Ảnh hưởng của kiểu gen đến tương tác thuốc có ý nghĩa lớn trên lâm sàng

Trungtamthuoc.com - Chương "Ảnh hưởng của kiểu gen đến tương tác thuốc" sẽ phân tích được ảnh hưởng của kiểu gen lên tần suất và ý nghĩa lâm sàng của tương tác thuốc khi xảy ra ở các cả thể khác nhau.
Trong những chương trước, chúng ta đã thấy được sự tương tác phức tạp và đa dạng giữa kiểu gen của người bệnh với thuốc, trong nhiều trường hợp gây ra sự dao động lớn về đáp ứng điều trị giữa các cá thể, có một mối quan hệ tương tác phức tạp khác xảy ra khi có hai hay nhiều thuốc được dùng đồng thời. Vậy mối quan hệ 3 chiều thuốc - gen - thuốc sẽ như thế nào? Chương này phân tích sự khác biệt về tần suất và ý nghĩa lâm sàng của tương tác thuốc xảy ra ở những cá thể bệnh nhân mang kiểu gen nhau.
1 Khái niệm tương tác thuốc
Tương tác thuốc xảy ra khi hoạt động hoặc tác dụng của một thuốc trong cơ thể bị thay đổi gây bởi sự có mặt của chất hóa học khác như thuốc, thức ăn hay các yếu tố môi trường.
Theo Dược thư quốc gia Việt Nam, tương tác thuốc là khả năng của một thuốc có thể làm thay đổi cường độ tác dụng dược lý của một thuốc khác khi sử dụng đồng thời. Kết quả cuối cùng có thể là một hoặc cả hai thuốc tăng hoặc giảm tác dụng hoặc xuất hiện một tác dụng mới không thấy có khi dùng riêng từng thuốc.
Dựa vào cơ chế gây tương tác, tương tác thuốc được chia thành ba loại chính: Tương tác dược lực học, tương tác dược học và tương tác dược động học. Trong số đó, các tương tác có liên quan đến sự khác biệt về di truyền chủ yếu là tương tác dược động học. Đây là các tương tác gây bởi tác dụng của một thuốc lên các quá trình dược động học của một thuốc khác trong cơ thể. Tương tác theo cơ chế dược động học chủ yếu bao gồm tương tác trong quá trình chuyển hóa thuốc và tương tác trong quá trình vận chuyển thuốc.
Các trường hợp tương tác thuốc có ý nghĩa lâm sàng quan trọng thường xảy ra khi:
- Thuốc có phạm vi điều trị hẹp.
- Có sự phụ thuộc chặt chẽ giữa nồng độ thuốc trong huyết tương với các đặc điểm lâm sàng như hiệu quả điều trị hoặc tác dụng không mong muốn.
- Quá trình vận chuyển hoặc chuyển hóa thuốc xảy ra chủ yếu bởi một con đường chính và con đường này chịu ảnh hưởng của yếu tố di truyền.
Thông thường, các hướng dẫn lâm sàng hiện hành chủ yếu mới quan tâm đến tương tác giữa các thuốc dùng đồng thời với nhau (drug - drug interaction hay DDI). Trong khi đó, yếu tố kiểu gen ảnh hưởng khá nhiều đến tần suất và ý nghĩa lâm sàng của tương tác thuốc, tạo nên một mối tương tác phức tạp giữa thuốc - thuốc - gen (drug - drug - gene interaction hay DDGI). Trong một nghiên cứu trên 23.000 bệnh nhân tại Mỹ đã phát hiện 6.900 tương tác thuốc, bao gồm 53% tương tác DDI, 22% tương tác DDGI, còn lại là tương tác giữa kiểu gen và 1 thuốc.
2 Ảnh hưởng của kiểu gen đến tương tác trong quá trình chuyển hóa thuốc
Tương tác dược động học dẫn đến thay đổi nồng độ trong máu/ mô của một thuốc hoặc của các dẫn chất chuyển hóa của thuốc đó. Mặc dù các tương tác dược động học có thể xảy ra trong quá trình hấp thu hay phân bố, nhưng các tương tác dược động học phổ biến và gây nhiều hậu quả lâm sàng nhất là tương tác thuốc xảy ra ở giai đoạn chuyển hóa.
Tương tác thuốc ở giai đoạn chuyển hóa có thể gây ra các biến chứng nguy hiểm và không được mong đợi trong quá trình sử dụng thuốc, đặc biệt thường hay xảy ra với các bệnh nhân nặng phải sử dụng nhiều thuốc và là đối tượng có tính nhạy cảm cao với tương tác thuốc. Ý nghĩa lâm sàng của một tương tác chuyển hóa thuốc phụ thuộc vào mức độ thay đổi nồng độ của dạng hoạt động (chất mẹ và/ hoặc chất chuyển hóa có hoạt tính) tại đích tác dụng và sự thay đổi chỉ số điều trị của thuốc. Sự chênh lệch giữa nồng độ tác dụng và nồng độ gây độc càng nhỏ (thuốc có phạm vi điều trị hẹp) thì khả năng xảy ra các tương tác thuốc nghiêm trọng càng lớn. Khả năng xuất hiện tương tác thuốc và ý nghĩa lâm sàng của tương tác phụ thuộc vào nhiều yếu tố như bản chất của các thuốc dùng đồng thời, liều dùng và đặc tính di truyền của mỗi cá thể bệnh nhân.
