Bệnh bạch cầu mạn dòng tủy: cơ chế bệnh sinh, biểu hiện, chẩn đoán và điều trị
Trungtamthuoc.com - Bệnh bạch cầu mạn dòng tủy (CML: chronic myelogenous leukemia) là bệnh của tế bào gốc đa năng đặc trưng bởi thiếu máu, sự gia tăng quá mức bạch cầu hạt và các giai đoạn bạch cầu hạt không trưởng thành, bạch cầu ưa kiềm trong máu, thường kèm theo tăng tiểu cầu và lách to. Trong bài viết này, Trung Tâm Thuốc Central Pharmacy sẽ chia sẻ đến bạn đọc về cơ chế bệnh sinh, triệu chứng, cách chẩn đoán và điều trị bệnh bạch cầu mạn tính dòng tủy.
1 ĐỊNH NGHĨA
Bệnh bạch cầu mạn dòng tủy (CML: chronic myelogenous leukemia) là bệnh của tế bào gốc đa năng đặc trưng bởi thiếu máu, sự gia tăng quá mức bạch cầu hạt và các giai đoạn bạch cầu hạt không trưởng thành, bạch cầu ưa kiềm trong máu, thường kèm theo tăng tiểu cầu và lách to. Các tế bào tạo máu chứa chuyển vị hỗ tương của hai nhiễm sắc thể (NST) 9 và 22 gặp trong hơn 90% bệnh nhân, dẫn đến nhánh dài của một NST 22 bị ngắn lại gọi là NST Philadelphia (NST Ph). Bệnh có khuynh hướng diễn tiến đến giai đoạn tiến triển và giai đoạn chuyển cấp kháng với điều trị.
2 DỊCH TỄ HỌC VÀ NGUYÊN NHÂN
2.1 Dịch tễ học
- CML chiếm 15% trong các bệnh bạch cầu. Ti lệ nam/nữ là 3/2 và thường gặp nhất ở lứa tuổi 40 đến 60
- Tại các nước châu Âu, tỉ lệ mắc bệnh là khoảng 1/100.000
- Hình thái và diễn tiến lâm sàng không khác nhau giữa nam và nữ
- Mặc dù CML cũng xảy ra ở trẻ em và thanh thiếu niên nhưng < 10% trường hợp CML gặp ở trẻ từ 5 đến 20 tuổi. CML ở trẻ em chiếm < 5% các bệnh bạch cầu ở trẻ em.
2.2 Nguyên nhân
- Tiếp xúc với phóng xạ liều rất cao làm tăng nguy cơ CML
- Đa số các trường hợp không tìm được nguyên nhân.
3 SINH LÝ BỆNH
3.1 Di truyền tế bào
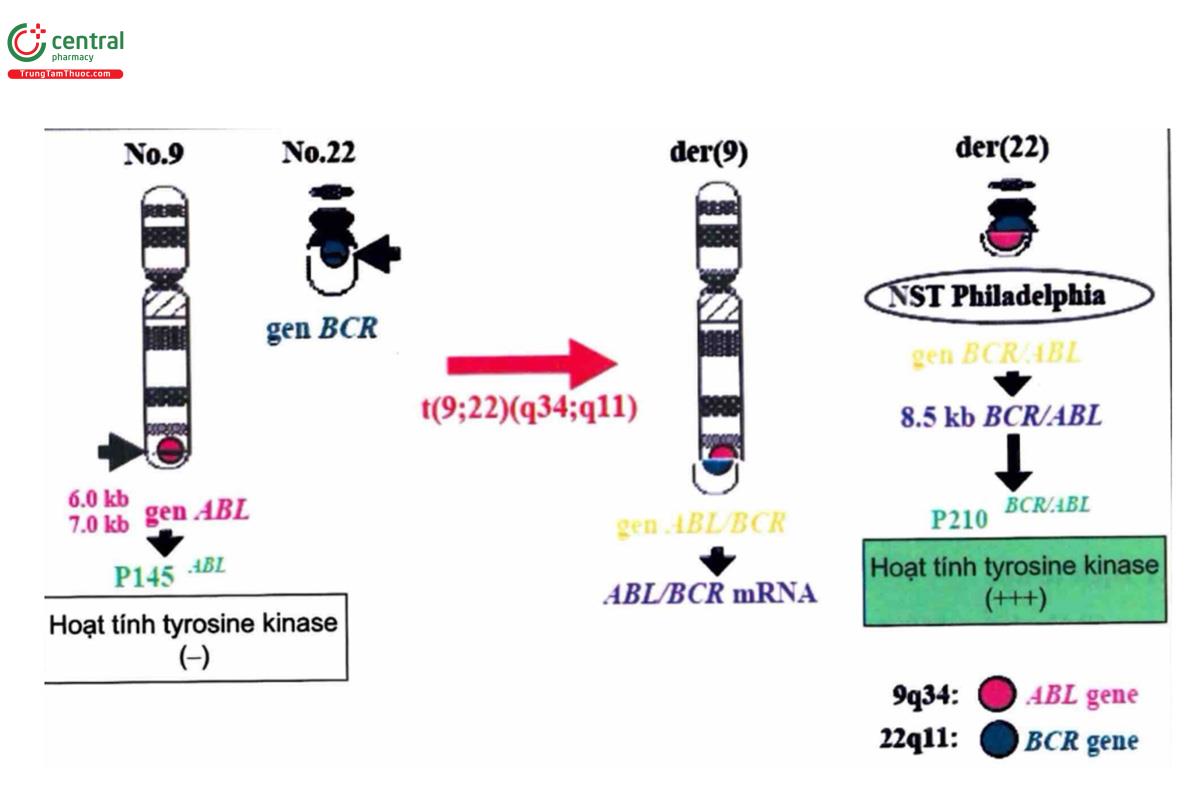
- Thay đổi quan trọng nhất và trở thành đặc hiệu cho CML là NST Ph. NST Ph là NST 22 có nhánh dài bị ngắn đi, được tạo ra do chuyển vị hỗ tương của nhánh dài NST 9 và nhánh dài của NST 22 và gọi là t(9;22)(q34;q11) (Hình 9.1).
