Hội chứng DiGeorge là gì, có di truyền không? Nguyên nhân, triệu chứng và cách điều trị

1 Định nghĩa
Rối loạn di truyền là tình trạng bệnh lý do bất thường trong DNA của một cá nhân gây ra. Trong đó, Hội chứng DiGeorge được coi là một trong những hội chứng di truyền phổ biến nhất với tỷ lệ mắc khoảng 1/2000 – 1/4000 trẻ sơ sinh, chỉ đứng sau hội chứng Down.
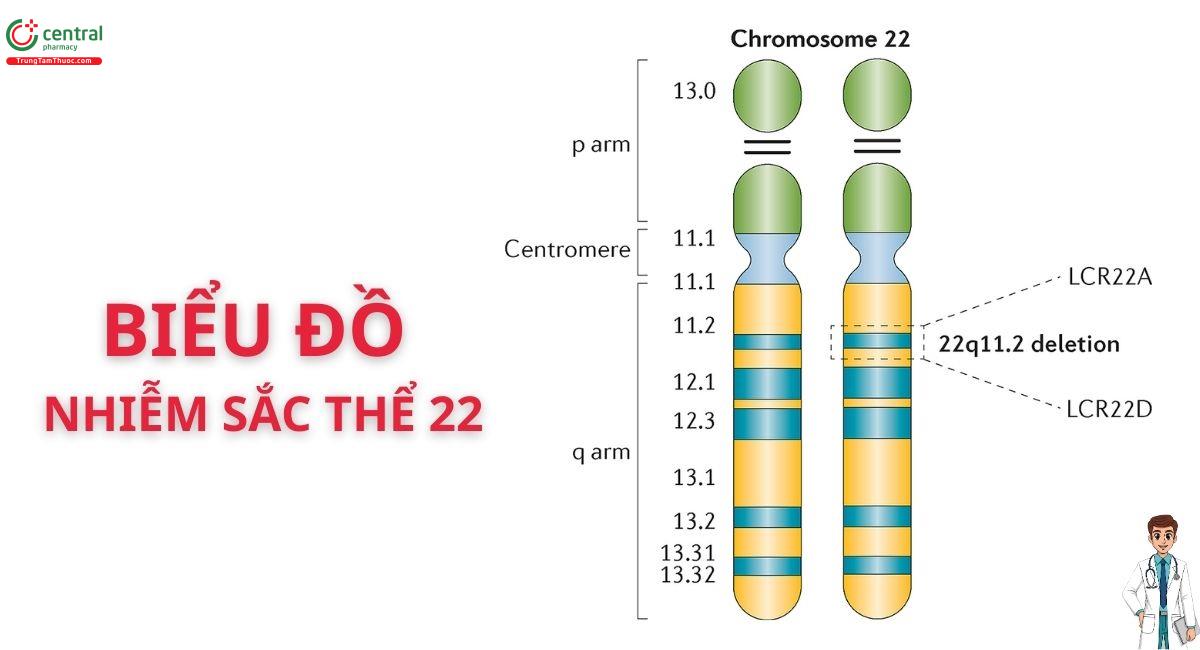
Hội chứng DiGeorge (DiGeorge Syndrome - DGS) là một rối loạn di truyền bẩm sinh có biểu hiện đa dạng, chủ yếu do mất đoạn nhiễm sắc thể 22 tại vị trí 22q11.2. Sự đột biến này gây ra sự phát triển bất thường của các túi hầu, ảnh hưởng đến sự hình thành của nhiều cơ quan và cấu trúc trong cơ thể, bao gồm tai ngoài, tai giữa, xương hàm trên và dưới, amidan khẩu cái, tuyến giáp, tuyến cận giáp, tuyến ức, cung động mạch chủ và các cấu trúc liên quan đến hệ tim mạch.[1]
2 Hội chứng DiGeorge nguyên nhân
Hội chứng DiGeorge (DGS) chủ yếu do mất đoạn nhiễm sắc thể 22, cụ thể hơn là trên cánh tay dài (q) tại locus 11.2 (22q11.2). Đột biến này thường phát sinh de novo, tức là một cách ngẫu nhiên trong quá trình hình thành trứng hoặc tinh trùng trong thụ thai. Điều này có nghĩa là đa số đột biến không được di truyền từ cha mẹ, và không có tiền sử gia đình mắc hội chứng DiGeorge.
Tuy nhiên, trong khoảng 10% trường hợp còn lại, mất đoạn 22q11.2 có thể được di truyền từ cha hoặc mẹ mắc hội chứng DiGeorge. Mặc dù những người này có thể không nhận ra mình mắc bệnh vì các triệu chứng có thể rất nhẹ, họ vẫn có thể truyền đột biến này cho con cái. Trong những trường hợp này, nguy cơ hội chứng DiGeorge tái phát ở con cái sẽ cao hơn.
3 Dịch tễ học
Hội chứng DiGeorge thường xảy ra do mất đoạn ở 22q11.2 và được mô tả lần đầu tiên vào năm 1968 như một tình trạng suy giảm miễn dịch nguyên phát do sự phát triển bất thường của túi hầu thứ ba và thứ tư trong giai đoạn phôi thai.
Hội chứng mất đoạn 22q11.2 (22q11.2DS) là hội chứng vi mất đoạn nhiễm sắc thể phổ biến nhất, với tỷ lệ mắc bệnh ước tính từ 1:3.000 đến 1:6.000 trẻ. Đa số các bệnh nhân (90–95%) mới được xác định mắc 22q11.2DS được phát hiện có đột biến mất đoạn de novo, nghĩa là đa số đột biến này phát sinh ngẫu nhiên, không di truyền từ cha mẹ. Tuy nhiên, với sự cải thiện về khả năng sống sót và sinh sản của bệnh nhân mắc hội chứng này, tỷ lệ mắc bệnh theo di truyền dự kiến sẽ tăng lên trong tương lai.
Hai nghiên cứu trước sinh đa trung tâm được công bố vào năm 2012 và 2015 đã báo cáo tình trạng mất đoạn 22q11.2 ở 1:347 và 1:992 thai nhi, sử dụng xét nghiệm trước sinh xâm lấn. Cả hai nghiên cứu đều bao gồm các phân tích về thai nhi có hoặc không có phát hiện siêu âm bất thường. Tỷ lệ mất đoạn 22q11.2 là khoảng 1:100 đối với thai nhi có dị tật cấu trúc lớn như CHD và là 1:1.000 ở thai nhi có vẻ bình thường về mặt giải phẫu (trong cả hai nghiên cứu).
Hội chứng này ảnh hưởng đến tất cả các giới và dân tộc, nhưng bệnh nhân không phải da trắng có thể ít được chẩn đoán hơn, có thể do các dấu hiệu lâm sàng khó nhận diện ở các nhóm dân tộc này.
