Ảnh hưởng của bụi - khói, khí độc hại và phản ứng của phế quản - phổi

Trungtamthuoc.com - Các chất gây ô nhiễm không khí bao gồm khói, bụi và các khí độc hại được thải ra mỗi ngày là nguyên nhân gây nên các bệnh mạn tính đường hô hấp như hen hoặc COPD. Bài viết dưới đây sẽ cho thấy ảnh hưởng của bụi - khói, khí độc hại đến đường hô hấp và phản ứng của phế quản - phổi
1 BỤI - KHÓI VÀ KHÍ ĐỘC HẠI VỚI PHỔI
Các chất gây ô nhiễm không khí bao gồm các hạt và khí độc hại được thải ra với số lượng lớn từ rất nhiều nguồn khác nhau. Các chất gây ô nhiễm chính bao gồm vật chất dạng hạt (particulate matter, PM), ozone (trioxygen,O3) nitơ dioxide (NO2) và sulfur dioxide (SO2). Các chất gây ô nhiễm sinh học như vi-rút, vi khuẩn, dị nguyên. Các hạt nhỏ hơn 10 µm nhưng lớn hơn 2 µm có thể đi vào hệ thống khí - phế quản, nhưng được loại bỏ thông qua khả năng thanh thải của niêm mạc. Các hạt nhỏ hơn có thể xâm nhập vào đường thở nhỏ và đến vùng phế nang của phổi. Ở vùng phế nang, các cytokine và chemokine hóa ứng động bạch cầu đa nhân trung tính và đại thực bào đến thực hiện thực bào các hạt lạ xâm nhập. Ozone trong không khí là một chất gây ô nhiễm môi trường khí thở có tác động đáng kể đến sức khỏe con người. Ozone có tính phản ứng cao và oxy hóa mạnh protein và lipid trong các khoang chứa chất lỏng của phổi [1,2], gây tổn thương tế bào biểu mô, cản trở đáp ứng viêm và làm sạch đường thở [3-7]. Dữ liệu dịch tễ học cho thấy những người mắc bệnh mạn tính đường hô hấp, như hen hoặc COPD, đặc biệt nhạy cảm với việc tiếp xúc với ozone và có tỷ lệ mắc bệnh tăng lên cũng như nguy cơ tử vong cao hơn khi phản ứng với ozone [1,2]. Chất ô nhiễm cũng có thể có tác động tới cấu trúc và chức năng phổi thông qua tổn thương tế bào qua trung gian phản ứng oxy hóa, stress oxy hóa và các phản ứng miễn dịch bẩm sinh hay thu được. Như vậy, có thể hiểu và đồng thuận rằng ô nhiễm khí thở (nhiễm trùng hay không nhiễm trùng) hoàn toàn có thể đẩy COPD vào trạng thái đợt cấp [8]. Trong khái niệm bụi - khói, khí độc hại còn có thuật ngữ bioaerosol để chỉ một loại hạt được giải phóng từ hệ sinh thái trên mặt đất hay trên biển vào khí quyển. Chúng bao gồm cả thành phần sống và không sống, thí dụ như nấm, phấn hoa, vi khuẩn và vi-rút. Bioaerosol thường trộn lẫn trong không khí và di chuyển thông qua sự nhiễu loạn của gió trên bề mặt. Khi ở trong khí quyển, chúng có thể được vận chuyển cục bộ hoặc toàn cầu thông qua gió, mây, bão. Do kích thước từ rất nhỏ, từ 0,001nm, bioaerosol dễ dàng lắng đọng ở phổi và thông qua phổi và hệ thống tuần hoàn để tới lắng đọng ở các bộ phận khác trong cơ thể. Nhìn một cách tổng thể, tuy tác động gây bệnh đối với phổi của bụi - khói, khí độc hại không quan trọng bằng khói thuốc lá [9] nhưng tác động làm tăng tần suất mắc và diễn biến nặng các bệnh phổi mạn tính là rõ ràng, nhất là khi nồng độ >10 µg/m3 và kích thước nhỏ hơn 2,5 µm [10].