- NST Ph được tìm thấy trong tất cả các tế bào máu, bao gồm cả tế bào B và tế bào T và nó hiện diện trong suốt quá trình bệnh, cả trong giai đoạn cấp.
- NST Ph được tìm thấy trong hơn 95% bệnh nhân CML. Ngoài ra NST Ph còn được tìm thấy trong Bệnh bạch cầu cấp dòng lympho (ALL: Acute lymphoblastic leukemia) với khoảng 30% ở người lớn, 3% ở trẻ em, khoảng 1% bệnh bạch cầu cấp dòng tủy (AML: acute myeloid leukemia) mới chẩn đoán.
3.2 Sinh học phân tử
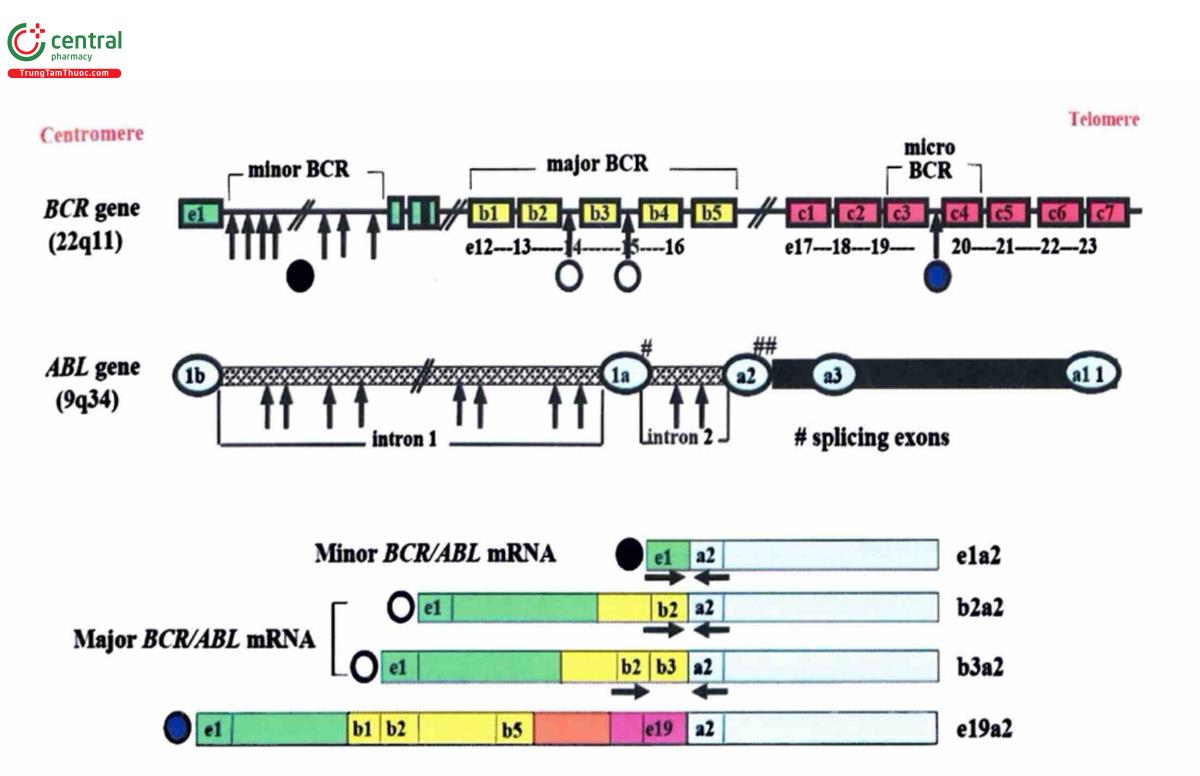
- Trong CML, t(9;22)(q34;q11) tạo ra tổ hợp gen BCR/ABL mà exon 1 của ABL được thay thế bằng những exon ở đầu 5' của BCR (Hình 9.2).