Trong giai đoạn chuyển hóa thuốc, các chuyển hóa qua hệ enzym CYP có liên quan nhiều nhất đến tương tác thuốc - thuốc trong lâm sàng bởi đây là họ enzym chuyển hóa nhiều thuốc, các enzym này rất dễ bị cảm ứng hoặc ức chế bởi nhiều tác nhân khác nhau, nhiều cơ chế ức chế khác nhau làm thay đổi hoạt tính của enzym chuyển hóa, từ đó làm thay đổi nồng độ, tác dụng và độc tính của các thuốc. Mặt khác, gen mã hóa cho các CYP thường có tính đa hình cao, do đó, ở các cá thể mang kiểu gen khác nhau, tương tác thuốc - thuốc thông qua CYP450 có thể xảy ra với tần suất và mức độ khác nhau, dẫn đến những đáp ứng thuốc đa dạng trong lâm sàng. Nhìn chung, sự thay đổi nồng độ của một thuốc do tương tác thuốc trong giai đoạn chuyển hóa thường xảy ra với mức độ rõ rệt hơn ở những người mang kiểu hình chuyển hóa bình thường (EM) của enzym chuyển hóa đó.
CYP2B6 không phải là một CYP chiếm tỷ lệ lớn ở gan nhưng cũng là một CYP được chú ý trong nghiên cứu Gen dược học, bởi đây là enzym chuyển hóa của nhiều thuốc có độc tính cao và phạm vi điều trị hẹp như các thuốc kháng retrovirus, thuốc ung thư, thuốc chống co giật... Gen mã hóa CYP2B6 nằm trên nhiễm sắc thể 19q. Đây là một gen có tính đa hình cao, dẫn đến sự khác biệt lớn giữa các cá thể về số lượng protein biểu hiện và về hoạt tính của protein enzym. Biến thể thường gặp nhất của gen CYP2B6 là alen CYP2B6*6 với hai SNP 516G>T và 7854>G, xuất hiện với tần suất khoảng 12 - 49% ở các quần thể khác nhau. Người mang biến thể này xuất hiện khiếm khuyết trên mARN, dẫn đến sự tạo thành phân tử protein giảm hoặc mất hoạt tính. Nói cách khác, hoạt tính enzym CYP2B6 giảm đi ở người mang kiểu gen CYP2B6*6, dẫn đến tăng nồng độ trong huyết tương cũng như tăng độc tính của các thuốc chuyển hóa qua CYP2B6, ví dụ như tăng độc tính trên thận của efavirenz và nevirapin ở bệnh nhân HIV. Người mang kiểu gen CYP2B6*6 đồng hợp tử có kiểu hình PM (chuyển hóa kém), dị hợp tử (CYP2B6*1/6) có kiểu hình chuyển hóa trung gian (IM) và kiểu gen CYP2B6*1/*1 có kiểu hình chuyển hóa bình thường (EM). Những bệnh nhân HIV bị nhiễm lao phải dùng đồng thời efavirenz và Rifampicin - một chất cảm ứng CYP2B6, do đó có thể xảy ra tương tác giữa hai thuốc này. Cơ chế gây cảm ứng CYP2B6 là do làm tăng biểu hiện gen, tăng cường tổng hợp các phân tử CYP2B6 trong tế bào, hậu quả là làm tăng chuyển hóa các thuốc, điều này có thể dẫn đến làm giảm nồng độ thuốc trong máu và giảm hiệu quả điều trị của thuốc, nếu không có sự điều chỉnh liều phù hợp. Tuy nhiên, nguy cơ xảy ra tương tác thuốc giữa rifampicin và các cơ chất của CYP2B6 còn tùy thuộc vào kiểu gen của từng bệnh nhân. Một nghiên cứu cho thấy các bệnh nhân mang kiểu gen wild type (CYP2B6*1/1) và kiểu gen chứa 1 alen biến thể 516 G>T (CYP2B6*1/6) (tương ứng với kiểu hình EM, IM) đều có nồng độ thuốc trong huyết tương giảm so với bình thường khi dùng đồng thời với rifampicin. Trong khi đó, những người mang kiểu gen CYP2B6*6/6 (kiểu hình PM) sự thay đổi nồng độ efavirenz do tương tác thuốc rifampicin - efavirenz không quá nhiều (Hình 8.1). Điều đó cho thấy khả năng cảm ứng enzym của rifampicin phụ thuộc vào kiểu gen của từng cá thể, kiểu gen biến thể tương ứng với kiểu hình PM ít chịu ảnh hưởng bởi tương tác rifampicin - efavirenz hơn các kiểu gen khác. Hiện tượng tương tự cũng được ghi nhận khi có sự phối hợp giữa efavirenz và một chất cảm ứng CYP2B6 khác là baicalin, một hoạt chất có nguồn gốc thảo dược.