22q11.2DS liên quan đến nhiều bệnh lý, đặc biệt là các dị tật tim bẩm sinh như tứ chứng Fallot (16%) và gián đoạn cung động mạch chủ (52%). Ngoài ra, bệnh cũng gây ra các vấn đề về phát triển trí tuệ (2–3%) và tâm thần (0,5–1%), cũng như các dị tật khác như hở hàm ếch và suy van hầu.
Tỷ lệ tử vong sớm ở trẻ sơ sinh mắc hội chứng này khoảng 4%, chủ yếu do các vấn đề như dị tật tim, hạ Canxi máu và tắc nghẽn đường thở với độ tuổi trung bình khi tử vong là 3–4 tháng. Tỷ lệ tử vong ở người lớn là khoảng 40 tuổi, với nguyên nhân có thể là đột tử không rõ nguyên nhân hoặc các bệnh lý tim mạch và tâm thần. Dù tỷ lệ tử vong đã giảm so với trước, nhưng vẫn cần các nghiên cứu thêm để làm rõ nguy cơ tử vong ở mọi lứa tuổi.[2]
4 Sinh lý bệnh
Hội chứng DiGeorge (DGS) là kết quả của vi mất đoạn nhiễm sắc thể 22 tại vị trí 22q11.2, mã hóa hơn 90 gen. Người mắc DGS có thể biểu hiện một loạt các kiểu hình khác nhau, trong đó những triệu chứng phổ biến nhất là dị tật tim, hạ canxi máu và thiểu sản tuyến ức.
Về mặt di truyền, yếu tố phiên mã hộp T 1 - TBX1 có liên quan chặt chẽ đến các kiểu hình đặc trưng của hội chứng này. Sự thất bại trong quá trình phát triển phôi thai của túi hầu, được điều khiển bởi TBX1, dẫn đến sự vắng mặt hoặc giảm sản tuyến ức và tuyến cận giáp. Các mô hình loại trừ gen TBX1 trên chuột và cá ngựa vằn đã được nghiên cứu để làm sáng tỏ cơ sở phôi thai của bệnh. Ở chuột, việc thiếu gen TBX1 gây ra các khiếm khuyết nghiêm trọng ở hầu, tim, tuyến ức và tuyến cận giáp, đồng thời dẫn đến các rối loạn hành vi. Các nghiên cứu trên cá ngựa vằn cũng cho thấy các dị tật ở tuyến ức, cung hầu và tai.
Ngoài ra, các nghiên cứu trên mô hình chuột loại bỏ gen 22q11.2 đã chỉ ra rằng những thay đổi phân tử và hành vi liên quan đến các bệnh lý như bệnh Parkinson, rối loạn phổ tự kỷ, rối loạn tăng động giảm chú ý và bệnh tâm thần phân liệt có thể xuất hiện ở những người mắc DGS. Những phát hiện này, cùng với các vấn đề mạch máu thần kinh tìm thấy ở chuột loại trừ gen TBX1, cho thấy cơ sở phân tử của các bệnh lý tâm thần liên quan đến DGS. Đặc biệt, những người mắc hội chứng DiGeorge có nguy cơ mắc bệnh tâm thần phân liệt cao gấp 30 lần so với dân số chung.1

5 Triệu chứng
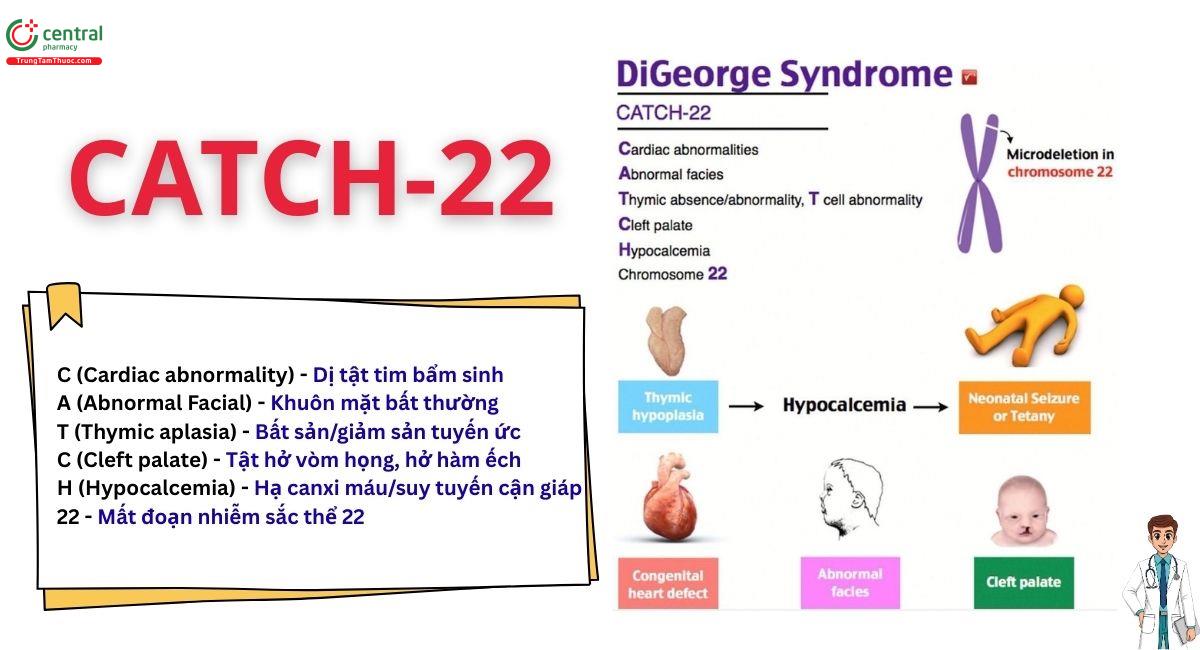
“CATCH-22” là một cách tóm tắt những đặc điểm lâm sàng chủ yếu của hội chứng DiGeorge, một rối loạn di truyền do mất đoạn vi nhiễm sắc thể 22q11.2, gây ra những ảnh hưởng đa dạng đến nhiều hệ thống trong cơ thể.
- C (Cardiac abnormality) - Dị tật tim bẩm sinh: Thường gặp nhất là các khuyết tật thân chung như tứ chứng Fallot, gián đoạn cung động mạch chủ loại B, động mạch thân chung và thông liên thất. Những khuyết tật này phát sinh do sự di chuyển bất thường của tế bào mào thần kinh ảnh hưởng đến sự phát triển của đường thoát tim.