- Tùy vào vị trí điểm gãy của BCR, những tổ hợp gen được tạo ra là ela2, b2a2 hoặc b3a2 và e19a2 mã hóa các dạng tổ hợp protein p190BCR/ABL, P210BCR/ABL và P230BCR/ABL tyrosine kinase tương ứng (Hình 9.2):
+ Hầu hết bệnh nhân CML có điểm gãy trên BCR ở vùng major (M-bcr) tạo ra b2a2 hoặc b3a2, mã hóa protein p210BCR/ABL
+ Khoảng 2/3 trường hợp ALL, rất hiếm trường hợp CML và AML có điểm gãy trên BCR ở vùng minor (m-bcr) tạo ra ela2, mã hóa protein p190BCR/ABL
+ Có một số ít trường hợp CML có điểm gãy trên BCR ở vùng µ-bcr tạo ra e19a2, mă hóa protein p230BCR/ABL. Những bệnh nhân này thường tăng ưu thế neutrophil
+ Ngoài ra, còn có một số vùng điểm gãy khác tạo ra các kiểu bản sao BCR/ABL hiếm gặp hơn như e6a2, b2a3, b3a3,...
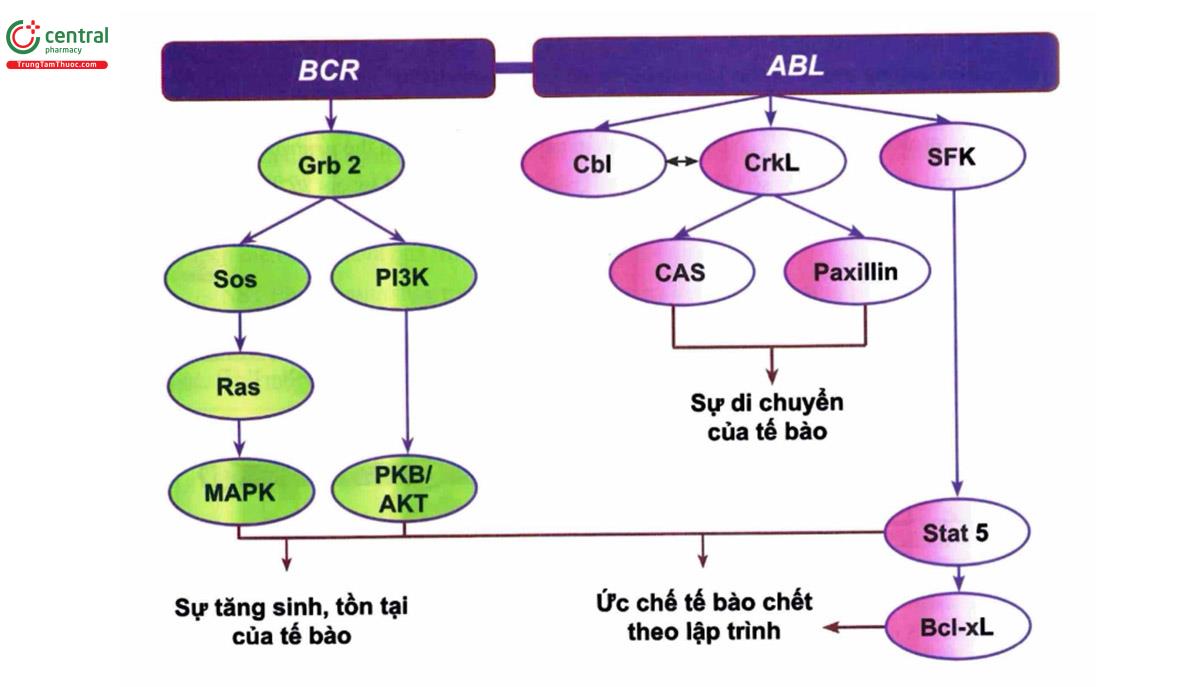
- Protein BCR/ABL nằm ở cytoskeleton và làm tăng hoạt tính tyrosine kinase gây ra sự tăng sinh tế bào (cell proliferation), ức chế tế bào chết theo chương trình dẫn tới sự lan rộng dòng tế bào ác tính của CML. Protein p210BCR/ABI, tương tác với một số yếu tố của các con đường truyền tín hiệu gây phosphoryl hóa hơn 20 protein nội bào có vai trò sinh ung thư (Hình 9.3).