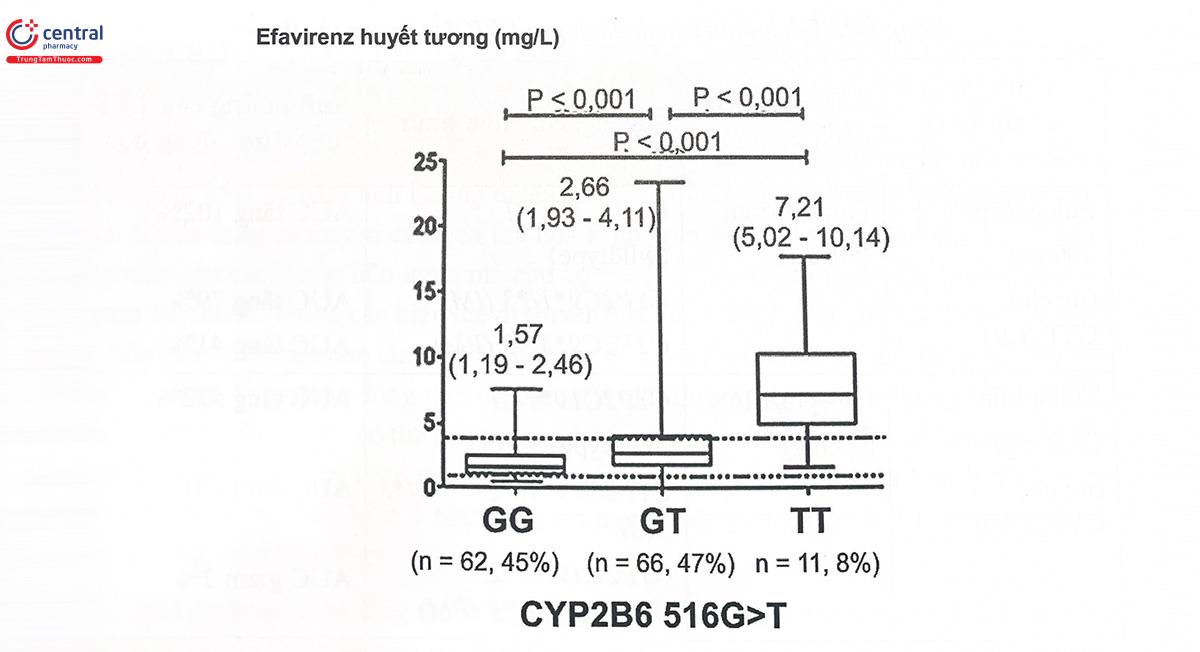
Hình 8.1. Sự thay đổi nồng độ efavirenz khi dùng đồng thời với rifampicin giữa những người mang kiểu gen CYP2B6 khác nhau
Các trường hợp tương tác thuốc liên quan đến các chất ức chế enzym CYP cũng xảy ra khá phổ biến. Omeprazol là một chất ức chế CYP2C19. Một trong những cơ chất của enzym này là moclobemid, một thuốc chống trầm cảm. Ở những người mang kiểu gen chuyển hóa bình thường - EM (CYP2C19*1/1), omeprazol ức chế đáng kể chuyển hóa của moclobemid, làm cho AUC tăng hơn 200%, trong khi ở người có kiểu gen chuyển hóa chậm - PM (CYP2C19*2/3 và CYP2C19*2/2) vốn chiếm 30% số người châu Á, tương tác với omeprazol hầu như không đáng kể.
Qua các ví dụ về hai CYP nói trên, có thể thấy ảnh hưởng của tương tác thuốc - thuốc rất khác nhau giữa những người mang kiểu gen khác nhau. Cho dù ở những người mang kiểu hình chuyển hóa chậm không chịu ảnh hưởng nhiều bởi tương tác thuốc nhưng nồng độ dạng thuốc còn hoạt tính trong máu cũng đều tăng lên ở cả hai trường hợp do tương tác thuốc ở người mang kiểu gen bình thường hay do ảnh hưởng của đa hình gen ở người mang kiểu gen đa hình.
Một số ví dụ khác về tương tác thuốc xảy ra ở những người mang kiểu gen CYP khác nhau được trình bày trong Bảng 8.1.
| Thuốc ảnh hưởng và cơ chế ảnh hưởng | Thuốc là cơ chất của CYP | Kiểu gen/ kiểu hình | Ảnh hưởng của TTT đến dược động học |
| Fluconazol 200 mg (ức chế CYP2C9) | Flurbiprofen (50 mg) | CYP2C9*1/*1 (wildtype) | AUC tăng 102% |
| CYP2C9*1/*3 (IM) | AUC tăng 79% | ||
| CYP2C9*3/*3 (PM) | AUC tăng 41% | ||
| Ticlopidin (200 mg) (ức chế CYP2C19) | Omeprazol (20 mg) | CYP2C19*1/*1 (wildtype) | AUC tăng 522% |
| CYP2C19*1/*2, *1/*3 (IM) | AUC tăng 401% | ||
| CYP2C19*2/*2, CYP2C19*2/3 (PM) | AUC giảm 2% | ||
| Rifampicin (300 mg) Cảm ứng CYP2C19 | Mephenytoin racemic (100 mg) | CYP2C19*1/*1 (wildtype) | Thải trừ dạng 4-OH mephenytoin tăng 203,9% |
| CYP2C19*1/*2, *1/*3 (IM) | Tăng 69,6% | ||
| CYP2C19*2/*3, *2/*2 (PM) | Tăng 80,1% | ||
| Quinidin (100 mg ngày 2 lần) ức chế CYP2D6 | S-venlafaxin (18,75 mg, ngày 2 lần) | CYP2D6*1/*4, *1/*1 (IM/ wild type) | AUC tăng 285% |
| CYP2D6*3/*4, *4/*4 (PM) | AUC tăng 15% | ||
| Quinidin (100 mg ngày 2 lần) ức chế CYP2D6 | R-venlafaxin (18,75 mg, ngày 2 lần) | CYP2D6*1/*4, *1/*1 (IM/ wild type) | AUC tăng 1118% |