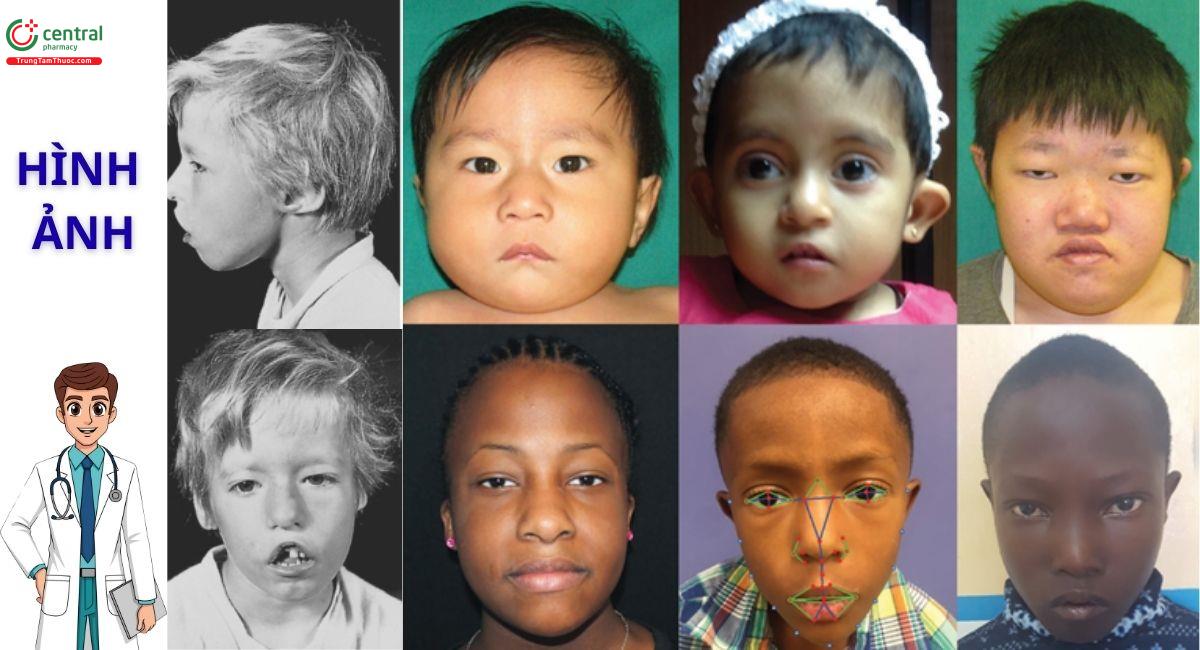
- A (Abnormal Facial) - Khuôn mặt bất thường: Các đặc điểm khuôn mặt đặc trưng bao gồm khuôn mặt dài, mí mắt sụp, mắt cách xa nhau (hai mắt cách xa nhau), sống mũi rộng, miệng nhỏ với môi trên mỏng và tai thấp. Những dị dạng tinh tế này phản ánh những bất thường về phát triển sọ mặt.
- T (Thymic aplasia) - Bất sản/giảm sản tuyến ức: Tuyến ức kém phát triển hoặc không có, gây suy giảm miễn dịch tế bào T. Điều này dẫn đến tăng nguy cơ nhiễm trùng và suy giảm miễn dịch qua trung gian tế bào.
- C (Cleft palate) - Tật hở vòm họng, hở hàm ếch: Bao gồm khe hở vòm miệng lộ rõ hoặc khe hở dưới niêm mạc, gây khó khăn cho việc ăn uống và phát âm. Tình trạng này phản ánh sự hợp nhất vòm miệng khiếm khuyết trong quá trình phôi thai.
- H (Hypocalcemia) - Hạ canxi máu/suy tuyến cận giáp: Do thiểu sản hoặc bất sản tuyến cận giáp, dẫn đến giảm tiết hormone tuyến cận giáp. Điều này gây ra hạ canxi máu ở trẻ sơ sinh hoặc trẻ nhỏ, biểu hiện bằng co giật.
- 22 - Mất đoạn nhiễm sắc thể 22: Hội chứng này là kết quả của mất đoạn nhiễm sắc thể ở 22q11.2 được phát hiện bằng kỹ thuật lai huỳnh quang tại chỗ (FISH) hoặc microarray. Khiếm khuyết di truyền này làm gián đoạn nhiều gen quan trọng cho sự phát triển của túi hầu.
6 Tiên lượng
6.1 Hội chứng DiGeorge hoàn toàn
Tiên lượng của Hội chứng DiGeorge hoàn toàn (cDGS) phụ thuộc vào sự thiếu hụt tế bào T do mất hoàn toàn tuyến ức, dẫn đến suy giảm miễn dịch nghiêm trọng và tiên lượng rất kém. Dưới đây là những điểm chính về tiên lượng của cDGS:
Nguy cơ tử vong: Ở bệnh nhân mắc cDGS, khi không có tuyến ức để phát triển tế bào T, khả năng miễn dịch bị thiếu hụt nghiêm trọng, dẫn đến nguy cơ tử vong do nhiễm trùng. Khoảng 2/3 bệnh nhân tử vong trước 1 tuổi, và phần còn lại thường tử vong trước 2 tuổi nếu không được điều trị kịp thời.
Các hội chứng tiềm ẩn: Khoảng 50% bệnh nhân mắc DGS hoàn toàn có dị hợp tử 22q11, trong khi khoảng 25% liên quan đến hội chứng CHARGE (hở, dị tật tim, teo lỗ mũi sau, chậm phát triển trí tuệ, dị tật sinh dục và tai). Khoảng 15% có liên quan đến bệnh tiểu đường trong thai kỳ.
Biểu hiện lâm sàng: Khoảng 80% bệnh nhân mắc DGS hoàn toàn cần bổ sung canxi và/hoặc Calcitriol do suy tuyến cận giáp. Mặc dù đa số bệnh nhân cần phẫu thuật tim, nhưng tỷ lệ mắc dị tật tim hình nón chỉ dưới 50%. Đặc biệt, những bệnh nhân không có dị hợp tử 22q11 có tỷ lệ mắc tim hình nón dưới 20%.