+ Sự hoạt hóa của phosphatidylinositol-3'-kinase (PI3K), con đường MAPK thông qua Grb2 và sự hoạt hóa STAT5 thông qua SKF gây ra sự tăng sinh các dòng tế bào có BCR/ABL
+ Sự hoạt hóa Paxillin và CAS thông qua CRKL gây ra khiếm khuyết về sự dính của các tế bào với chất nền, ảnh hưởng đến sự di chuyển của tế bào
+ Sự hoạt hóa của AKT thông qua PI3K và sự hoạt hóa của STAT5 gây ức chế sự chết tế bào theo lập trình.
4 TRIỆU CHỨNG
4.1 Triệu chứng lâm sàng
4.1.1 Triệu chứng cơ năng
Triệu chứng lâm sàng phát triển rất chậm và hầu hết bệnh nhân được phát hiện ở giai đoạn mạn. Ngày nay, nhiều trường hợp được phát hiện sớm khi khám sức khỏe định kỳ. Lúc đầu, triệu chứng mơ hồ và không đặc hiệu. Khoảng 70% bệnh nhân có triệu chứng lúc chẩn đoán và thường than phiền nhất là:
- Mệt mỏi, biếng ăn, sụt cân, giảm thể lực và ra mồ hôi nhiều
- Bệnh nhân cảm thấy khó chịu, căng tức ở hạ sườn trái và ăn mau no: là triệu chứng của lách lớn.
4.1.2 Triệu chứng thực thể
Khám lâm sàng có thể thấy bệnh nhân xanh xao và lách to. Lách to thường hiện diện trong khoảng 90% bệnh nhân, tuy nhiên, tỉ lệ này giảm dần do bệnh ngày càng được phát hiện sớm hơn.
Những triệu chứng không thường gặp:
- Triệu chứng của tăng chuyển hóa: ra mồ hôi về đêm, không chịu được nóng
- Viêm khớp gout cấp nghĩ do tăng Urea máu
- Gan to và hạch to nhưng hiếm khi kích thước hạch lớn hơn 1 cm
- Nhồi máu lách có thể gặp
- Chảy máu do rối loạn số lượng và chất lượng tiểu cầu.
4.2 Triệu chứng sinh học
4.2.1 Huyết đồ
Chẩn đoán sơ bộ bệnh bạch cầu mạn dòng hạt có thể dựa vào kết quả số lượng bạch cầu và phết máu ngoại biên.
- Bạch cầu tăng trong khoảng từ 20×109/L đến hơn 500×109/L, với khoảng trung bình từ 134×109/L đến 225×10/L trong hầu hết các nghiên cứu.
- Hiện diện tất cả các giai đoạn của quá trình biệt hóa bạch cầu hạt từ myeloblast đến segment neutrophil. Không có “khoảng trống bạch cầu".
- Bạch cầu ưa kiềm tăng về số lượng, nhưng tỉ lệ không quá 10 – 15% trong giai đoạn mạn
- Tiểu cầu tăng trong khoảng 50% các trường hợp, có khi tăng trên 1.000×10° /L. Hiếm khi số lượng tiểu cầu giàm dưới 100×109/L. Tiểu cầu giàm lúc chẩn đoán là dấu hiệu của giai đoạn tiến triển
- Thiếu máu nhẹ và vừa thường thấy lúc mới chẩn đoán, thường là thiếu máu đẳng sắc, đẳng bào
- Hồng cầu lưới phần nhiều là bình thường.
4.2.2 Tủy đồ
- Tủy xương rất giàu tế bào, ưu thế là các giai đoạn của dòng bạch cầu hạt. Hình thái tế bào trong mỗi giai đoạn thì bình thường. Tỉ lệ blast thường không vượt quá 5%. Tăng dòng bạch cầu ưa acid và ưa kiềm.
- Mẫu tiểu cầu gia tăng điển hình trên tủy hút và thỉnh thoảng tạo thành từng đám mẫu tiểu cầu. Sọ với mẫu tiểu cầu bình thường, mẫu tiểu cầu trong CML nhỏ hơn và có thể gặp micromegakaryocyte trong một số trường hợp.
- Số lượng tế bào đầu dòng của dòng hồng cầu có thể tăng, bình thường hoặc giảm.
- Collagen type III tăng (reticulin fibrosis: xơ hóa tủy) có thể gặp trong gần một nửa số bệnh nhân lúc chẩn đoán và nó liên quan đến một phần mẫu tiểu cầu trong tủy. Sự gia tăng xơ hóa cũng liên quan đến kích thước lớn hơn của lách, thiếu máu nặng hơn và tăng tỉ lệ blast trong máu và tủy
4.2.3 Những xét nghiệm di truyền học phân tử
Nhiễm sắc thể đồ
Ưu điểm:
+ Phát hiện được t(9;22) và những bất thường NST khác đi kèm trong giai đoạn mạn giúp đánh giá tiên lượng
+ Phát hiện được những bất thường NST đi kèm như iso(17q), + NST Ph, trisomy 8, trisomy 19,... chiếm 70 – 80% trong giai đoạn tiến triển hoặc chuyển cấp.