| CYP2D6*3/*4, *4/*4 (PM) | AUC giảm 1% | ||
| Paroxetin (20 mg) ức chế CYP2D6 | Desipramin (100 mg) | Wild type | Độ thanh thải giảm 78% |
| PM | Độ thanh thải giảm 20% |
3 Ảnh hưởng của kiểu gen đến tương tác trong quá trình vận chuyển thuốc qua màng sinh học
Như đã trình bày ở trên, một trong những loại protein vận chuyển thuốc được nghiên cứu nhiều nhất và chịu ảnh hưởng nhiều của sự đa hình di truyền là P-glycoprotein (Pgp). Sự đa dạng di truyền của gen mã hóa P-gp (gen MDRI) không chỉ ảnh hưởng đến đáp ứng của các thuốc liên quan mà còn có thể đóng vai trò trong sự xuất hiện nguy cơ tương tác thuốc. Trong các biến thể di truyền của gen MDR1, biến thể C3435T ở exon 26 có liên quan đến việc làm tăng Sinh khả dụng và giảm tốc độ thải trừ qua thận của Digoxin đường uống. Việc dùng đồng thời digoxin với clarithromycin, một chất ức chế bơm tống thuốc P-gp về lý thuyết, có thể làm tăng sinh khả dụng của digoxin. Tuy nhiên, hiện tượng tương tác này này chỉ thực sự biểu hiện ở những người mang kiểu gen bình thường (3435CC), không xuất hiện ở những người mang kiểu gen CT hoặc TT, tương ứng với kiểu hình ức chế bơm P-gp.
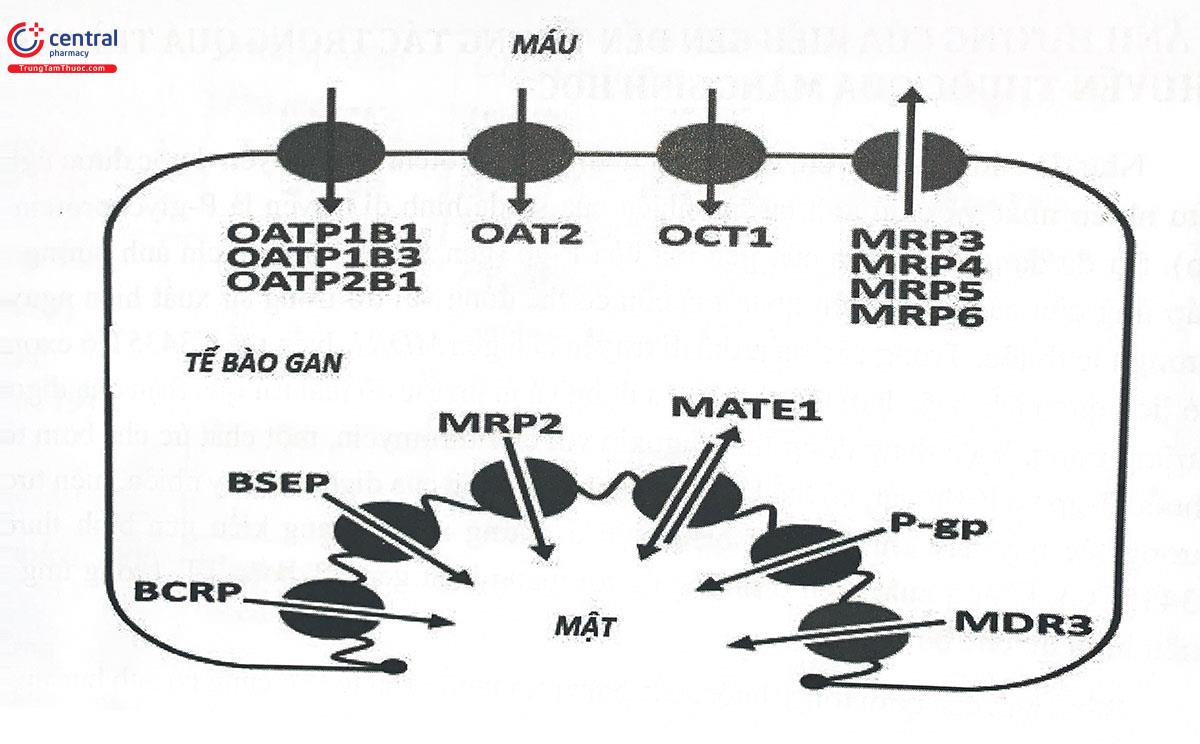
Bên cạnh các bơm tống thuốc, các bơm đưa thuốc vào tế bào cũng có ảnh hưởng lớn đến số phận của thuốc. Trong số đó, bơm OATP1B1 (organic anion transporting peptide 1B1) có chức năng bơm thuốc vào bên trong tế bào gan (Hình 8.2). Các thuốc statin hoạt động với cơ chế ức chế enzym tổng hợp cholesterol ở gan, do đó, tác dụng của các thuốc này phụ thuộc rất nhiều vào hoạt động của bơm OATP1B1. Trong số hơn 40 biến thể của gen OATP1B1, biến thể 521T>C(Val174Ala) làm giảm hoạt động của bơm, dẫn đến giảm lượng statin đến tế bào gan để phát huy tác dụng. Mặt khác, có một tương tác thuốc nghiêm trọng liên quan đến các statin là tương tác với Gemfibrozil và cyclosporin, hai thuốc này có khả năng ức chế các enzym chuyển hóa statin ở gan và ức chế OATP1B1, hậu quả là làm tăng nồng độ statin trong máu, giảm lượng thuốc ở gan, có nghĩa là giảm tác dụng của thuốc, đồng thời tăng độc tính toàn thân với các tác dụng không mong muốn điển hình là đau cơ, tiêu cơ. Các nghiên cứu cho thấy, tần suất và mức độ nghiêm trọng của tương tác statin - gemfibrozil, Cyclosporin là cao nhất ở nhóm người mang kiểu gen wildtype của OATP1B1.