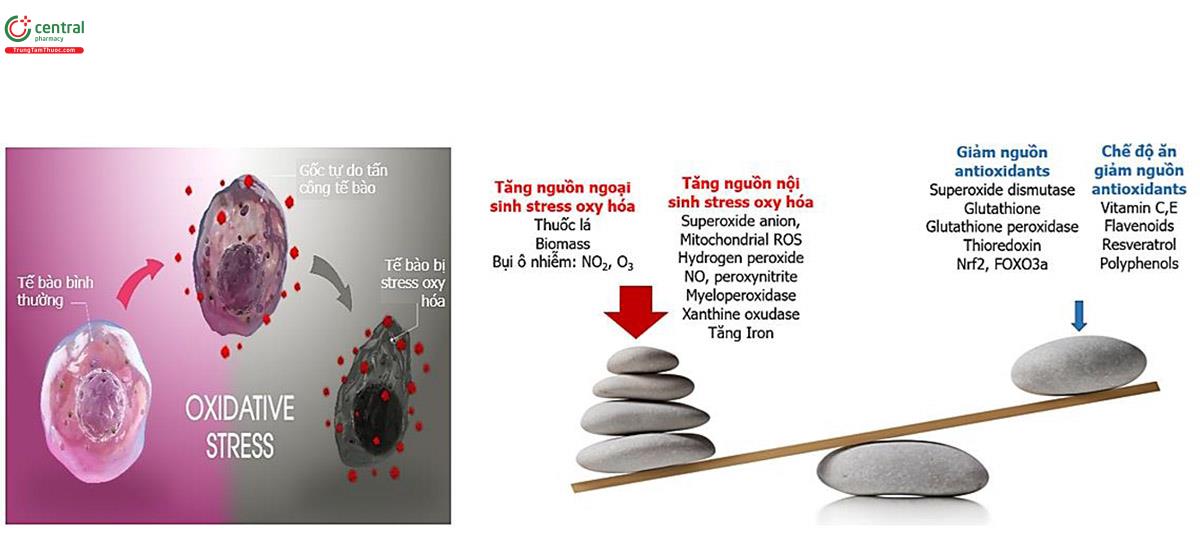
Stress oxy hóa nên được hiểu thế nào?. Các phân tử mang oxy có số lượng electron không đồng đều được gọi là gốc tự do. Con số không đồng đều này cho phép chúng phản ứng với các phân tử khác và gây ra các phản ứng hóa học chuỗi lớn. Chất chống oxy hóa là các phân tử có thể nhường electron cho gốc tự do mà không làm cho chúng mất tính ổn định, đồng thời làm cho gốc tự do ổn định và ít phản ứng hơn. Các tế bào của cơ thể tạo ra các gốc tự do trong quá trình trao đổi chất. Mặt khác, tế bào cũng sản xuất ra chất chống oxy hóa giúp vô hiệu hóa tác động của các gốc tự do trên và như vậy cơ thể thường có thể tự quản lý sự cân bằng giữa cả hai. Sự mất cân bằng giữa chất oxy hóa và chất chống oxy hóa là cơ sở cho stress oxy hóa. Mất thăng bằng oxy hóa và chống oxy hóa, nhất là khi kéo dài, dẫn tới thoái hóa tế bào, viêm mạn tính, đột biến.
Tăng viêm tương quan với tăng tình trạng tắc nghẽn trong COPD. Hút thuốc lá là yếu tố nguy cơ chính cho sự phát triển của bệnh phổi tắc nghẽn mạn tính (COPD). Tuy nhiên, lý do tại sao chỉ có 15 đến 20% số người nghiện thuốc lá nặng bị hạn chế luồng khí mạn tính [11,12] vẫn chưa được biết. Khái niệm cho rằng vị trí gây ra hạn chế luồng khí trong COPD là đường thở ngoại vi đã được xác định rõ ràng [13,14], tuy nhiên, một số nghiên cứu đã chỉ ra rằng đường hô hấp lớn cũng bị ảnh hưởng bởi quá trình viêm ở người hút thuốc [15-18]. Các nghiên cứu cho thấy quá trình viêm khác biệt ở những người hút thuốc bị hạn chế luồng khí mạn tính so với ở những người hút thuốc không bị hạn chế luồng khí mạn tính và cho thấy số lượng tế bào lympho T và đại thực bào tăng lên ở những người đã hút thuốc trước đây [18]. Như vậy, có thể kết luận rằng ở những người hút thuốc, mức độ nặng của hạn chế luồng khí tương quan với mức độ nghiêm trọng của tình trạng viêm đường thở và hạn chế luồng khí nặng có liên quan đến việc tăng số lượng bạch cầu đa nhân trung tính, đại thực bào, tế bào lympho NK (natural killer) và tế bào MIP-1α+ (Macrophage inflammatory protein-1α) ở niêm mạc phế quản [19].