- Khuyết điểm:
+ Đòi hỏi tế bào phân chia (tế bào metaphase)
+ Chỉ nhận ra bất thường khi băng bất thường đó ≥ 10 Mb (độ nhạy để phát hiện clone bất thường là 5 – 10%)
+ Kết quả thường có trong vòng 2 tuần, sớm nhất là 3 ngày
+ Khoảng 5% trường hợp không phát hiện được NST Ph bằng NST đồ do tái sắp xếp BCR/ABL ẩn hoặc không quan sát được dưới kính hiển vi
+ Phân tích khoảng 20 tế bào.
Kỹ thuật lai tại chỗ phát huỳnh quang (FISH)
Ưu điểm:
+ Phát hiện NST Ph trên tế bào metaphase và tế bào không phân chia (tế bào interphase)
+ Cho kết quả nhanh trong vòng 24 giờ
+ Dùng đoạn mồi đặc hiệu cho BCR và ABL nên có thể phát hiện được tái sắp xếp BCR/ABL ẩn hoặc không quan sát được dưới kính hiển vi mà NST đồ không phát hiện được
+ Phân tích được nhiều tế bào hơn: từ 200 đến 500 tế bào (độ nhạy để phát hiện clone bất thường là 1%).
- Khuyết điểm:
+ Chỉ phát hiện được NST Ph và không phát hiện được bất thường đi kèm vì không quan sát được cấu trúc của bộ NST.
Những kỹ thuật phân tử khác
- Tổ hợp gen BCR/ABL có thể được phát hiện bằng Southern blot analysis hoặc bằng RT-PCR (reverse transcriptase-polymerase chain reaction)
- Kỹ thuật PCR có thể được sử dụng để định tính hoặc định lượng để theo dõi đáp ứng với điều trị.
4.2.4 Các kết quả xét nghiệm khác
- Acid uric tăng 2 đến 3 lần lúc chẩn đoán. Nếu điều trị làm ly giải tế bào nhanh có thể gây tắt đường niệu do kết tủa acid uric. Sỏi urat đường niệu thường gặp trên bệnh nhân CML và một số bệnh nhân phát triển viêm khớp gout cấp hoặc bệnh thận do acid uric.
- Protein gắn kết Vitamin B12 huyết thanh tăng, trung bình hơn 10 lần so với bình thường (nhất là transcobalamin-1) vì neutrophil chứa protein này. Nồng độ tỉ lệ thuận với số lượng bạch cầu trong những bệnh nhân CML chưa điều trị.
- Histamin máu toàn phần tăng trên bệnh nhân CML giai đoạn mạn (khoảng 5.000 ng/mL) so với người bình thường (khoảng 50 ng/mL). Sự tăng histamin máu có liên quan đến tăng số lượng tế bào basophil. Một số trường hợp tăng histamin máu cao sẽ gây ra ngứa, mề đay và tăng tiết dịch dạ dày.
- Lactic acid dehydrogenase (LDH) huyết thanh tăng.
5 TIẾN TRIỂN VÀ TIÊN LƯỢNG
5.1 Tiến triển
Diễn tiến từ lúc xuất hiện NST Ph đến khi có triệu chứng là khoảng 6 năm. Khi hiện diện tăng bạch cầu đến khi số lượng bạch cầu được 100×109/L là khoảng 19 tháng và lách lớn phát hiện được khi số lượng bạch cầu > 40×10/L (từ 30×109/L đến 90×109/L).
CML gồm ba giai đoạn: mạn, tiến triển và chuyển cấp:
- Giai đoạn mạn (Chronic phase)
Bệnh hầu hết được chẩn đoán ở giai đoạn này với các triệu chứng của một CML điển hình. Đáp ứng rất tốt với thuốc ức chế men tyrosine kinase như imatinib, nilotinib,... Chất lượng cuộc sống thay đổi không nhiều. Tử vong trong giai đoạn này rất hiếm, thường do những nguyên nhân không liên quan đến bệnh CML.
- Giai đoạn tiến triển (Accelerated phase)
+ Nguyên tùy bào (Myeloblast) ở chiếm ≥ 15% nhưng <30% trong máu ngoại biên
+ Tổng nguyên tủy bào và tiền tùy bào (Promyelocyte) ≥ 30% trong máu ngoại biên
+ Bạch cầu ưa kiềm (Basophil) > 20% trong máu
+ Số lượng tiểu cầu < 100×10/L (Không do điều trị)
+ Bất thường di truyền tế bào khác trên dòng tế bào có Ph.
Hiện diện từ một tiêu chuẩn trên được chẩn đoán là CML giai đoạn tiến triển. Trong giai đoạn này cần tìm đột biến kháng thuốc để chuyển sang điều trị với thuốc ức chế men tyrosine kinase thế hệ 2 như dasatinib, bosutinib hoặc tăng liều các thuốc đang sử dụng hoặc ghép tế bào gốc tạo máu.