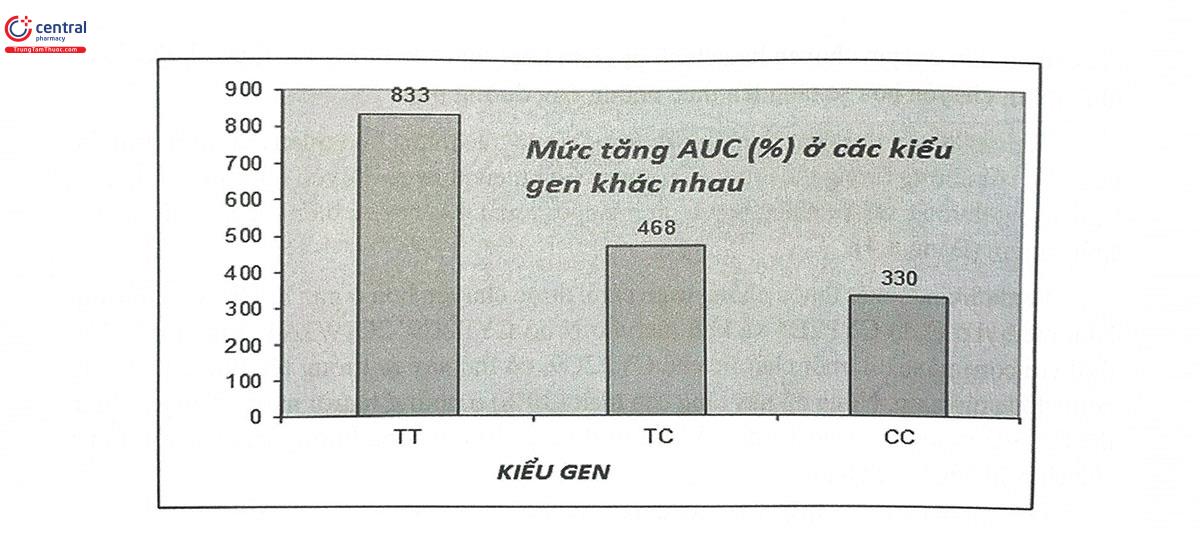
Một chất ức chế biểu hiện gen OATP1B khác là rifampicin cũng có thể gây tương tác thuốc với các statin, làm tăng nồng độ statin trong máu, gây phản ứng đau cơ, tiêu cơ vân cấp. Một nghiên cứu cũng cho thấy tương tác này chỉ thực sự có ý nghĩa lâm sàng, làm tăng Cmax của atorvastatin nhiều nhất ở người mang kiểu gen wildtype của gen OATPIB1. Người mang ít nhất một alen biến thể (C thay thế cho T, xét tại vị trí 521) mặc dù vẫn có nồng độ atorvastatin cao trong máu do bơm OATP1B1 bị ức chế, nhưng mức tăng không cao bằng so với kiểu gen wildtype. (Bảng 8.2 và Hình 8.2)
| Cmax của atorvastatin (ng/ml) | |||
| Kiểu gen | TT | TC | CC |
| Atorvastatin + Placebo | 3,4 ± 0,9 | 5,7 ± 1,6 | 12,1 ± 8,5 |
| Atorvastatin + Rifampicin | 50,5 ± 15,1 | 42,9 ± 27,4 | 90,8 ± 31,4 |
| Mức tăng Cmax atorvastatin do tương tác thuốc với rifampicin (%) | 1485,3 | 752,6 | 750,4 |
4 Ảnh hưởng của kiểu gen đến tương tác thuốc liên quan đến nhiều gen/protein khác nhau
Chỉ riêng tương tác giữa các thuốc dùng đồng thời hoặc tương tác giữa gen với một thuốc đã rất phức tạp và có nhiều kiểu ảnh hưởng đa dạng trên lâm sàng. Trong thực tế, đáp ứng của một thuốc phụ thuộc vào nhiều protein/ gen khác nhau ở nhiều khâu trong quá trình dược động học và dược lực học. Do vậy, khi tương tác thuốc liên quan đến nhiều gen/ protein khác nhau trong cùng một cơ thể càng trở nên phức tạp hơn rất nhiều và hậu quả trong lâm sàng cũng nặng nề hơn. Trường hợp thường gặp là tương tác thuốc xảy ra khi một thuốc là cơ chất của nhiều enzym chuyển hóa khác nhau khi dùng đồng thời với một thuốc ức chế hoặc cảm ứng một trong những enzym chuyển hóa đó, sự thay đổi về một enzym chuyển hóa sẽ dẫn đến sự thay đổi trong chuyển hóa thuốc bởi một hoặc nhiều enzym khác như một hiện tượng bù trừ. Tuy nhiên, sự thay đổi đáp ứng điều trị sẽ rất khác nhau ở những người mang kiểu gen khác nhau của một hoặc nhiều enzym chuyển hóa khác.