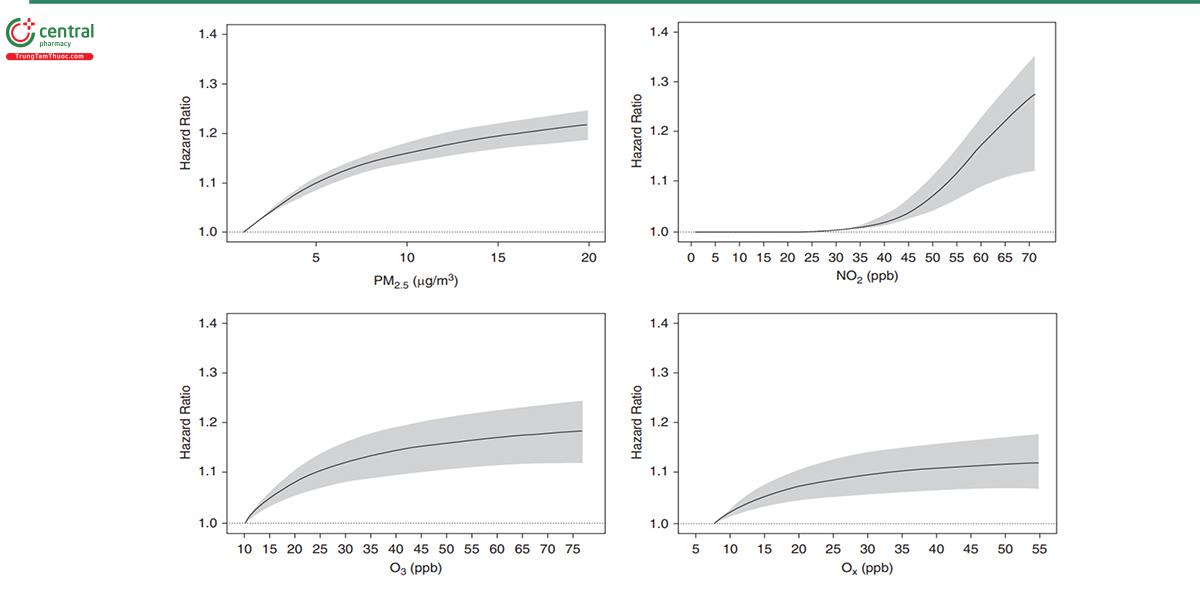
Vấn đề ô nhiễm và COPD đã được nhiều nghiên cứu đề cập. Trong một theo dõi trên 15 năm Shin và cs [20] ghi nhận mức phơi nhiễm ô nhiễm không khí trung bình trong 3 năm với độ trễ 1 năm, nhiều chất gây ô nhiễm ngoài trời, bao gồm chất dạng hạt mịn (PM2,5), nitơ dioxide (NO2) và ozone (O3), có liên quan đến tỷ lệ mắc COPD cao hơn và tăng COPD tương quan thuận với nồng độ ô nhiễm (hình 1).
1.1 Khói thuốc lá
Khói thuốc lá là một hỗn hợp phức tạp của các hợp chất hóa học, bao gồm các gốc tự do và các chất oxy hóa khác có khả năng gây tổn thương mô [21]. Bề mặt biểu mô của đường hô hấp và các khoảng không khí là tuyến phòng thủ đầu tiên của phổi chống lại các chất độc hại khi hít vào. Các hiện tượng xảy ra trong đường hô hấp có thể liên quan đến cơ chế bệnh sinh của các bệnh về đường hô hấp do hút thuốc lá, như bệnh phổi tắc nghẽn mạn tính. Những thay đổi quan sát được trong đường thở của những người hút thuốc dễ mắc bệnh COPD có thể làm sáng tỏ cơ chế bệnh sinh của bệnh. Tăng nồng độ yếu tố hoại tử u α (TNF-α) và Interleukin 8 (IL-8) đã được phát hiện trong đờm của bệnh nhân COPD so với người không hút hoặc hút thuốc nhưng không bị COPD [22]. Các tế bào biểu mô và đại thực bào có trong lòng đường thở có thể là nguồn IL-8, một yếu tố hóa ứng động mạnh đối với bạch cầu đa nhân trung tính, trong khi TNF-α được biết là có tác dụng điều chỉnh tăng sự biểu hiện của các phân tử dính. Cũng đã có nghiên cứu ghi nhận khói thuốc lá làm tăng phản ứng đường thở với kích thích (thông qua test methacholine) mà không phụ thuộc vào đáp ứng miễn dịch type 2 (thông qua IgE toàn bộ và đặc hiệu) [23]. Hút thuốc lá là yếu tố nguy cơ chính từ môi trường của bệnh phổi tắc nghẽn mạn tính. Tuy nhiên, mặc dù nó gây ra những thay đổi rõ rệt về phiên mã và mã hóa gen (epigenetic) [24] nhưng không phải tất cả những người hút thuốc đều mắc bệnh [25] do những lý do vẫn còn chưa được biết rõ ràng [26]. Quá trình methyl hóa DNA, bao gồm việc bổ sung nhóm methyl vào bazơ cytosine liền kề với bazơ guanine (vị trí CpG), điều chỉnh biểu hiện gen [27]. Các nghiên cứu trước đây đã báo cáo các mô hình methyl hóa khác biệt liên quan đến hút thuốc lá [28] và COPD [29,30]. Sandra Casas-Recasens và cs thực hiện nghiên cứu phân tích quá trình methyl hóa DNA trên những người phơi nhiễm khói thuốc lá bị COPD cho thấy đặc điểm methyl hóa ở phổi khác nhau tùy theo mức độ nặng của tình trạng tắc nghẽn và một số thay đổi mã hóa gen được xác định có liên quan đến chuyển thành những thay đổi thực sự biểu hiện gen cụ thể trong phổi [31].
