Hội chứng Ehlers - Danlos (Ehlers - Danlos syndrome): dịch tễ, biểu hiện, cách chẩn đoán

Trungtamthuoc.com - Hội chứng Ehlers - Danlos (Ehlers - Danlos syndrome) là một bệnh di truyền do đột biến gen với dấu hiệu chính là da giãn quá mức, khớp lỏng và các vết bầm tím. Bài viết dưới đây sẽ giúp bạn đọc hiểu nguyên nhân, biểu hiện và cách chẩn đoán hội chứng này.
Chương 4. BỆNH DA HIẾM GẶP DO DI TRUYỀN, HỘI CHỨNG EHLERS - DANLOS (Ehlers - Danlos syndrome), trang 96-101, Sách BỆNH DA HIẾM GẶP
Nhà xuất bản Y học Hà Nội - 2024
Chủ biên: Thầy thuốc Nhân dân, Giáo sư, Tiến sĩ Trần Hậu Khang - Giảng viên Cao cấp Trường Đại học Y Hà Nội
Tải bản PDF TẠI ĐÂY
1 LỊCH SỬ BỆNH
Hội chứng Ehlers - Danlos (EDS cũng được gọi là bệnh Ehlers - Danlos disease - EDD) là một bệnh cổ nhất mà lịch sử y học ghi nhận. Bệnh được Hippocrates mô tả lần đầu tiên vào năm 400 trước công nguyên. Tuy nhiên mãi đến năm 1892, Tschenogobow mới báo cáo hai trường hợp nghi là EDS với các triệu chứng ở da và khớp.
Năm 1901, nhà Da liễu học người Đan Mạch Edvard Ehers báo cáo chi tiết toàn bộ các biểu hiện của một bệnh nhân với dấu hiệu chính là da giãn quá mức, khớp lỏng và các vết bầm tím. Đến năm 1908, Henri - Alexandre Dalos, bác sĩ người Pháp tổng hợp các triệu chứng của hội chứng này với các biểu hiện ở da, khớp, sụn, mạch máu và một số cơ quan khác.
Năm 1998, Beighton công bố bảng phân loại EDS theo phân loại bệnh của Villefranche.
2 CĂN SINH BỆNH HỌC
EDS là một bệnh di truyền do đột biến gen mã hoá các sợi Collagen type I, III và V. Ngoài ra người ta cũng đã xác định được sự đột biến gen mã hoá các enzyme có liên quan tới chuyển hoá và sửa chữa các sợi collagen.
Chính vì đột biến gen liên quan đến sự tổng hợp, sửa chữa collagen nên các cơ quan tổ chức chứa collagen bị tổn thương như da, khớp, sụn, mạch máu, đặc biệt là hiện tượng chùng da, lỏng khớp, xuất huyết là đặc điểm chung của bệnh.
3 PHÂN LOẠI
Có nhiều cách phân loại song bảng phân loại năm 1998 của Beighton được sử dụng nhiều hơn:
Bảng phân loại EDS theo Beighton 1998
STT | Thể bệnh | Kiểu di truyền | Gen đột biến | Biểu hiện lâm sàng |
1 | EDS cổ điển | AD | COL5A1 | Da giãn, khớp lỏng, sẹo teo |
2 | EDS tăng vận động (Type III) | AD | COL5A2 | - Khớp lỏng, tăng vận động - Bầm tím da, da mềm, sẹo teo |
3 | EDS mạch (Type IV) | AD | COL3A1 | Da mỏng, mạch dễ vỡ: mạch ruột, tử cung |
4 | EDS gù, vẹo (Type VI) (Kyphoscoliosis) | AR | PLOD | - Triệu chứng giống thể cổ điển - Gù, vẹo, biến chứng mắt - Giảm trương lực (Hypotonia) |
5 | EDS thể A, B (Type VII) (Arthrochalasis) | AD | COL1A1 COL1A2 | - Tăng vận động khớp nặng - Chảy máu - Giảm trương lực - Vẹo cột sống |
6 | Dermatosparaxis | AR | Collagen N-peptidase | - Da mỏng, chùng, chảy xệ - Bầm tím |
Ghi chú: AD = Autosomal dominant: di truyền trội AR = Autosomal recessive: di truyền lặn | ||||
4 DỊCH TỄ BỆNH
Bệnh rất hiếm gặp, tỷ lệ lưu hành tuỳ thuộc vào thể bệnh và từng quốc gia:
- Thể VI (Kyphoscoliosis) thường hay gặp ở các nước như Thổ Nhĩ Kỳ, Hy Lạp, Trung Đông.
- Thể Dermatosparaxis: cực kỳ hiếm gặp, chỉ có một vài trường hợp trên thế giới được báo cáo.
- Thể mạch (Type IV): 1/100.000.