Voriconazol là một thuốc chống nấm nhóm triazol được chuyển hóa bởi 3 CYP (CYP3A4, CYP2C19 và CYP2C9). Alen CYP2C19*2 tương ứng với kiểu hình PM làm AUC tăng gấp 3 lần và giảm tốc độ thải trừ voriconazol so với bình thường. Mặt khác, một tương tác thuốc thường gặp giữa voriconazol với Ritonavir do hiện tượng ức chế CYP3A4 bởi ritonavir. Tương tác này gây ảnh hưởng nặng nề nhất ở cá thể mang alen CYP2C19*2, làm giảm đến 86% độ thanh thải của voriconazol, trong khi ở các kiểu gen chuyển hóa bình thường (EM), mức giảm trung bình là 34%. Như vậy, khi chuyển hóa qua CYP2C19 giảm (do đa hình gen) sẽ làm CYP3A4 nhạy cảm hơn với tác dụng ức chế của ritonavir. Đó cũng là một quy luật chung, đối với thuốc là cơ chất của nhiều CYP khác nhau, khi enzym chuyển hóa chính bị giảm hoạt tính (do tương tác thuốc hoặc do đa hình gen), chuyển hóa sẽ tăng lên theo những con đường phụ.
Ngược lại với quy luật thường gặp trong tương tác thuốc liên quan đến một protein/ gen, đối với những tương tác thuốc liên quan đến nhiều enzym chuyển hóa, sự ảnh hưởng trầm trọng thường xảy ra nhiều hơn ở những người mang kiểu gen biến thể làm giảm hoạt tính enzym (Bảng 8.4).
Venlafaxin là một thuốc chống trầm cảm, được chuyển hóa ở gan bởi hai con đường: Khử methyl ở O do CYP2D6 và khử metyl ở N do CYP2C9/ 2C19/3A4. Khi dùng đồng thời với cotrimoxazol, một chất ức chế CYP2C9, có thể xảy ra tương tác thuốc làm tăng nồng độ venlafaxin. Nồng độ này tăng cao nhất (30%) ở những người mang kiểu gen biến thể làm giảm hoặc mất hoạt tính CYP2C19 do hiện tượng cộng hưởng của hai yếu tố ức chế chuyển hóa khác nhau.
Mối quan hệ trong tương tác đa chiều thuốc - gen - thuốc - gen còn phức tạp hơn nữa trong trường hợp đa hình gen làm tăng nồng độ của thuốc gây tương tác thuốc. Tacrolimus là một thuốc được chuyển hóa bởi CYP2C19 và CYP3A4. Khi dùng cùng với itraconazol, một chất ức chế CYP3A4, tương tác thuốc xảy ra làm tăng nồng độ tacrolimus. Khi dùng đồng thời Tacrolimus và itraconazol, nồng độ tacrolimus càng tăng cao hơn (đến 200%) ở những người có kiểu gen biến thể làm giảm hoạt tính CYP2C19. Nhưng thậm chí, mức tăng còn gấp đôi khi thay itraconazol bằng một thuốc kháng nấm tương tự là voriconazol. Điều này được giải thích là do voriconazol vừa là chất ức chế CYP3A4 như itraconazol nhưng đồng thời lại là cơ chất của CYP2C19. Những người bị giảm hoạt tính CYP2C19 có nồng độ voriconazol cao hơn dẫn đến tương tác thuốc xảy ra ở mức độ nghiêm trọng hơn nữa, làm tăng 1500% nồng độ tacrolimus.
| Thuốc ảnh hưởng và cơ chế ảnh hưởng | Thuốc là cơ chất của CYP | Enzym chuyển hóa thứ hai | Kiểu gen/ Kiểu hình của enzym chuyển hóa chính | Ảnh hưởng của TTT đến dược động học |
| Clarithromycin (500 mg, ngày 2 lần) (40 mg) ức chế CYP3A4 | Esomeprazol (40mg) | CYP3A4 | CYP2C19*1/*1 (Wild type) | AUC tăng 70% |
CYP2C19*2/2 CYP2C19*2/3 (PM) | AUC tăng 114% | |||
| Troleandomycin (500 mg) ức chế CYP3A4 | Omeprazol (20mg) | CYP3A4 | CYP2C9*1/*1 (Wild type) | AUC tăng 28% |
| CYP2C9*1/*2, *1/*3 (IM) | AUC tăng 26% | |||
| CYP2C9*2/*2, 3/*3 (PM) | AUC tăng 81% | |||
| Clarithromycin (úrc chế CYP3A4) và paroxetin (ức chế CYP2D6) | Venlafaxin (75mg) | CYP3A4 | CYP2D6*1/*1, *1/*2 (wild type) | Sau khi dùng clarithromycin: AUC tăng 8% Sau khi dùng clarithromycin và paroxetin: AUC tăng 328% |
| CYP2D6*10/10 (IM) | Sau khi dùng clarithromycin: AUC tăng 31% Sau khi dùng Clarithromycin và paroxetin: AUC tăng 124% | |||
| Itraconazol (200 mg) ức chế CYP3A4 | Risperidone (2-8 mg) | CYP3A4 | CYP2D6*1/*1 (wild type) | AUC tăng 55% |
| CYP2D6 *10/*10 (IM) | AUC tăng 99% |
Tính chất phức tạp của tương tác thuốc - thuốc liên quan đến nhiều gen/ protein khác nhau được thể hiện ở Hình 8.4, mô tả các khả năng có thể xảy ra ở những người mang kiểu gen khác nhau. Với cùng một cách phối hợp điều trị, nồng độ thuốc dạng ban đầu và nồng độ các sản phẩm chuyển hóa sẽ rất khác nhau ở những người mang kiểu gen khác nhau, dẫn đến ý nghĩa lâm sàng của tương tác thuốc rất khác nhau.