Mặc dù hút thuốc lá gây ra phản ứng viêm trong phổi của tất cả những người hút thuốc nhưng phản ứng này không chỉ tiếp tục tồn tại mà thậm trí còn tăng lên sau khi ngưng hút thuốc ở những người bị COPD [32]. Điều này gợi ý rằng ở những người hút thuốc bị COPD có sự điều hòa bất thường phản ứng viêm ở phổi. Các yếu tố nhạy cảm với khói thuốc ở người COPD vẫn chưa được hiểu rõ và có thể liên quan đến yếu tố di truyền và kiểu hình có khả năng di truyền (epigenesis), thay đổi quy trình điều hòa miễn dịch, suy giảm khả năng giải quyết tình trạng viêm và tạo ra cơ chế sửa chữa bất thường [33]. Tuy nhiên, mối quan hệ giữa phản ứng viêm ở phổi và sự suy giảm nhanh chóng của FEV1, đặc trưng cho tình trạng tắc nghẽn trong COPD, vẫn chưa rõ ràng. Thật vậy, giả thuyết rằng tình trạng viêm giải thích cho tất cả các đặc điểm của quá trình sinh bệnh học liên quan đến sự phá hủy phế nang và tái cấu trúc đường thở trong COPD đã đơn giản hóa quá mức tính chất đa hình và đa yếu tố của tình trạng viêm trong bệnh lý này và do đó, nó đã không dẫn đến những tiến bộ lớn trong điều trị bệnh COPD [34]. Rõ ràng rằng COPD là một bệnh không đồng nhất, trong đó sự liên quan đến đường hô hấp lớn và nhỏ (viêm phế quản/viêm tiểu phế quản) và nhu mô phổi (khí phế thũng) rất khác nhau giữa các bệnh nhân. Cơ chế gây ra những thay đổi bệnh lý này cũng có thể khác nhau, trong đó có sự tham gia tiềm năng của một số quá trình sinh bệnh học, đặc biệt liên quan đến sự tham gia tiềm năng của các quá trình sinh học liên quan đến lão hóa, tăng cường phá hủy mô liên quan đến chết chương trình (apoptosis) tế bào phế nang. Các quá trình này có tương tác mật thiết để tạo ra các vòng xoắn dẫn đến sự phân giải cấu cấu trúc nền ngoại bào quá mức (tức là mất cân bằng protease/anti-protease) và stress oxy hóa.
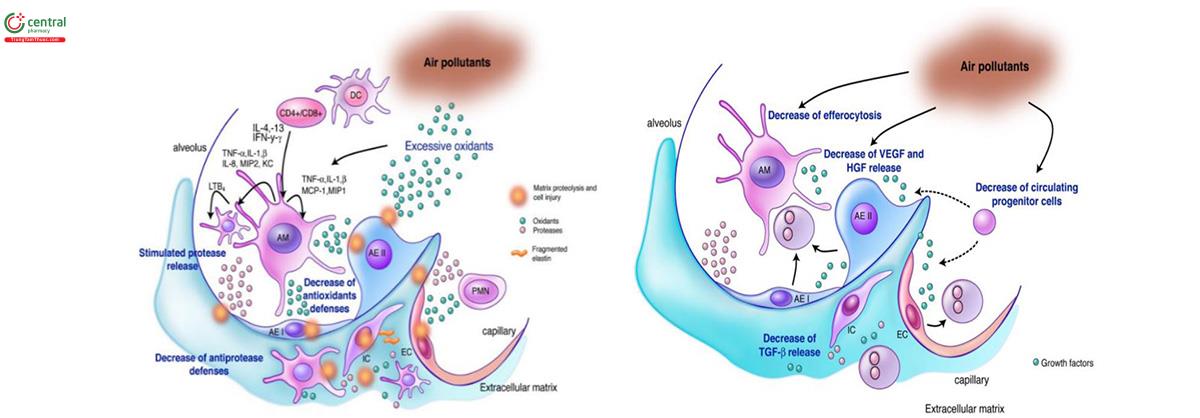
Stress oxy hóa tạo ra quá mức các phân tử có khả năng phản ứng cao, những phân tử có khả năng phá hủy tất cả các thành phần tế bào, bao gồm cả protein, lipid và DNA. Stress oxy hóa còn có khả năng gây độc thông qua việc tạo ra các gốc tự do và các peroxide (hình 3). Những hiểu biết này cung cấp một cái nhìn mới cho các nghiên cứu về sinh bệnh học của COPD, trong đó tình trạng viêm có thể được định dạng như một cơ chế phức tạp hơn làm cơ sở cho việc tái tạo và phá hủy mô trong COPD. Khói thuốc lá không chỉ trực tiếp gây viêm làm tổn thương mô phế nang mà còn khuếch đại quá trình viêm, có khả năng dẫn đến tự miễn dịch và kích thích tự duy trì để tăng cường dòng