Kiểu hình lâm sàng: Có 2 kiểu hình trong DGS hoàn toàn:
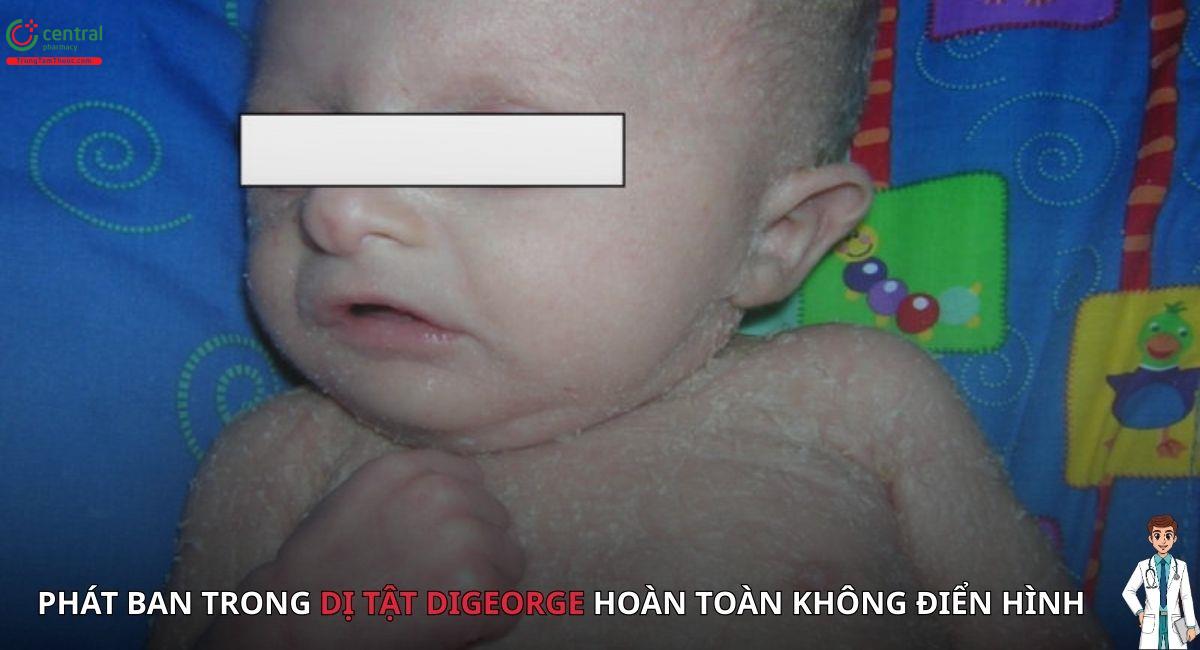
- Hội chứng DiGeorge hoàn toàn điển hình: Khoảng 2/3 bệnh nhân có ít tế bào T và không có triệu chứng phát ban hoặc hạch to.
- Hội chứng DiGeorge hoàn toàn không điển hình: Khoảng 1/3 bệnh nhân có phát ban và hạch to, liên quan đến tế bào T "chủ" oligoclonal. Tình trạng này giống với hội chứng Omenn, và ức chế miễn dịch là cần thiết để kiểm soát tế bào T oligoclonal.
Kết quả điều trị ghép tuyến ức: Ghép tuyến ức là một trong những phương pháp điều trị chủ yếu cho DGS hoàn toàn. Một nghiên cứu trên 50 bệnh nhân cho thấy có 36 bệnh nhân sống sót đến hai năm sau khi ghép tuyến ức, với sự phát triển của các tế bào T ngây thơ (tế bào T đã biệt hóa trong tuyến ức) và một kho tế bào T đa dạng.[3]
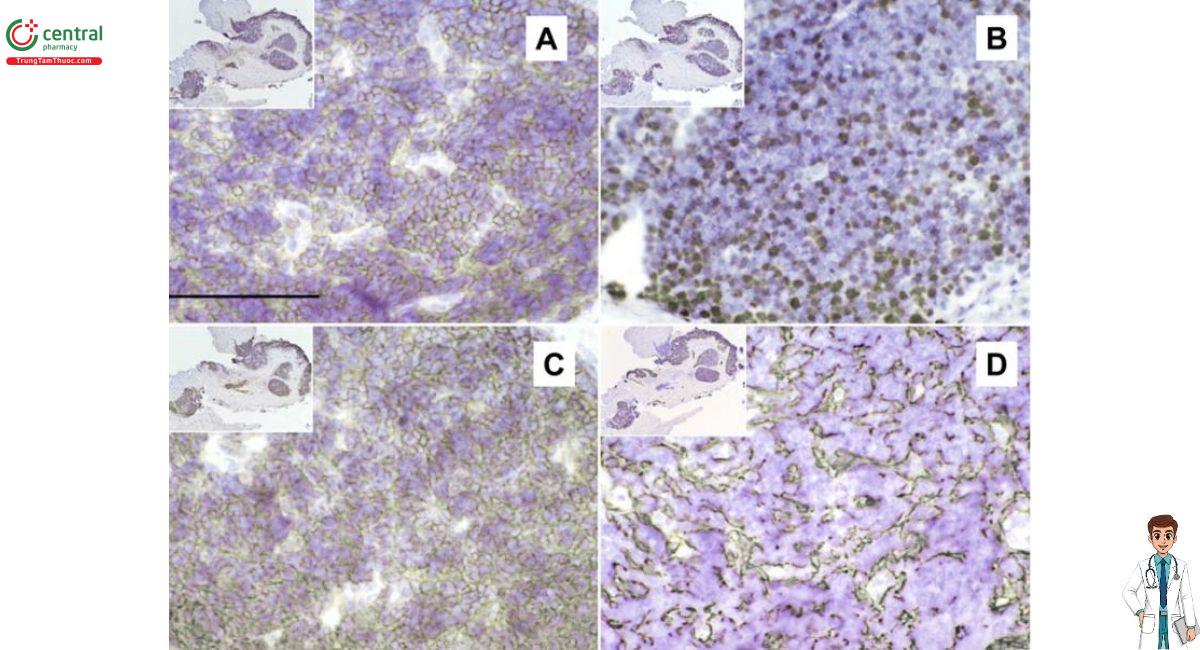
Tế bào T biểu hiện CD1, đặc trưng của tế bào tuyến ức vỏ. Cytokeratin có dạng ren, một mô hình bình thường.
6.2 Hội chứng DiGeorge một phần
Tiên lượng của DGS một phần thay đổi tùy vào mức độ nghiêm trọng của các biến chứng và không có mốc thời gian rõ ràng. Mặc dù một số bệnh nhân không sống qua giai đoạn sơ sinh do dị tật tim, nhiều người sống sót và phát triển bình thường đến tuổi trưởng thành. DGS đôi khi bị chẩn đoán muộn, và nhiều người trưởng thành mắc bệnh chưa được phát hiện, mặc dù có dị tật nhẹ và khiếm khuyết trí tuệ hoặc xã hội. Các tiến bộ trong chẩn đoán di truyền có thể cải thiện hiểu biết về bệnh này trong tương lai.
7 Quy trình quản lý bệnh
7.1 Sàng lọc lâm sàng
Xét nghiệm chẩn đoán vi mất đoạn 22q11.2 trong thai kỳ
Xét nghiệm chẩn đoán vi mất đoạn 22q11.2 trong thai kỳ có thể được thực hiện thông qua các phương pháp như chọc ối (amniocentesis) hoặc lấy mẫu sinh thiết võng mạc (CVS). Về thời điểm lấy mẫu để xét nghiệm chẩn đoán xác định, thời điểm này là từ tuần thứ 10 đến tuần thứ 13 của thai kỳ đối với sinh thiết gai nhau (CVS) và sau 15 tuần tuổi thai đối với chọc ối. Đây là các xét nghiệm xâm lấn giúp xác định chính xác sự hiện diện của mất đoạn 22q11.2, một bệnh lý di truyền có thể dẫn đến các dị tật tim, mặt, và các vấn đề khác cho thai nhi. Các xét nghiệm này thường được thực hiện sau khi có chỉ định từ bác sĩ chuyên khoa, đặc biệt trong trường hợp có nghi ngờ về sự hiện diện của vi mất đoạn hoặc khi gia đình có tiền sử bệnh lý này.