Giai đoạn chuyển cấp (Blast phase)
+ Blast trong tùy và/hoặc máu ngoại biên > 30%
+ Có biểu hiện xâm lấn ngoài tủy (Extramedullary infiltrates of leukemic cells).
Hiện diện từ một tiêu chuẩn trên được chẩn đoán là CML giai đoạn chuyển cấp.
Bệnh có thể chuyển cấp dòng lympho hoặc dòng tủy. Trong giai đoạn này bệnh đáp ứng kém với thuốc ức chế men tyrosine kinase và hóa trị liệu. Ghép tế bào gốc tạo máu có thể kéo dài được thời gian sống nhưng tiên lượng cũng xấu.
5.2 Tiên lượng
Có nhiều thang điểm tiên lượng được đề nghị trong CML, bao gồm hệ thống Sokal, Hasford và ELTS (EUTOS long-term survival score) cho bệnh nhân lúc chẩn đoán và the European Bone Marrow Transplantation Consortium Risk Score cho bệnh nhân dị ghép tế bào gốc. Phân nhóm nguy cơ trung bình – cao được khuyến cáo điều trị ưu tiên với TKI thế hệ sau từ đầu thay vì imatinib.
Bảng 9.1. Phân nhóm nguy cơ CML giai đoạn mạn
Thang điểm | Công thức tính | Phân nhóm nguy cơ |
Sokal score | Tổng = 0,0116 x (tuổi - 43,4) + 0,0345 x (lách - 7,51) + 0,188 × [(Tiểu cầu/700)2- 0,563] + 0,0887 x (Blast - 2,10) | Thấp: < 0,8 Trung gian: 0,8-1,2 Cao: > 1,2 |
score (EURO) Hasford | (0,6666 x tuổi [0 nếu <50 tuổi; còn lại là 1] + 0,042 x lách (cm dưới bờ sườn] + 0,0584 x % Blast + 0,0413x % eosinophil + 0,2039 x basophil [0 nếu basophil <3%; còn lại là 1] + 1,0956 x Tiểu cầu [0 nếu tiểu cầu < 1500 × 109/L: còn lại là 1] × 1.000 | Thấp: ≤ 780 Trung gian: > 780 đến ≤ 1.480 Cao: > 1.480 |
EUTOS long-term survival (ELTS) score | 0,0025 x (Tuổi/10)3 + 0,0615 × lách (cm dưới bờ sườn) + 0,1052 x blast ngoại biên + 0,4104 × (Số lượng tiểu cầu/1.000)-05 | Thấp: ≤ 1,5680 Trung gian: >1,5680 đến ≤ 2,2185 Cao: > 2,2185 |
6 CHẨN ĐOÁN PHÂN BIỆT
6.1 Bạch cầu mạn dòng tủy mono (Chronic Myelomonocytic Leukemia – CMML)
- Khoảng 50% trường hợp có bệnh cảnh tăng bạch cầu, lách to
- Tăng monocyte ≥ 10% (trong CML chỉ tăng khoảng 2-3%)
- Có loạn sản một hay nhiều dòng tế bào tủy
- Không có hiện diện NST Ph hoặc BCR/ABL.
6.2 Bạch cầu mạn dòng tủy mono ở trẻ em (Juvenile Myelomonocytic Leukemia - JMML)
- Bệnh điển hình gặp ở trẻ em ≤ 3 tuổi. Khoảng 10% trường hợp kết hợp với bệnh u xơ thần kinh
- Tăng sinh dòng tủy và dòng mono (monocyte > 1×109/L), đi kèm giảm tiểu cầu và thiếu máu. Blast < 20% trong tủy
- Không có hiện diện NST Ph hoặc BCR/ABL
- Khoảng 40% có bất thường NST, thường gặp là monosomy 7
- Cần phân biệt với bệnh cảnh nhiễm Epstein-Barr virus ở trẻ em
- Bệnh diễn tiến nhanh giống bệnh cảnh AML, đáp ứng kém với điều trị. Dị ghép tế bào gốc có thể đưa đến lui bệnh hoàn toàn.
6.3 Chronic Neutrophilic Leukemia (CNL)
- Là một bệnh rất hiếm
- Bệnh cảnh lâm sàng rất giống CML
- Hiện diện ưu thế bạch cầu hạt trưởng thành trong tủy xương
- Không có hiện diện NST Ph hoặc BCR/ABL.
6.4 CML không điển hình
- Bệnh cảnh lâm sàng rất giống CML
- Thường gặp ở người lớn tuổi
- Tỉ lệ của blasts, promyelocyte và myelocyte từ 10% đến 20% trong máu. Không tăng ưu thế bạch cầu ưa kiềm
- Thường có loạn sản các dòng tế bào
- Không có hiện diện NST Ph hoặc BCR/ABL
- Bệnh tiến triển nhanh và thời gian sống trung bình khoảng 2 năm.