tế bào viêm vào phổi bị tổn thương [35]. Nhiều nghiên cứu đã ghi nhận sự biểu hiện gia tăng các dấu hiệu stress oxy hóa trong phổi của bệnh nhân COPD so với những người khỏe mạnh hoặc những người hút thuốc nhưng không bị COPD [36]. Nhận định này được minh họa bằng biểu hiện của 4-hydroxy-2-nonenal (4HNE), một sản phẩm cuối cùng của quá trình peroxid hóa lipid có khả năng phản ứng cao trong phổi COPD. 4HNE phản ứng nhanh với protein ngoại bào để tạo thành các dạng chất kết hợp (form adducts). Những dạng chất kết hợp này đã được chứng minh là hiện diện với số lượng lớn hơn trong các tế bào biểu mô và nội mô đường thở trong phổi của bệnh nhân COPD so với những người hút thuốc nhưng không bị COPD [37]. 4HNE có thể hoạt động như một chất hóa ứng động với bạch cầu đa nhân trung tính [38] và cũng tham gia vào nhiều chức năng của tế bào, chẳng hạn như ức chế/tăng sinh tế bào [39], thoái hóa tế bào T [40] và kích hoạt các con đường truyền tín hiệu khác nhau [41]. Có nhiều tác động của tình trạng stress oxy hóa được cho là đóng vai trò trong sinh bệnh học COPD. Vai trò đó bao gồm làm bất hoạt tác dụng của anti-proteases (thí dụ như α1-antitrypsin hoặc chất ức chế protease do bạch cầu tiết ra) [42] hoặc hoạt hóa metalloproteases [43], từ đó tạo ra tình trạng mất thăng bằng protease/anti-protease ở phổi. Đây là cơ chế được cho là có vai trò chính trong sự hình thành khí phế thũng [44]. Chất oxy hóa có thể trực tiếp phá hủy các thành phần của cấu trúc nền ngoại bào phổi (ví dụ Elastin và Collagen) và cũng có thể cản trở quá trình tổng hợp và sửa chữa elastin [36].
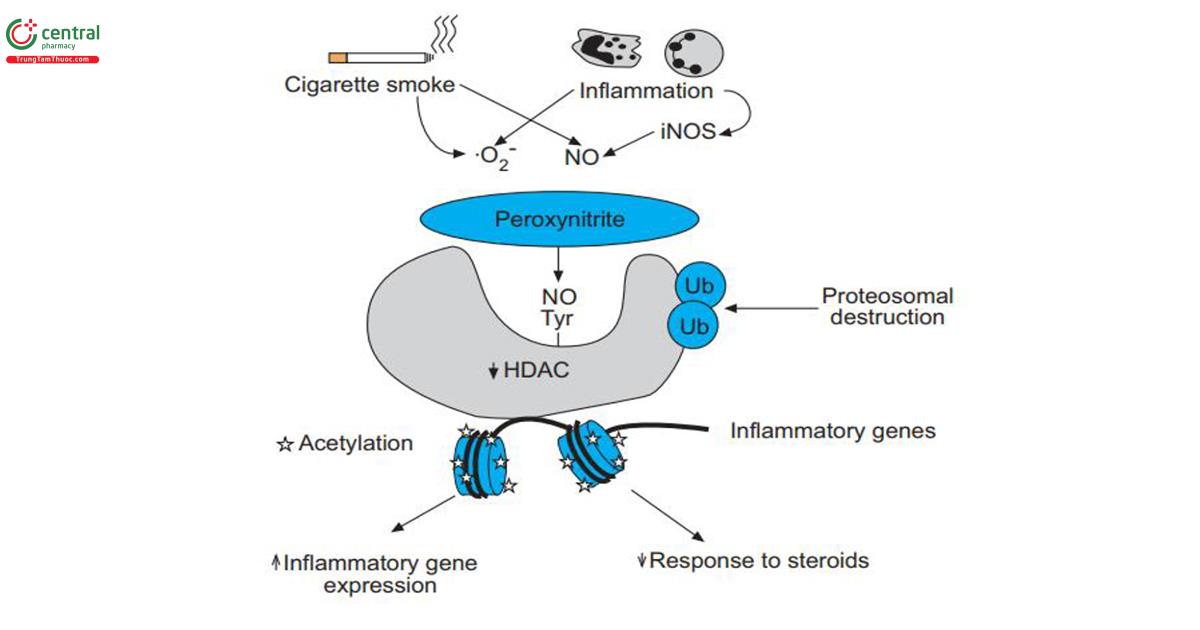
Stress oxy hóa cũng tác động lên cơ chế phân tử liên quan đến trình diện gen gây viêm. Sự kích hoạt gen bằng các yếu tố phiên mã được điều hòa bởi một số yếu tố, trong đó có sự tái cấu trúc DNA phụ thuộc vào sự cân bằng acetyl hóa/deacetyl hóa lõi histone [45], trạng thái cân bằng được kiểm soát bởi hoạt động của histone acetyl-transferase (HAT) và histone deacetylase (HDAC). DNA trong tế bào, ở trạng thái nghỉ, cuộn quanh lõi nucleosome của các gốc histone. Cấu hình này ngăn chặn khả năng tiếp cận của các yếu tố phiên mã như yếu tố nhân-kB (nuclear factor-kB ) đối với trình tự DNA cùng gốc. Ở người hút thuốc lá hay trên động vật thí nghiệm phơi nhiễm khói thuốc, hoạt động của histone deacetylase trong đại thực bào bị giảm [46]. Trong COPD, tình trạng giảm HDAC2 tăng lên theo mức độ nặng của bệnh [34,47]. Giảm HDAC đồng nghĩa với tăng acetyl-transferase, tăng