường hợp chẩn đoán không rõ ràng hoặc bệnh nhân trẻ chưa biểu hiện đầy đủ triệu chứng, gia đình có người mắc hội chứng Marfan.
6 Biện pháp điều trị hội chứng Marfan
Hiện tại không có phương pháp điều trị khỏi hội chứng Marfan do đó để kiểm soát tốt bệnh và kéo dài sự sống cho bệnh nhân mắc rối loạn này cần phát hiện sớm, theo dõi và điều trị tích cực các biểu hiện ở nhiều hệ cơ quan, điều trị các suy giảm thứ phát liên quan rất cần thiết và việc theo dõi và điều trị phải tiến hành suốt đời.
6.1 Sử dụng thuốc
Các thuốc điều trị chủ yếu là thuốc tim mạch giúp giảm ảnh hưởng của bệnh đến tim, đặc biệt là động mạch chủ và van tim. Chẳng hạn như:
- Thuốc chẹn beta: làm chậm quá trình giãn động mạch chủ ở bệnh nhân do tác dụng giảm nhịp tim và áp lực trong động mạch chủ.
- Thuốc ức chế men chuyển: có tác dụng giảm huyết áp, bảo vệ mạch máu
6.2 Phẫu thuật
Phẫu thuật thay động mạch chủ: Thay thế đoạn phình hoặc yếu của động mạch chủ bằng mảnh ghép nhân tạo khi đường kính động mạch chủ vượt quá 5.0-5.5 cm hoặc tăng nhanh (>0.5 cm/năm). Điều trị sa van hai lá hoặc thay thế van trong trường hợp có hở van tim nghiêm trọng.
Phẫu thuật chỉnh hình: khi bệnh nhân bị biến dạng xương như vẹo cột sống, lồng ngực lõm hoặc nhô, và khớp lỏng lẻo.
Phẫu thuật mắt : Phẫu thuật thay thế thủy tinh thể nhân tạo nếu trật gây ảnh hưởng đến tầm nhìn. Đặc biệt khi bong võng mạc cần can thiệp phẫu thuật khẩn cấp để tránh mất thị lực.
6.3 Thay đổi lối sống
Tránh các hoạt động thể lực mạnh, đặc biệt là các môn thể thao cường độ cao như cử tạ, bóng đá, hoặc chạy đường dài, vì chúng làm tăng áp lực lên tim và mạch máu thay vào đó tập thể dục nhẹ nhàng như đi bộ, yoga hoặc bơi lội.
Có chế độ ăn giàu dinh dưỡng, duy trì cân nặng hợp lý để giảm áp lực lên khớp và cột sống.
Kiểm soát huyết áp thận trọng, duy trì trong giới hạn bình thường để giảm nguy cơ phình và bóc tách động mạch chủ
7 Tư vấn di truyền và sàng lọc hội chứng Marfan
Việc sàng học sớm hội chứng Marfan rất quan trọng để phát hiện và quản lý triệu chứng trong quá trình phát triển, giảm nguy cơ gặp biến chứng. Đối với gia đình có tiền sử mắc bệnh, việc tư vấn di truyền là điều cần thiết thực hiện, giúp lựa chọn những xét nghiệm di truyền phù hợp đánh giá nguy cơ di truyền cho con cái.
Xác định được hội chứng Marfan bằng xét nghiệm di truyền thường được chỉ định trong trường hợp gia đình có tiền sử mắc bệnh hoặc kết quả siêu âm nghi ngờ có triệu chứng bất thường. Nguyên nhân là các xét nghiệm di truyền thực hiện sẽ là xét nghiệm chuyên sâu cần xâm lấn vào thai nhi gồm sinh thiết nhau thai, chọc dò ối, sàng lọc phôi.
Tuy nhiên sàng lọc hội chứng Marfan vẫn có nhiều hạn chế vì có tới hơn 3000 đột biến gen FBN1 và không phải tất cả các đột biến đều phát hiện được. Bên cạnh đó khoảng 25% người mắc hội chứng này do đột biến mới, khi gia đình không có tiền sử mắc bệnh.
8 Hội chứng Ehlers-Danlos và hội chứng Marfan
Hội chứng Ehlers-Danlos và hội chứng Marfan là các rối loạn đa hệ thống chủ yếu ảnh hưởng đến các mô liên kết mềm. Tuy nhiên hai hội chứng này có thể phân biệt dựa trên nguyên nhân, triệu chứng và đặc điểm di truyền.
| Đặc điểm | Hội chứng Ehlers-Danlos | Hội chứng Marfan |
| Nguyên nhân | Liên quan đến nhiều gen, phổ biến là: COL5A1, COL5A2 (dạng cổ điển), COL3A1 (dạng mạch máu), giảm tổng hợp hoặc bất thường collagen | Đột biến ở gen FBN1 (Fibrillin-1) gây tổn thương protein fibrillin-1, ảnh hưởng đến độ bền của mô liên kết, giảm khả năng chịu lực của mô liên kết |
| Kiểu di truyền | Hầu hết là trội trên nhiễm sắc thể thường, một số dạng lặn trên nhiễm sắc thể thường | Trội trên nhiễm sắc thể thường, 25% trường hợp do đột biến mới |
| Triệu chứng lâm sàng | Da đàn hồi quá mức Không cao bất thường Chủ yếu ở dạng EDS mạch máu, có nguy cơ cao Vấn đề thị lực không điển hình | Da bình thường, không đàn hồi. Cao bất thường, tay chân dài Rất phổ biến, đặc biệt ở gốc động mạch chủ. Trật thủy tinh thể, cận thị nặng phổ biến |
| Chẩn đoán | Khám lâm sàng: Đánh giá độ đàn hồi da, tính lỏng lẻo khớp. Xét nghiệm di truyền: Xác định đột biến gen Collagen (COL5A1, COL5A2, COL3A1,..). | Khám lâm sàng: Dựa trên tiêu chuẩn Ghent sửa đổi (2010), bao gồm: Giãn/phình gốc động mạch chủ, trật thủy tinh thể, các đặc điểm xương đặc trưng. Xét nghiệm di truyền: Đột biến gen FBN1. |
9 Kết luận
Hội chứng Marfan là bệnh lý di truyền hiếm gặp có biểu hiện đa dạng trên nhiều bộ phận, nếu không được chẩn đoán và kiểm soát bệnh sớm có thể gây nguy hiểm đến tính mạng. Mong rằng bài viết đã cung cấp những thông tin hữu ích cho người đọc về hội chứng Marfan.
Tài liệu tham khảo
- ^ Tác giả Lynn Y. Sakai và cộng sự (ngày đăng 10 tháng 10 năm 2016) FBN1: The disease-causing gene for Marfan syndrome and other genetic disorders. Sciencedirect. Truy cập ngày 18 tháng 11 năm 2024
- ^ Tác giả Irim Salik; Prashanth Rawla (ngày đăng 23 tháng 1 năm 2023) Marfan Syndrome. Pubmed. Truy cập ngày 18 tháng 11 năm 2024
- ^ Tác giả Bart L Loeys và cộng sự (ngày đăng tháng 7 năm 2010) The revised Ghent nosology for the Marfan syndrome. Pubmed. Truy cập ngày 18 tháng 11 năm 2024