Một số ví dụ về tương tác thuốc xảy ra ở những người mang kiểu gen CYP khác nhau được trình bày trong Bảng 8.4 và Hình 8.4.
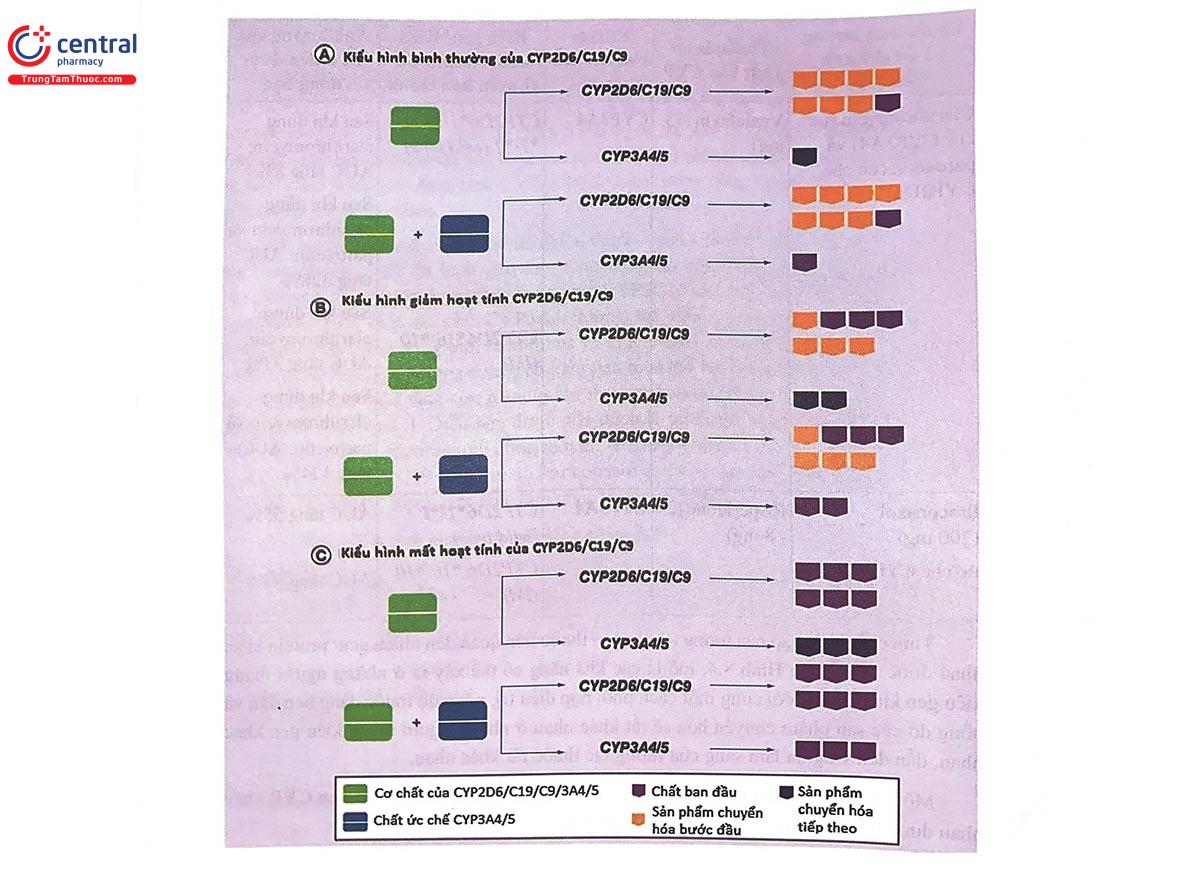
A: Ở người mang kiểu gen bình thường của CYP2D6/CYP2C19/CYP2C9:
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5: Qua chuyển hóa bởi CYP2D6/CYP2C19/CYP2C9, phần lớn sẽ thành sản phẩm chuyển hóa bước đầu, chỉ một phần nhỏ ở dạng ban đầu, qua chuyển hóa bởi CYP3A4/A5 sẽ thành sản phẩm chuyển hóa tiếp theo.
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5 đồng thời với một thuốc ức chế CYP3A4/A5: Qua chuyển hóa bởi CYP2D6/CYP2C19/ CYP2C9, phần lớn sẽ thành sản phẩm chuyển hóa bước đầu, chỉ một phần nhỏ ở dạng ban đầu, quá trình chuyển hóa bởi CYP3A4/A5 bị ức chế, do đó thuốc vẫn ở dưới dạng ban đầu.