phiên mã gen sinh tổng hợp protein tiền viêm mà trong đó có IL-8 (yếu tố hóa ứng động bạch cầu đa nhân trung tính) là IL-8 mRNA, từ đó gây ra tình trạng viêm kéo dài trong bệnh lý này (hình 3). Stress oxy hóa có liên quan mật thiết đến sự chết của tế bào, bao gồm cả quá trình chết theo chương trình của tế bào phế nang trong bệnh cảnh khí phế thũng ở người và thực nghiệm [48-51]. Hơn nữa, stress oxy hóa là nền tảng cho một số cơ chế được cho là có liên quan đến quá trình lão hóa, có thể làm giảm ngưỡng gây tổn thương phổi do khói thuốc lá [52]. Các nghiên cứu đã ghi nhận rằng chết theo chương trình (apoptosis) tế bào phổi xảy ra ở phổi bị khí thũng, chủ yếu liên quan đến các tế bào nội mô trong thành phế nang, so với phổi của những người bình thường hoặc của những người hút thuốc không mắc COPD [49]. Khí thũng thực nghiệm ở động vật có thể được tạo ra do giảm yếu tố tăng trưởng nội mô mạch máu (vascular endothelial growth factor, VEGF) hoặc tín hiệu VEGF [53]. Các nghiên cứu ở phổi người đã chứng minh sự giảm biểu hiện của VEGF và biểu hiện thụ thể VEGF2 có liên quan đến khí phế thũng [49]. Mối liên quan tổng thể của con đường truyền tín hiệu này, trong tổn thương phổi do khói thuốc lá, đã được nhấn mạnh trong các nghiên cứu thực nghiệm tiếp theo mà trong đó khói thuốc lá làm giảm mức độ biểu hiện của thụ thể VEGF2 [54]. Tổng hợp những dữ liệu đã thu được từ các nghiên cứu như trên đã dẫn đến khái niệm về chương trình bảo trì phế nang cần thiết để bảo tồn cấu trúc của phổi. Khói thuốc lá được cho là nguyên nhân phá hủy chương trình bảo trì này, do đó gây ra khí phế thũng. Sự phá hủy mô phổi xảy ra do sự tương tác lẫn nhau giữa các quá trình chết theo chương trình của tế bào phế nang, stress oxy hóa và mất cân bằng protease/antiprotease [55]. Khái niệm này được hỗ trợ bởi các quan sát cho thấy rằng việc điều trị bằng chất chống oxy hóa đã ngăn ngừa cả apoptosis và khí thũng gây ra do sự điều chỉnh giảm của VEGF [56] và các phát hiện rằng bệnh khí phế thũng do khói thuốc lá thực nghiệm đòi hỏi phải có mặt cathepsin-S (một protease nội bào), vì chuột bị loại bỏ cathepsin-S được bảo vệ khỏi bệnh và tình trạng tế bào phế nang chết theo chương trình [57]. Theo chiều ngược lại, tổn thương phế nang lại làm kích thích và khuếch đại tình trạng viêm để tạo thành một vòng xoắn bệnh lý. Hơn nữa, nhiễm vi-rút có thể tham gia phối hợp với khói thuốc lá làm tăng quá trình phá hủy nhu mô phổi, tăng chết theo chương trình tế bào phế nang để hình thành khí phế thũng [58].