6.5 Nhóm bệnh tân sinh tăng sinh tủy không biểu hiện BCR/ABL
- Huyết đồ và tùy đồ giúp phân biệt từng bệnh gồm: đa hồng cầu, tăng tiểu cầu nguyên phát và xơ tùy nguyên phát
- Không có hiện diện NST Ph hoặc BCR/ABL
- Có hiện diện một trong các đột biến gen JAK2, CALR và MPL.
7 ĐIỀU TRỊ
Tùy theo giai đoạn.
7.1 Giai đoạn mạn
7.1.1 Hóa trị
Thuốc ức chế BCR-ABL tyrosine kinase: Imatinib mesylate (Gleevec, Glivec).
- Imatinib là dẫn xuất của 2-phenylaminopyrimidine, ức chế đặc hiệu ABL tyrosine kinase bằng cách cạnh tranh với ATP trong túi kinase của P210BCR-ABL, làm ức chế hoạt tính tyrosine kinase của protein này, vì vậy ức chế sự tăng sinh tế bào.
- Thuốc dung nạp tốt bằng đường uống
- Liều: 400 – 600 mg/ngày
- Tác dụng phụ: thường gặp là phù, buồn nôn, co thắt cơ, nổi mẩn ngứa, mệt mỏi và tiêu chày. Tăng men gan và ức chế tùy có thể gặp.
Hydroxyurea (Hydrea)
- Liều: 1 – 6 g/ngày, tùy vào số lượng bạch cầu
- Hấp thu rất tốt theo đường uống
- Độc tính: suy tủy nhưng phục hồi nhanh sau khi ngưng thuốc, nôn ói, tiêu chảy, loét miệng và loạn dưỡng móng.
Interferon alpha (Roferon A, Intron A)
- Là một loại cytokin ức chế sự phát triển của tế bào ác tính, điều hòa miễn dịch, kích hoạt tế bào giết tự nhiên (NK cell: Natural killer cell), thay đổi kháng thể và kích hoạt kháng nguyên HLA.
- Interferon alpha kéo dài thời gian sống và tạo ra lui bệnh hoàn toàn về di truyền tế bào.
- Liều: 5 triệu đơn vị/m²/ngày, tiêm dưới da.
- Độc tính:
+ Suy tủy nhẹ thoáng qua, buồn nôn, nôn và hội chứng giống cảm cúm thường thấy và gia tăng theo liều.
+ Triệu chứng toàn thân: mệt mỏi, chán ăn, sụt cân gặp khi dùng lâu dài.
+ Triệu chứng thần kinh: nhức đầu, lừ đừ, ào giác và rối loạn tiểu não.
Thuốc ức chế tyrosine kinase thế hệ mới
- Nilotinib là thuốc ức chế đặc hiệu ABL tyrosine kinase, liều dùng là 400 – 600 mg/ngày
- Dasatinib là thuốc ức chế Src/ABL tyrosine kinase, liều dùng là 100 mg x 2 lần/ngày.
Cả hai loại trên đều có tác dụng mạnh hơn imatinib nhiều lần và dung nạp tốt bằng đường uống.
7.1.2 Ghép tủy
- Ghép tế bào gốc đồng loài: trước đây là phương pháp duy nhất có khả năng chữa khỏi bệnh, tuy nhiên, hiện nay được chỉ định khi bệnh nhân CML không dung nạp với thuốc, kháng thuốc hoặc có biểu hiện bệnh tiến triển.
- Tế bào gốc máu cuống rốn cũng là một nguồn quan trọng được sử dụng trong ghép tế bào gốc đồng loài, đặc biệt là ở trẻ em.
7.2 Giai đoạn tiến triển
Khi bệnh bắt đầu chuyển sang giai đoạn tiến triển, tùy vào kiểu đột biến BCR-ABL kháng thuốc để quyết định chuyển sang các thuốc thệ hệ mới hơn như nilotinib, dasatinib, bosutinib, Ponatinib,... hoặc tiến hành ghép tế bào gốc đồng loài cho bệnh nhân.
7.3 Giai đoạn chuyển cấp
- CML có thể chuyển cấp dòng tủy hoặc dòng lympho. Thời gian sống thêm trung bình đối với chuyển cấp dòng tủy là 6 tháng và chuyển cấp dòng lympho là 12 tháng.
- Điều trị phối hợp thuốc như bệnh bạch cầu cấp và phối hợp dùng thuốc ức chế tyrosine kinase thế hệ mới.
8 THEO DÕI VÀ ĐÁNH GIÁ ĐIỀU TRỊ
8.1 Theo dõi bằng phương pháp phân tử và khảo sát tồn lưu tế bào ác tính
- Mặc dù lui bệnh trên lâm sàng nhưng ước tính vẫn còn 109 – 1010 tế bào ác tính.
- Hiện nay, trọng tâm để theo dõi điều trị CML là real-time PCR. Xác định tỉ lệ tồn lưu tế bào ác tính gián tiếp thông qua chỉ số BCR-ABL115 (Đo tỉ lệ bản sao BCR-ABL còn “sót” lại), với độ nhạy < 1/105.