Sàng lọc trước sinh không xâm lấn (NIPS) cho các vi mất đoạn 22q11.2
Sàng lọc trước sinh không xâm lấn (NIPS) được thương mại hóa từ năm 2011 và đã trở thành một lựa chọn tiêu chuẩn để sàng lọc trước sinh về dị bội. Việc sàng lọc các vi mất đoạn được chọn lọc, bao gồm vi mất đoạn 22q11.2, đã có sẵn từ năm 2015. Đây là phương pháp sàng lọc bằng cách phân tích phần DNA tự do của thai nhi (cfDNA) trong huyết tương mẹ. Đây là một công cụ hữu ích trong việc phát hiện các bất thường di truyền, bao gồm vi mất đoạn 22q11.2. NIPS có độ chính xác cao trong việc phát hiện tình trạng mất đoạn 22q11.2 nếu người cha mang đoạn mất này. Tuy nhiên, nếu mẹ mang vi mất đoạn, NIPS không thể phát hiện chính xác tình trạng này. NIPS có thể giúp các gia đình có nguy cơ cao phát hiện sớm và đưa ra quyết định kịp thời về các bước tiếp theo trong thai kỳ.
Sàng lọc trước sinh - Hình ảnh siêu âm trong vi mất đoạn 22q11.2
Hình ảnh siêu âm là công cụ quan trọng trong việc sàng lọc các bất thường trước sinh, đặc biệt là trong việc phát hiện các dị tật liên quan đến 22q11.2DS. Các nghiên cứu đã chỉ ra rằng các dị tật tim, xương, sọ mặt, và các vấn đề về tuyến ức có thể được phát hiện qua siêu âm. Siêu âm có thể giúp bác sĩ xác định các bất thường như dị tật tim (ví dụ: ToF, IAA), thiểu sản tuyến ức, bàn chân khoèo, hoặc các dị tật về xương và sọ mặt. Các bất thường này có thể được phát hiện ngay từ tam cá nguyệt đầu tiên hoặc thứ hai của thai kỳ, giúp gia đình có thông tin sớm về tình trạng của thai nhi.
Siêu âm tam cá nguyệt đầu tiên
Siêu âm trong tam cá nguyệt đầu tiên là một phần quan trọng trong sàng lọc trước sinh, với mục đích phát hiện sớm các dị tật như ToF, IAA, và các dị tật bẩm sinh khác. Siêu âm ở giai đoạn này có thể phát hiện những dấu hiệu ban đầu của bất thường về tim hoặc các dị tật khác. Mặc dù siêu âm tam cá nguyệt đầu tiên chủ yếu được sử dụng để đánh giá tình trạng của thai nhi, nó cũng có thể giúp đánh giá tuyến ức, một dấu hiệu quan trọng trong việc chẩn đoán vi mất đoạn 22q11.2.
Siêu âm tam cá nguyệt thứ hai
Siêu âm trong tam cá nguyệt thứ hai giúp phát hiện các dị tật bẩm sinh rõ rệt hơn, bao gồm các vấn đề về tim (như ToF), dị tật xương (bàn chân khoèo), và các dị tật sọ mặt (sứt môi, hở hàm ếch). Đây cũng là giai đoạn quan trọng để đánh giá tuyến ức, một dấu hiệu chẩn đoán vi mất đoạn 22q11.2. Siêu âm thứ hai cung cấp thông tin chi tiết hơn về cấu trúc và sự phát triển của các cơ quan trong cơ thể thai nhi, giúp bác sĩ đưa ra quyết định chính xác hơn về việc tiếp tục theo dõi hay xét nghiệm thêm.
Siêu âm tam cá nguyệt thứ ba
Siêu âm trong tam cá nguyệt thứ ba giúp theo dõi sự phát triển của thai nhi và xác định các vấn đề liên quan đến sự phát triển kém trong tử cung (IUGR). Đây cũng là giai đoạn quan trọng để phát hiện các dị tật chưa được phát hiện ở các tam cá nguyệt trước. Các bất thường như đa ối, nước ối ít, hoặc các vấn đề liên quan đến phát triển tim mạch hoặc hệ thống tiêu hóa có thể được phát hiện trong giai đoạn này. Đặc biệt, đa ối có thể là một dấu hiệu gián tiếp của các vấn đề về hệ tiêu hóa hoặc vòm miệng, hai vấn đề phổ biến trong vi mất đoạn 22q11.2.
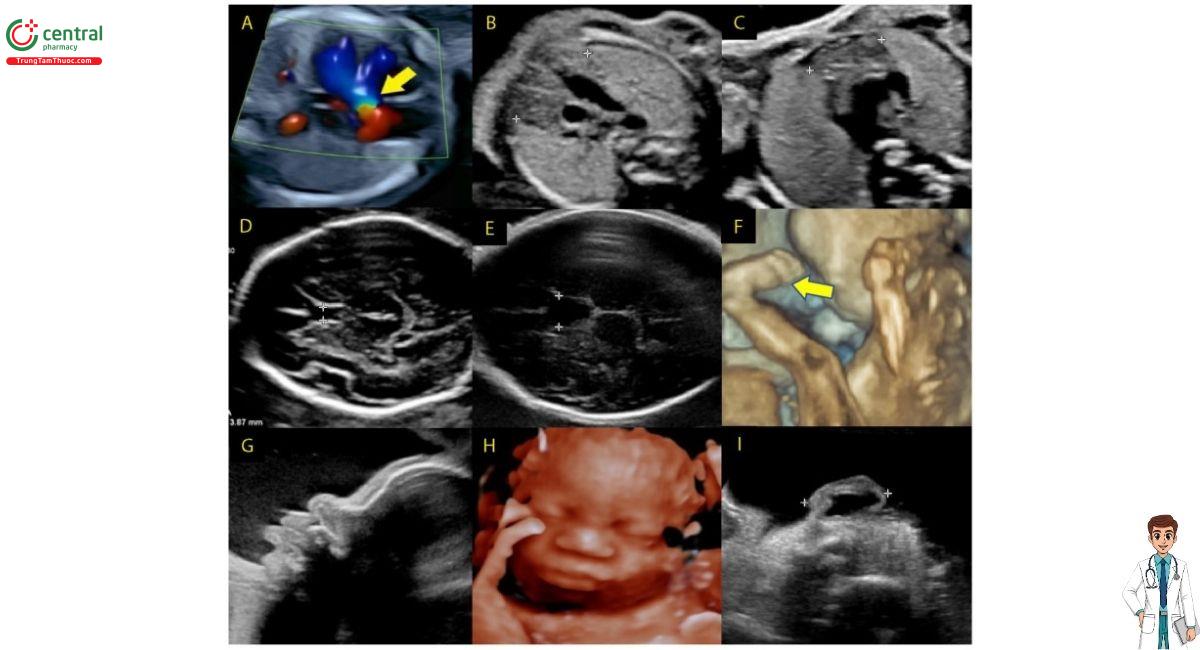
( A ) Tứ chứng Fallot, động mạch chủ đè lên (mũi tên),
( B ) tuyến ức bình thường, ( C ) tuyến ức thiểu sản,
( D ) vách ngăn trong suốt (CSP) bình thường,
( E ) CSP mở rộng, ( F ) bàn chân khoèo (mũi tên),
( G ) nhìn nghiêng với mũi củ hành, ( H ) khuôn mặt với mũi củ hành,
( I ) tai nhỏ với xoắn ốc dày gấp lại. ( B đến E —các vùng quan tâm được chỉ ra bằng compa).