Trong một tổng quan năm 2009 [34], William MacNee và cs cho rằng có nhiều bằng chứng về các đặc điểm chung giữa khí thũng phổi và lão hóa phổi nên có thể cho rằng cả hai tình trạng này đều có chung các cơ chế cơ bản, bao gồm stress oxy hóa, viêm và apoptosis [59,60]. Stress oxy hóa cũng làm rút ngắn telomere (là đoạn nucleotide ở đầu các nhiễm sắc thể có chức năng duy trì tính ổn định của nhiễm sắc thể trong quá trình phân bào. Telomere ngắn lại sau mỗi lần phân chia, mất dần khả năng kiểm soát nhân lên và đột biến của DNA) giống như trong quá trình lão hóa [61]. Tốc độ rút ngắn telomera tương ứng với số lượng hút thuốc lá đã hút [62]. Do đó, việc đưa COPD vào danh sách một trong những bệnh liên quan đến lão hóa “sớm” là điều rất hấp dẫn [34].
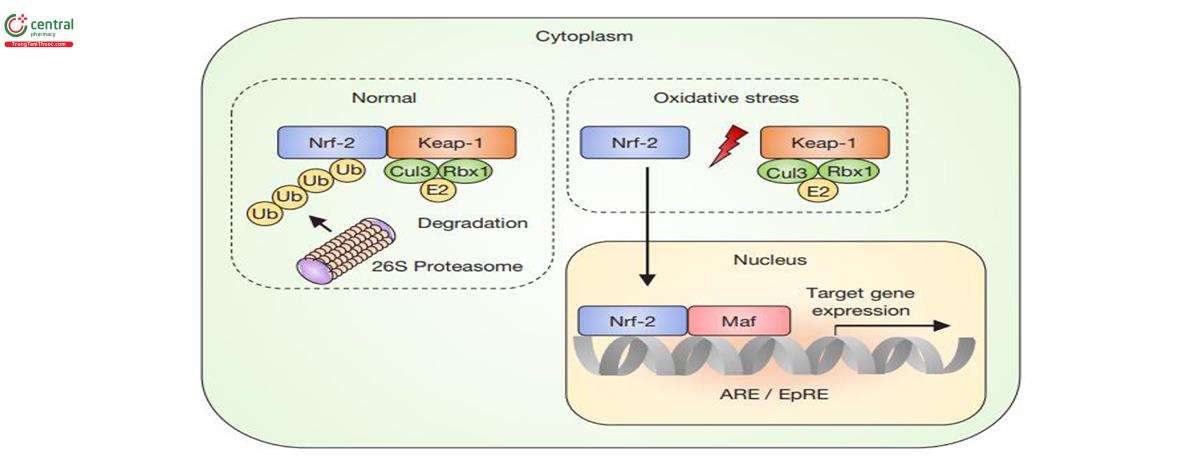
Yếu tố phiên mã Nrf2 (Nuclear factor E2-related factor 2) tạo điều kiện cho sự trình diện gen chống oxy hóa [63]. Trong điều kiện không stress, Nrf2 được giữ lại trong tế bào chất bằng cách liên kết trực tiếp với Kelch-like ECH-associated protein 1 (Keap1) (hình 4). Khi có các kích thích từ các chất có gốc oxy phản ứng (ROS) liên kết giữ Nrf-2 yếu đi, cho phép Nrf-2 di chuyển tới nhiễm sắc thể để thực hiện quá trình phiên mã, sinh tổng hợp protein chống viêm. Có hơn 500 gen được điều hòa bởi Nrf2, bao gồm các gen chống oxy hóa, chống viêm, chuyển hóa xenobiotic, chết theo chương trình và chết tự thực tế bào (autophagy, tự phá hủy tế bào có lập trình) [64,65]. Thuốc lá cũng đã được xác định là nguyên nhân của hiện tượng chết tế bào tự thực, một quá trình khác với chế tế bào theo chương trình, tạo ra nhiều yếu tố viêm góp phần duy trì tình trạng viêm đường hô hấp dai dẳng. Tuy nhiên, tự thực tế bào cũng là hiện tượng xảy ra tự nhiên trong quá trình lão hóa [66].
Bạch cầu đa nhân trung tính dường như đóng một vai trò quan trọng trong sự hình thành và tiến triển của bệnh COPD ở người hút thuốc. Tăng bạch cầu đa nhân trung tính biểu mô đã được ghi nhận ở những người hút thuốc với FEV1 giảm quá mức theo thời gian [67,68]. Đặc biệt, nghiên cứu dài hạn của Stanescu và cs [67] đã xác định xem liệu quá trình viêm đường hô hấp có khác nhau hay không ở những người hút thuốc dễ mắc bệnh COPD so với những người hút thuốc “không bị COPD” bằng phân tích đờm và cho thấy tỷ lệ bạch cầu đa nhân trung tính trong đờm cao hơn ở những người hút thuốc mắc COPD so với những người hút thuốc không có triệu chứng và có tương quan với mức giảm FEV1 hàng năm. Ngoài ra, ở những người hút thuốc mắc bệnh COPD, bạch cầu đa nhân trung tính trong đờm có sự gia tăng biểu hiện của phân tử dính CD11b/CD18, phối tử ICAM-1 (phân tử dính giữa các tế bào loại 1) và biểu hiện này có liên quan đến mức độ tắc nghẽn đường thở [67]. Một số nghiên cứu cũng đã xác nhận sự gia tăng bạch cầu đa nhân trung tính ở đường thở ở bệnh nhân COPD so với những người hút thuốc khỏe mạnh [69-71].