- Các mốc thời gian theo dõi và mức độ đáp ứng được thể hiện như sau:
Bảng 9.2. Sơ đồ các mốc theo dõi đáp ứng điều trị CML
Tỉ lệ BCR-ABLIS | 3 tháng | 6 tháng | 12 tháng |
> 10% | Có thể kháng TKI | Kháng TKI | |
>1% - 10% | Nhạy TKI | Có thể kháng TKI |
|
> 0,1%- 1% | Nhạy TKI |
|
|
Lưu ý khi ngờ hoặc đánh giá kháng thuốc TKI:
- Cần đánh giá lại sự tuân thủ TKI và khả năng tương tác với các loại thuốc khác người bệnh đang sử dụng đồng thời
- Sàng lọc đột biến kháng thuốc BCR/ABL kinase domain
- Cân nhắc khả năng đổi thuốc TKI và ghép tế bào gốc đồng loài.
8.2 Sàng lọc đột biến BCR/ABL kinase domain
- Chỉ định: khi không đạt được các mốc đáp ứng tối ưu hoặc mất đáp ứng với ức chế tyrosin kinase.
- Tùy theo vị trí và loại đột biến mà quyết định hướng điều trị kế tiếp: tăng liều, đổi thuốc ức chế tyrosine kinase thế hệ mới hoặc ghép tế bào gốc đồng loài.
9 CÂU HỎI TỰ LƯỢNG GIÁ
1. Thiếu máu trong bệnh bạch cầu mạn dòng tủy thường là:
A. Thiếu máu nhược sắc, hồng cầu nhỏ
B. Thiếu máu đẳng sắc, hồng cầu nhỏ
C. Thiếu máu ưu sắc, hồng cầu nhỏ
D. Thiếu máu đằng sắc, đằng bào
E. Thiếu máu đẳng sắc, hồng cầu to
2. Số lượng tiểu cầu trong bệnh bạch cầu mạn dòng tủy:
A. Bình thường
B. Tăng
C. Giàm
D. Có thể bình thường hoặc tăng
E. Có thể bình thường hoặc giảm
3. Phân tử trong con đường truyền tín hiệu gây tăng sinh tế bào của bệnh bạch cầu mạn dòng tủy:
A. PI3K
B. CAS
С. СЫ
D. MLL
E. Crkl
4. Phân từ trong con đường truyền tín hiệu gây ức chế tế bào chết theo lập trình của bệnh bạch cầu mạn dòng tủy:
A. CAS
B. STAT5
С. СЫ
D. MLL
E. Crkl
5. Phân từ trong con đường truyền tín hiệu gây khiếm khuyết về sự dính của các tế bào bạch cầu mạn dòng tùy với chất nền tủy xương:
A. Grb2
B. Ras
C. MAPK
D. Paxillin
E. Bcl-xl
10 ĐÁP ÁN
1D 2D 3A 4B 5D
11 TÀI LIỆU THAM KHẢO
- Suzanne Monivong Cheanh Beaupha (Thanh Thanh), Lê Quốc Bảo, Đinh Gia Khánh, Quách Thanh Lâm, Trần Thị Thanh Loan, Huỳnh Văn Mẫn, Huỳnh Nghĩa, Nguyễn Vũ Hải Sơn, Nguyễn Quốc Thành, Mai Phương Thảo, Lại Thị Thanh Thảo, Nguyễn Bình Thư, Phan Thị Xinh. "DÒNG BẠCH CẦU". Giáo trình HUYẾT HỌC CƠ SỞ. Trường Đại học Y Dược Thành phố Hồ Chí Minh, Module Huyết học. Chương III. Bài 9-BỆNH BẠCH CẦU MẠN DÒNG TỦY. Trang 115-127. Tải bản PDF TẠI ĐÂY
- Chereda B, Melo JV. (2015), Natural course and biology of CML, Ann Hematol, 94, Suppl 2:S107- 21.
- Lichman MA., Liesveld JL. (2006), Chronic myelogenous leukemia and related disorders, Williams Hematology, Seventh edition, McGraw-Hill Medical, Inc., New York, pp. 1237-1268.
- Melo JV. (1996), The diversity of BCR-ABL fusion proteins and their relationship to leukemia phenotype, Blood, 88:2375-84.
- Reichard KK, Larson RS. and Wolff SN. (2009), Chronic myeloid leukemia, Wintrobe's clinical hematology, 12th edition, Lippingcott Williams & Wilkins, Philadelphia, Electric book.
- Schwartz LC, Mascarenhas J. (2018), Current and evolving understanding of atypical chronic myeloid leukemia, Blood Rev, Jul 29.
- Shah NP, Bhatia R. (2022). "Chronic Myeloid Leukemia, Version 1.2023, NCCN Clinical Practice Guidelines in Oncology.