- Thể cổ điển: 1/20.000 - 1/40.000.
- Tỷ lệ lưu hành chung là 1/5000.
5 BIỂU HIỆN LÂM SÀNG
Các biểu hiện lâm sàng của các thể riêng biệt được trình bày trong bảng phân loại của Beighton.
Các triệu chứng sau đây hay gặp nhất của các thể:
- Khớp: lỏng, vận động quá dễ và theo bất kỳ vị trí nào: xoay vòng, gập, duỗi trước sau, ngược chiều.
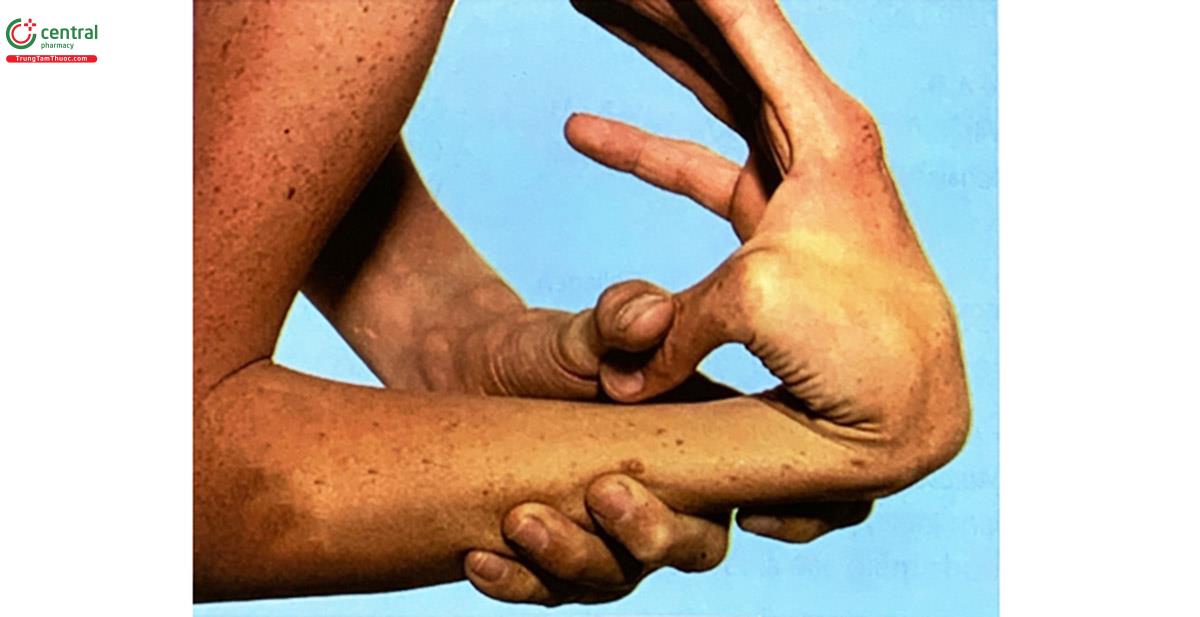
- Da: mỏng mịn như nhung, chùng, mềm và dễ rạn, thương tổn, khó hình thành sẹo.
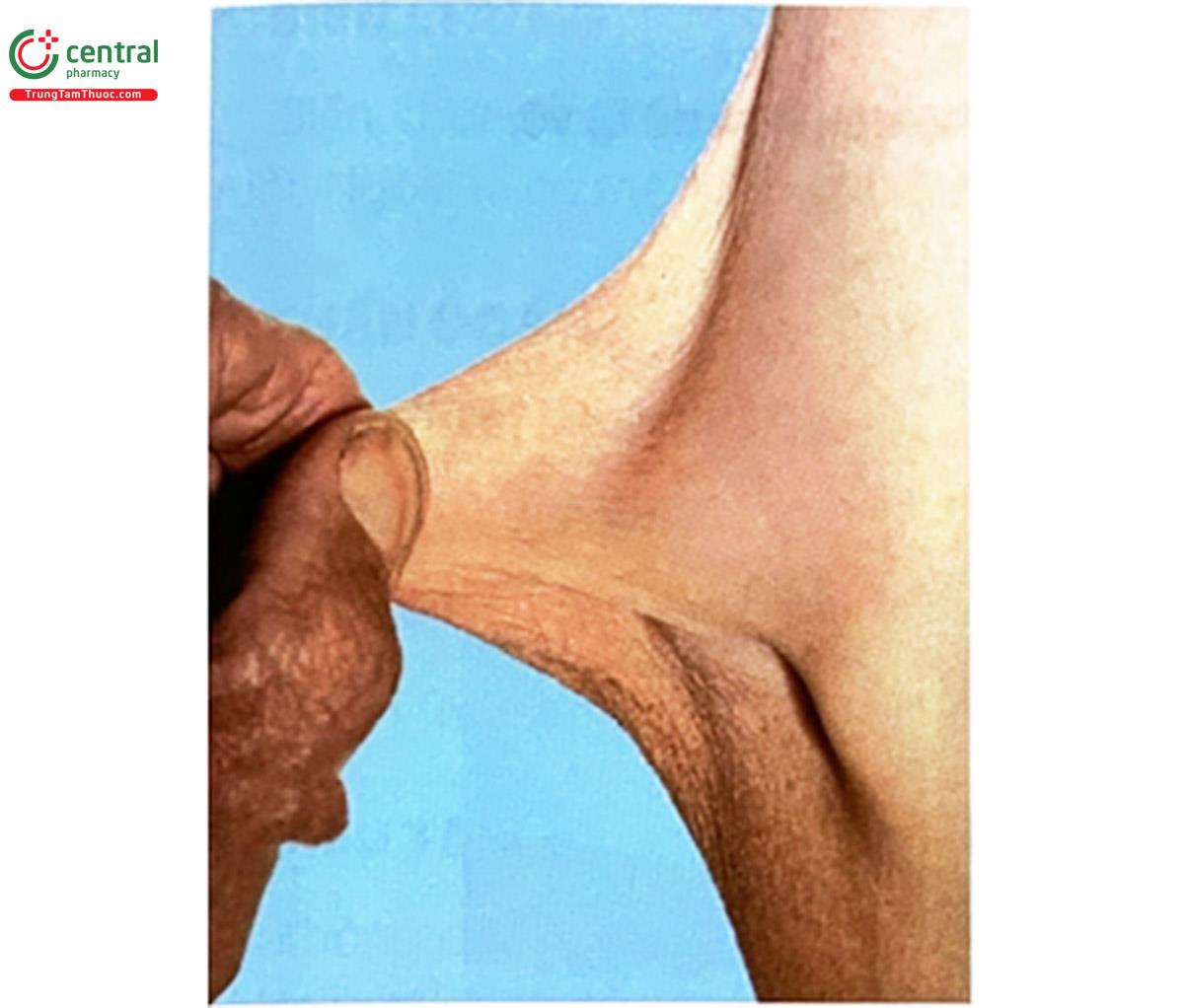
- Da bị rạn, bầm tím: do xuất huyết.
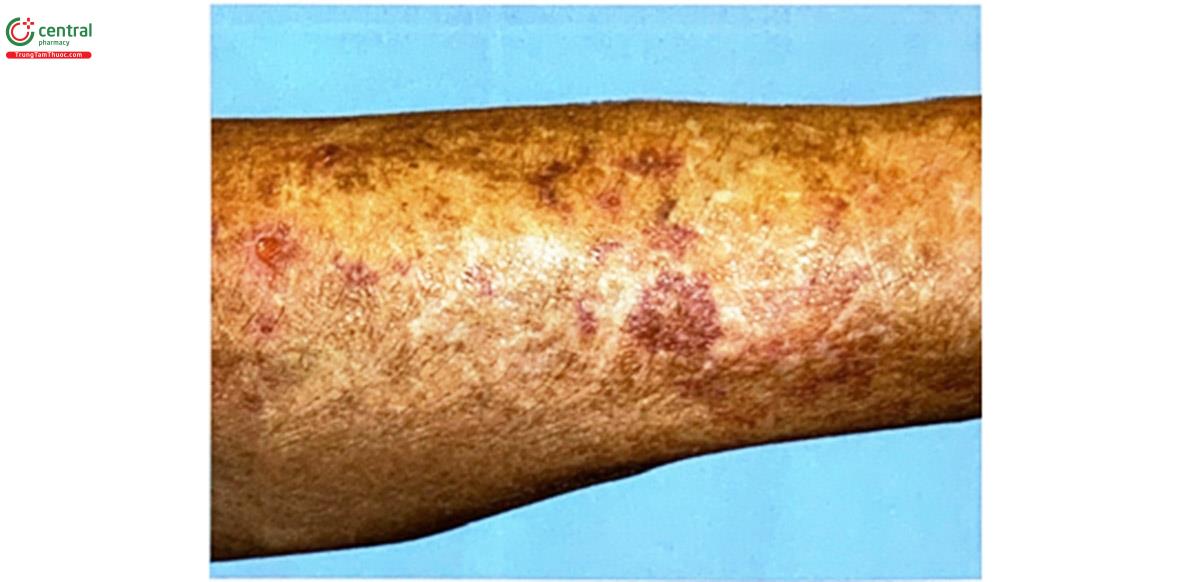
Thể mạch máu type IV có các triệu chứng đặc biệt sau đây:
- Mặt điển hình:
+ Mũi nhỏ, mỏng.