B: Ở người mang kiểu gen tương ứng kiểu hình giảm hoạt tính của CYP2D6/ CYP2C19/CYP2C9:
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5: Quá trình chuyển hòa bởi CYP2D6/CYP2C19/CYP2C9 giảm, chỉ một phần chuyển thành sản phẩm chuyển hóa bước đầu, phần còn lại ở dạng ban đầu, qua chuyển hóa bởi CYP3A4/A5 sẽ thành sản phẩm chuyển hóa tiếp theo, quá trình chuyển hóa bởi CYP3A4/A5 tăng lên do giảm quá trình chuyển hóa bởi CYP2D6/CYP2C19/CYP2C9.
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5 đồng thời với một thuốc ức chế CYP3A4/A5: Quá trình chuyển hóa bởi CYP2D6/ CYP2C19/CYP2C9 giảm, chỉ một phần chuyển thành sản phẩm chuyển hỏa bước đầu, phần còn lại ở dạng ban đầu, quá trình chuyển hóa bởi CYP3A4/A5 sẽ thành sản phẩm chuyển hóa tiếp theo, quá trình chuyển hóa bởi CYP3A4/A5 bị ức chế, do đó thuốc vẫn ở dưới dạng ban đầu nhiều hơn trường hợp A.
C: Ở người mang kiểu gen tương ứng kiểu hình mất hoạt tính của CYP2D6/ CYP2C19/CYP2C9:
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5: Quá trình chuyển hóa bởi CYP2D6/CYP2C19/CYP2C9 không xảy ra, thuốc vẫn tồn tại chủ yếu ở dạng ban đầu, quá trình chuyển hóa bởi CYP3A4/A5 tăng lên nhiều hơn trường hợp B do giảm quá trình chuyển hóa bởi CYP2D6/CYP2C19/CYP2C9.
- Nếu dùng một thuốc là cơ chất của CYP2D6/CYP2C19/CYP2C9/ CYP3A4/A5 đồng thời với 1 thuốc ức chế CYP3A4/A5: Cả hai quả trình chuyển hóa bởi CYP2D6/ CYP2C19/CYP2C9 và CYP3A4/A5 đều không xảy ra, thuốc còn lại nguyên vẹn ở dạng ban đầu.
Trong thực tế, tương tác thuốc - gen - thuốc có thể liên quan đồng thời tới nhiều enzym khác nhau của chuyển hóa thuốc pha I và và pha II, thậm chí, liên quan tới cả các protein vận chuyển thuốc. Ví dụ như một trường hợp tương tác thuốc khi dùng haloperidol đồng thời với ciprofloxacin. Haloperidol được chuyển hóa pha 1 chủ yếu bởi CYP2D6 và một phần bởi CYP3A4, chuyển hóa pha II bởi UDP-Glucuronosyltransferase-2B7 (UGT2B7). Một bệnh nhân mang kiểu gen CYP2D6*6/*6, CYP3A4*1/*1 và UGT2B7 -161 C/T, tương ứng với kiểu hình của 3 enzym CYP2D6, CYP3A4 và UGT2B7 lần lượt là mất hoạt tính, bình thường và giảm hoạt tính. Khi đó, chuyển hóa haloperidol phụ thuộc chủ yếu vào enzym CYP3A4. Tuy nhiên, việc dùng đồng thời với Ciprofloxacin, một chất ức chế CYP3A4 đã dẫn đến tăng nồng độ haloperidol trong máu, với hậu quả là TDKMM hội chứng ngoại tháp nghiêm trọng.
5 Kết luận
Chỉ riêng sự tương tác qua lại giữa hai hay nhiều thuốc dùng đồng thời đã có thể tạo ra những thay đổi rất lớn về đáp ứng điều trị. Do vậy, sự ảnh hưởng của đa hình gen giữa những cá thể dùng thuốc càng làm cho mối quan hệ 3 chiều thuốc - gen - thuốc thêm phức tạp, dẫn đến thay đổi đáng kể về hiệu quả điều trị và tác dụng không mong muốn. Nói cách khác, mức độ ảnh hưởng của tương tác thuốc khác nhau giữa các cá thể và phụ thuộc đáng kể vào yếu tố khác biệt di truyền.
6 Tài liệu tham khảo
- Phùng Thanh Hương, Đỗ Hồng Quảng, Nguyễn Văn Rư, Nguyễn Thị Lập, Nguyễn Quốc Bình. Giáo trình GEN DƯỢC HỌC - ẢNH HƯỞNG CỦA GEN ĐẾN ĐÁP ỨNG THUỐC. Trường Đại học Dược Hà Nội, Bộ môn Hóa Sinh - Khoa Công Nghệ Sinh Học. CHƯƠNG 8. Trang 106-118. Tải PDF sách TẠI ĐÂY
- Bahar MA, Setiawan D, Hak E, Wilffert B, Pharmacogenetics of drug-drug interaction and drug-drug-gene interaction: a systematic review on CYP2C9, CYP2C19 and CYP2D6, Pharmacogenomics. 2017 May; 18(7):701-739. doi: 10.2217/pgs-2017-0194. Epub 2017 May 8.
- Brian Thomas Hocum, Cytochrome P-450 gene and drug interaction analysis in patients referred for pharmacogenetic testing, Am J Health-Syst Pharm Vol 73(2), 2016.
- Shabbir Ahmed, Zhan Zhou, Jie Zhou, and Shu-Qing Chen, Pharmacogenomics of Drug Metabolizing Enzyms and Transporters: Relevance to Precision Medicine, Genomics Proteomics Bioinformatics. 2016 Oct; 14(5): 298-313.