Sàng lọc trước sinh- Chụp MRI trong vi mất đoạn 22q11.2
Chụp MRI thai nhi là một công cụ sàng lọc hữu ích trong việc đánh giá cấu trúc các cơ quan như não, tim, và tuyến ức. MRI cung cấp hình ảnh chi tiết hơn so với siêu âm truyền thống và có thể giúp phát hiện các bất thường mà siêu âm có thể bỏ sót, đặc biệt là các dị tật tim phức tạp hoặc các vấn đề về tuyến ức. Tuy nhiên, vì MRI yêu cầu các thiết bị chuyên dụng và không phải lúc nào cũng có sẵn ở mọi cơ sở y tế, nên phương pháp này thường được sử dụng tại các trung tâm chuyên sâu để hỗ trợ chẩn đoán.
Các lựa chọn sinh sản cho cha mẹ có vi mất đoạn 22q11.2
Các bậc cha mẹ mang đoạn mất 22q11.2 (hay còn gọi là hội chứng 22q11.2, 22q11.2DS) cần được tư vấn di truyền kỹ càng để hiểu rõ các lựa chọn sinh sản và các nguy cơ liên quan khi quyết định có con. Dưới đây là các lựa chọn sinh sản có thể được xem xét:
- Không xét nghiệm di truyền trước sinh: Một số cặp vợ chồng có thể lựa chọn không thực hiện bất kỳ xét nghiệm di truyền trước sinh nào. Điều này có nghĩa là họ không thực hiện xét nghiệm sàng lọc hoặc chẩn đoán trong suốt thai kỳ, và sẽ chỉ biết nếu có vấn đề khi đứa trẻ được sinh ra.
- Xét nghiệm di truyền trước khi làm tổ (PGT-SR): Đây là một phương pháp được thực hiện khi thụ tinh trong ống nghiệm (IVF). Các phôi sẽ được sinh thiết và xét nghiệm di truyền để phát hiện các thay đổi nhiễm sắc thể, chẳng hạn như vi mất đoạn 22q11.2, trước khi chúng được cấy vào tử cung. Phương pháp này bao gồm các kỹ thuật như FISH (Fluorescence in situ hybridization) hoặc các công nghệ hiện đại như aCGH (array comparative genomic hybridization) hoặc SNP (single nucleotide polymorphism). PGT-SR là lựa chọn chính để phát hiện vi mất đoạn như 22q11.2, vì các phương pháp xét nghiệm di truyền trước khi làm tổ thông thường (PGT-A) không thể phát hiện được những thay đổi nhỏ như vậy.
- Xét nghiệm di truyền trong thai kỳ: Trong trường hợp cặp vợ chồng lựa chọn mang thai tự nhiên hoặc không sử dụng PGT-SR, họ có thể xét nghiệm di truyền trong thai kỳ. Các phương pháp như chọc ối (amniocentesis) hoặc lấy mẫu tua màng đệm (CVS) có thể giúp phát hiện liệu thai nhi có mang đoạn mất 22q11.2 hay không. Tuy nhiên, những phương pháp này đều có tính xâm lấn và đôi khi có thể có rủi ro.
- Sàng lọc không xâm lấn (NIPS): Đây là một lựa chọn sàng lọc không xâm lấn thông qua xét nghiệm máu của mẹ. Xét nghiệm này có thể phát hiện các nguy cơ di truyền, bao gồm 22q11.2DS nếu người cha mang đoạn mất 22q11.2. Tuy nhiên, xét nghiệm này không thể phát hiện chính xác khi mẹ mang 22q11.2DS, vì vậy không phải lúc nào cũng hữu ích nếu người mẹ có đoạn mất này.
- Thụ tinh nhân tạo bằng tinh trùng hoặc trứng hiến tặng: Nếu người cha hoặc mẹ mang đoạn mất 22q11.2, có thể xem xét thụ tinh nhân tạo bằng tinh trùng hoặc trứng hiến tặng để giảm nguy cơ con cái bị ảnh hưởng. Điều này có thể là một giải pháp nếu cặp vợ chồng không muốn truyền tải gen này cho con cái.
- Tư vấn di truyền trước khi thụ thai: Trước khi quyết định có con, cặp vợ chồng nên tham khảo tư vấn di truyền để hiểu rõ về các nguy cơ và lựa chọn sinh sản. Tư vấn này sẽ giúp họ đánh giá khả năng di truyền của tình trạng 22q11.2DS và quyết định phương án tốt nhất cho gia đình.
Các lựa chọn sinh sản này có thể có những thách thức và tác động tâm lý lớn đối với các cặp vợ chồng. Việc thụ tinh trong ống nghiệm và PGT-SR có chi phí khá cao và không đảm bảo 100% thành công. Tuy nhiên, chi phí này có thể được cân nhắc với chi phí và tác động lâu dài nếu đứa trẻ bị ảnh hưởng bởi 22q11.2DS. Hơn nữa, phương pháp IVF/PGT-SR đôi khi có thể gây căng thẳng tâm lý cho cặp vợ chồng vì không có gì đảm bảo rằng quá trình mang thai sẽ thành công.[4]
7.2 Chẩn đoán
Bác sĩ lâm sàng đưa ra chẩn đoán xác định DGS ở những cá nhân có vi mất đoạn nhiễm sắc thể 22 tại locus 22q11.2. Các phương pháp đánh giá bất thường di truyền cổ điển, chẳng hạn như thể tam bội, bao gồm kỹ thuật gắn dải Giemsa, không thể phát hiện vi mất đoạn. Do đó, vi mất đoạn gây ra DGS được phát hiện bằng kỹ thuật lai huỳnh quang tại chỗ (FISH), khuếch đại đầu dò phụ thuộc vào liên kết đa đoạn (MLPA), mảng đa hình nucleotide đơn (SNP), mảng lai so sánh bộ gen (CGH) hoặc phản ứng chuỗi polymerase định lượng (qPCR). Tính khả dụng và chi phí của các kỹ thuật này có thể làm chậm trễ việc chẩn đoán, đặc biệt là ở những nơi thiếu nguồn lực.