+ Môi trên nhỏ.
+ Dái tai nhỏ.
+ Mắt lồi.
+ Các mao mạch nổi dưới da.
+ Động mạch chủ và các mạch tử cung, hệ tiêu hoá dễ bị vỡ.
- Triệu chứng khác:
+ Đau đầu, thoát vị đĩa đệm.
+ Vẹo cột sống.
+ Rối loạn thị lực.
+ Hội chứng dạ dày, tiêu hoá.
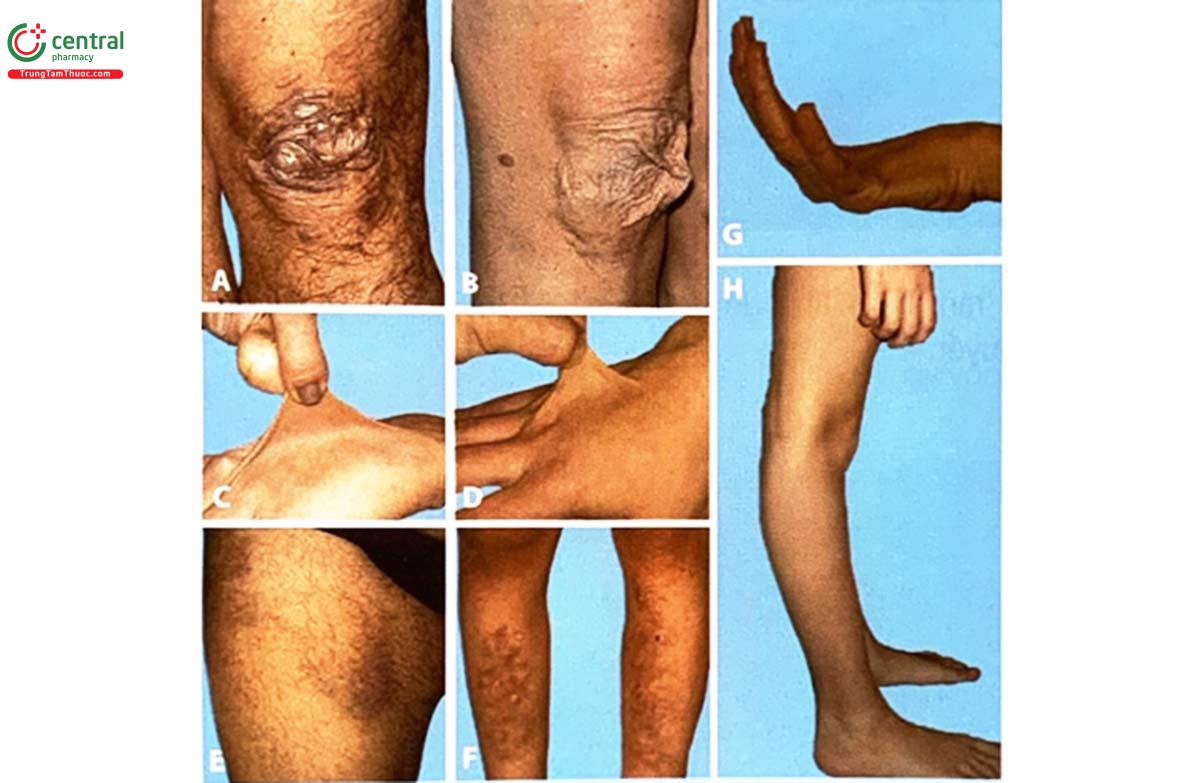
A, B. Sẹo vùng đầu gối ở người trưởng thành mắc hội chứng Ehlers-Danlos cổ điển
C, D. Các mức độ rạn da khác nhau
E, F. Bầm tím, xuất huyết do mao mạch dễ vỡ
G, H. Tăng vận động khớp
(Nguồn: Marco Castori và cộng sự, 2014, Iranian Journal of Neurology)
6 BIẾN CHỨNG
Biến chứng nguy hiểm nhất là vỡ động mạch tử cung, hệ tiêu hoá: dạ dày, ruột. Ngoài ra có thể gây một số biến chứng khác như: trật khớp, sẹo, mù...
7 CHẨN ĐOÁN
Chẩn đoán bệnh dựa vào các yếu tố:
- Tiền sử gia đình.
- Biểu hiện lâm sàng đặc trưng của từng thể bệnh.
- Xét nghiệm di truyền phát hiện gen đột biến.
8 ĐIỀU TRỊ
Không có phương pháp điều trị khỏi bệnh này. Tuy nhiên cần theo dõi để xử lý kịp thời các triệu chứng có nguy cơ gây biến chứng trầm trọng.
Mời bạn đọc xem thêm về Hội chứng Leopard (Leopard syndrome) TẠI ĐÂY