Bệnh nhân được chẩn đoán hoặc nghi ngờ mắc DGS nên được đánh giá toàn diện, đặc biệt nếu có các khiếm khuyết về tim mạch hoặc miễn dịch đe dọa tính mạng. Các xét nghiệm sau đây nên được xem xét:
- Siêu âm tim để đánh giá các bất thường ở nón
- Công thức máu toàn phần với phân biệt
- Bảng phân nhóm tế bào lympho T và B
- Đo lưu lượng tế bào để đánh giá khả năng của tế bào T
- Mức độ globulin miễn dịch
- Nồng độ vắc-xin để đánh giá phản ứng với vắc-xin
- Nồng độ canxi và phốt pho ion hóa trong huyết thanh
- Mức độ hormone tuyến cận giáp
- Chụp X-quang ngực để đánh giá bóng tuyến ức
- Siêu âm thận để phát hiện các khiếm khuyết có thể có ở thận và tiết niệu sinh dục
- Creatinin huyết thanh
- TSH
- Kiểm tra tình trạng thiếu hụt hormone tăng trưởng1
7.3 Điều trị
Điều trị Hội chứng DiGeorge yêu cầu sự chăm sóc chuyên môn và theo dõi thường xuyên.
- Phần lớn bệnh nhân chỉ bị suy giảm miễn dịch nhẹ, với chức năng tế bào T bảo tồn dù sản xuất tế bào T giảm. Tuy nhiên, trẻ sơ sinh mắc suy giảm miễn dịch hoàn toàn (cDGS) cần được điều trị bằng cách cách ly, tiêm IgG tĩnh mạch, kháng sinh dự phòng, và ghép tế bào tuyến ức hoặc tế bào tạo máu (HSCT).
- Các phác đồ tiêm chủng, tiêm nhắc lại, tiêm globulin miễn dịch tĩnh mạch và kháng sinh dự phòng nên dựa trên các giá trị xét nghiệm của từng bệnh nhân. Nồng độ kháng thể của vaccine cần được kiểm tra lại sau 6-12 tháng. Mặc dù vaccine sống như MMR, bại liệt uống và rotavirus có tranh cãi, nhưng chúng đã chứng minh hiệu quả và an toàn cho trẻ trên 1 tuổi nếu số lượng tế bào CD8 lớn hơn 300 và số lượng tế bào CD4 lớn hơn 500. Tuy nhiên, vaccine rotavirus không nên tiêm cho trẻ sơ sinh có tế bào T giảm.
- Dị tật tim: Nếu không phát hiện trong siêu âm thai, có thể gây bệnh sau sinh, cần phẫu thuật khẩn cấp. Việc đánh giá phẫu thuật tim lồng ngực nhi khoa có thể được yêu cầu khẩn cấp. Nếu cần, các chế phẩm máu nên được chiếu xạ, xét nghiệm CMV âm tính và giảm bạch cầu để ngăn ngừa bệnh ghép chống vật chủ liên quan đến truyền máu. Các biện pháp này cũng nhằm mục đích giảm tổn thương phổi, đặc biệt trong các trường hợp phẫu thuật cần tuần hoàn ngoài cơ thể.
- Hở hàm ếch: Cần phẫu thuật chỉnh sửa sớm để cải thiện khả năng ăn uống, phát âm và giảm tỷ lệ nhiễm trùng xoang phổi.
- Hạ canxi máu: Điều trị bằng bổ sung canxi và Vitamin D, hoặc PTH tái tổ hợp cho bệnh nhân không đáp ứng liệu pháp tiêu chuẩn.
- Bệnh tự miễn: Các bệnh như viêm khớp dạng thấp, thiếu máu tự miễn và bệnh Graves thường gặp ở bệnh nhân DGS, cần theo dõi các triệu chứng tự miễn.
- Thính lực: Đánh giá thính lực và can thiệp sớm cho trẻ gặp khó khăn phát triển nhận thức và hành vi. Liệu pháp ngôn ngữ và chăm sóc tâm thần cho các bệnh nhân có triệu chứng trầm cảm hoặc loạn thần là cần thiết.
- Tư vấn di truyền: Được khuyến cáo cho các bậc phụ huynh có con mắc DGS hoặc những bệnh nhân DGS muốn có con, vì khả năng di truyền có thể lên tới 50%.
Các phương pháp tiếp cận tiên tiến để quản lý trẻ em mắc dị tật DiGeorge hoàn toàn:
- Trong trường hợp cDGS, khi trẻ không có chức năng tuyến ức và không thể phát triển tế bào T, trẻ thường tử vong trước 2 tuổi do nhiễm trùng nặng. Ghép tế bào gốc tạo máu (HSCT) là phương pháp điều trị chính, với tỷ lệ sống sót cao hơn khi ghép từ anh chị em ruột phù hợp. Mới đây, FDA đã phê duyệt ghép tuyến ức là phương pháp chăm sóc tiêu chuẩn, giúp tăng cường sản xuất tế bào T non. Tỷ lệ sống sót lâu dài đạt tới 75%, mặc dù có thể gặp các di chứng tự miễn như viêm tuyến giáp, tan máu tự miễn và giảm tiểu cầu.1
8 Các câu hỏi thường gặp
8.1 Mất đoạn NST số 22 gây bệnh gì?
Hội chứng DiGeorge (DGS): Đây là một rối loạn di truyền gây ra bởi mất một đoạn nhỏ của nhiễm sắc thể 22 ở vị trí 22q11.2. Các triệu chứng của hội chứng DiGeorge bao gồm thiểu sản hoặc bất sản tuyến ức, bệnh tim bẩm sinh, và suy tuyến cận giáp. Bệnh nhân có thể gặp các vấn đề về hệ miễn dịch, dễ nhiễm trùng, chậm phát triển, và các dị tật về mặt thể chất như sứt môi, hở hàm ếch. Hội chứng này có thể dẫn đến tử vong sớm nếu không được điều trị kịp thời, đặc biệt ở các trường hợp bị suy giảm miễn dịch nặng.
Hội chứng Phelan-McDermid (mất đoạn 22q13.3): Đây là một bệnh lý di truyền gây ra bởi mất đoạn nhiễm sắc thể 22 tại vị trí 22q13.3. Các triệu chứng của hội chứng này bao gồm chậm phát triển, khuyết tật trí tuệ từ trung bình đến nặng, giảm trương lực cơ, mất hoặc chậm nói, và các vấn đề hành vi tương tự rối loạn phổ tự kỷ. Bệnh nhân có thể có các đặc điểm hình thể đặc trưng như đầu dài và hẹp, tai vểnh, cằm nhọn, và mắt trũng sâu. Một số người cũng gặp các vấn đề sức khỏe như giảm khả năng đổ mồ hôi, nôn mửa chu kỳ, và trào ngược dạ dày thực quản.
8.2 Hội chứng DiGeorge có di truyền không?
Hội chứng DiGeorge (DGS) có thể có yếu tố di truyền, nhưng phần lớn các trường hợp là do đột biến mới (de novo) chứ không phải di truyền từ cha mẹ.
Khoảng 90% trường hợp DGS là do mất đoạn nhiễm sắc thể 22q11.2, và đột biến này thường xảy ra một cách ngẫu nhiên trong quá trình phát triển phôi thai, mà không có tiền sử di truyền trong gia đình. Những đột biến này là "de novo", nghĩa là chúng không được thừa hưởng từ cha mẹ mà phát sinh mới trong cặp nhiễm sắc thể.
Tuy nhiên, trong khoảng 10% các trường hợp, DGS có thể di truyền từ cha mẹ. Nếu cha mẹ mang đột biến 22q11.2 (mặc dù họ có thể không có triệu chứng rõ ràng), có khả năng 50% con cái sẽ thừa hưởng đột biến và có nguy cơ mắc DGS.
8.3 Hội chứng DiGeorge có thể chữa khỏi không?
Hiện tại, chưa có phương pháp chữa khỏi Hội chứng DiGeorge. Tuy nhiên, can thiệp sớm, quản lý y tế và các liệu pháp hỗ trợ có thể giúp những người mắc các bệnh lý này có cuộc sống tốt hơn.
9 Hội chứng DiGeorge và Hội chứng Down khác nhau như thế nào?

| Tiêu chí | Hội chứng DiGeorge (22q11.2 deletion syndrome) | Hội chứng Down (Trisomy 21) |
| Nguyên nhân | Mất đoạn nhiễm sắc thể 22 tại vị trí 22q11.2. | Thừa một bản sao của nhiễm sắc thể 21 (Trisomy 21). |
| Tỷ lệ mắc | Phổ biến thứ 2, khoảng 1/2.000 đến 1/4.000 trẻ. | Phổ biến thứ nhất, khoảng 1/1.000 đến 1/1.100 trẻ sinh sống. |
| Di truyền | Thường là ngẫu nhiên, có thể di truyền trong một số trường hợp. | Do sự hiện diện thêm một bản sao nhiễm sắc thể 21, thường là do sai sót ngẫu nhiên. |
| Đặc điểm di truyền | Mất một đoạn nhỏ nhiễm sắc thể 22. | Thừa một bản sao nhiễm sắc thể 21. |
| Triệu chứng thể chất | - Dị tật tim bẩm sinh (chẳng hạn như Tứ chứng Fallot, dị tật vách tim). - Suy giảm miễn dịch (do tuyến ức không phát triển đầy đủ). - Các vấn đề về xương mặt (môi, vòm miệng), bàn chân khoèo. - Rối loạn phát triển thần kinh, động kinh. | - Khuôn mặt phẳng, mắt xếch, mũi nhỏ, lưỡi thè ra. - Vóc dáng thấp bé, trương lực cơ giảm. - Mắt xếch, các đặc điểm khuôn mặt đặc trưng. - Thường có khuyết tật trí tuệ và chậm phát triển ngôn ngữ. |
| Phát hiện trước sinh | Có thể phát hiện qua xét nghiệm di truyền như FISH, NIPS, CVS, chọc ối. | Có thể phát hiện qua xét nghiệm NIPS, chọc ối, xét nghiệm gen. |
| Can thiệp y tế và điều trị | - Phẫu thuật tim nếu có dị tật tim. - Tiêm phòng và điều trị các vấn đề miễn dịch nếu cần. | - Trị liệu ngôn ngữ, vật lý trị liệu, trị liệu nghề nghiệp để hỗ trợ phát triển. - Có thể cần phẫu thuật cho các dị tật tim và các vấn đề khác. |
| Tiên lượng và chất lượng cuộc sống | Phụ thuộc vào mức độ nghiêm trọng của các triệu chứng, nhiều người có thể sống cuộc sống bình thường với sự chăm sóc y tế thích hợp. | Với sự can thiệp kịp thời, trẻ em mắc hội chứng Down có thể có cuộc sống viên mãn và hòa nhập xã hội tốt. |
9.1 Hội chứng DiGeorge và hội chứng Down có thể xảy ra cùng lúc ở cùng một cá nhân không?
Mặc dù một người có thể mắc nhiều rối loạn di truyền, nhưng Hội chứng DiGeorge và Hội chứng Down là do những bất thường di truyền khác nhau gây ra. Tuy nhiên, cũng có những trường hợp hiếm gặp khi một người được chẩn đoán mắc cả hai tình trạng.
10 Kết luận
Hội chứng DiGeorge là một rối loạn di truyền phức tạp, đòi hỏi sự nhận diện và điều trị kịp thời để giảm thiểu tác động đến sức khỏe và phát triển của bệnh nhân. Với tiến bộ trong chẩn đoán và điều trị, việc quản lý hội chứng này ngày càng hiệu quả hơn. Hy vọng bài viết đã cung cấp thông tin hữu ích, giúp độc giả hiểu rõ hơn về hội chứng DiGeorge.
Tài liệu tham khảo
- ^ Lackey AE, Muzio MR (Ngày cập nhật: Ngày 8 tháng 8 năm 2023, DiGeorge Syndrome, Pubmed. Truy cập ngày 9 tháng 12 năm 2025
- ^ McDonald-McGinn DM, Sullivan KE, Marino B, Philip N, Swillen A, Vorstman JA, Zackai EH, Emanuel BS, Vermeesch JR, Morrow BE, Scambler PJ, Bassett AS (Ngày đăng: Ngày 19 tháng 11 năm 2015), 22q11.2 deletion syndrome, Pubmed. Truy cập ngày 9 tháng 12 năm 2025
- ^ Markert ML, Devlin BH, Chinn IK, McCarthy EA (Ngày đăng: Năm 2009), Thymus transplantation in complete DiGeorge anomaly, Pubmed. Truy cập ngày 9 tháng 12 năm 2025
- ^ Blagowidow N, Nowakowska B, Schindewolf E, Grati FR, Putotto C, Breckpot J, Swillen A, Crowley TB, Loo JCY, Lairson LA, Óskarsdóttir S, Boot E, Garcia-Minaur S, Cristina Digilio M, Marino B, Coleman B, Moldenhauer JS, Bassett AS, McDonald-McGinn DM (Ngày đăng: Ngày 6 tháng 1 năm 2023), Prenatal Screening and Diagnostic Considerations for 22q11.2 Microdeletions, Pubmed. Truy cập ngày 9 tháng 12 năm 2025