Sự phân ly giữa ưu thế của bạch cầu trung tính trong lòng đường thở (được đánh giá bằng dịch rửa phế quản phế nang hoặc đờm) [67-71] so với không tăng các tế bào này trong lớp dưới biểu mô từ bệcnh phẩm sinh thiết phế quản đã được quan sát thấy ở bệnh nhân COPD nhẹ tới trung bình. Có khả năng bạch cầu đa nhân trung tính tích tụ vào trong lòng đường thở bằng cách huy động từ hệ tuần hoàn. Sự điều hòa của E-selectin trên các mạch máu ở lớp dưới niêm mạc và tăng biểu hiện ICAM-1 biểu mô trên các tế bào đáy ở bệnh nhân COPD, kết hợp với hoạt tính hóa ứng động của IL-8, gợi ý một cơ chế thu hút các tế bào này từ tuần hoàn và tạo điều kiện cho chúng di chuyển vào lòng đường thở [73]. Số lượng bạch cầu đa nhân trung tính tăng lên trong biểu mô phế quản của cả đường hô hấp trung tâm và đường hô hấp ngoại biên trên bệnh nhân COPD [74,75]. Sự mất cân bằng giữa cytokine tiền viêm và kháng viêm có thể hỗ trợ cho quá trình di chuyển này. Nồng độ IL-10 ức chế giảm trái ngược với tăng nồng độ IL-8 và TNF-α trong COPD [65,76].
Tình trạng viêm phế quản phổi trong COPD khác với tình trạng viêm trong hen, ngay cả khi một số bệnh nhân mắc COPD có biểu hiện tăng bạch cầu ái toan ở đường thở ở một mức độ nào đó [70,71,77-80]. Vẫn còn tranh luận rằng liệu tăng bạch cầu ái toan trong COPD có liên quan đến các đặc điểm của hen hay không. Chanez và cs [77] đã báo cáo rằng những bệnh nhân COPD đáp ứng với điều trị bằng corticosteroid có số lượng bạch cầu ái toan trong đường thở nhiều hơn đáng kể và tăng độ dầy cấu trúc lưới ở màng đáy.
Thành đường dẫn khí bên dưới bề mặt biểu mô niêm mạc trên bệnh nhân COPD nhẹ tới trung bình biểu hiện tình trạng viêm tế bào đơn nhân với sự gia tăng đại thực bào, tế bào lympho T [81-84]. Đặc biệt, sự thay đổi trong sự cân bằng của tỷ lệ tế bào CD4+/CD8+, nghiêng về tế bào CD8+, đã được thấy trong một số nghiên cứu. Hình ảnh thâm nhiễm tế bào CD8+ hiện diện ở một số khoang của đường hô hấp trung tâm [74,83,85], đường hô hấp ngoại vi [75,81] và nhu mô phổi [86]. Khác với tình trạng viêm với sự có mặt của bạch cầu đa nhân trung tính ở bề mặt niêm mạc đường hô hấp, quá trình viêm tế bào CD8+ dường như phân bố rộng rãi trong phổi. Vai trò của các tế bào này vẫn chưa rõ ràng, nhưng mối tương quan giữa số lượng tế bào CD8+ xâm nhập vào đường dẫn khí lớn, nhỏ và nhu mô phổi với mức độ tắc nghẽn luồng khí cho thấy chúng có thể liên quan đến sự tiến triển của bệnh. Ngoài chức năng được biết đến là tế bào gây độc tế bào, tế bào CD8+ còn có khả năng gây tổn thương phổi khi xuất hiện quá mức để đáp ứng với tình trạng nhiễm trùng đường hô hấp [87]. Có bằng chứng cho thấy các tế bào lympho CD8+ biểu hiện các chức năng khác nhau tùy thuộc vào các dạng phản ứng cytokine khác nhau [88] và chúng có thể tạo ra các cytokine liên quan đến phản ứng viêm type 1 hoặc viêm type 2 [89,90]. Do đó, tế bào CD8+ có thể có vai trò trong việc điều chỉnh phản ứng viêm đối với khói thuốc lá thông qua việc giải phóng các sản phẩm của chúng [91]. Một số lymphokine hóa ứng động đối với tế bào này có thể liên quan đến cơ chế bệnh sinh của COPD bằng cách thu hút bạch cầu đa nhân trung tính [92,93]. IL-13 được quan sát thấy trên mô hình gây bệnh khí phế thũng [94] và tạo dịch nhầy thực nghiệm trên động vật [95]. Dữ liệu trên người cũng được ghi nhận về vai trò của IL-13 ở những người hút thuốc dễ bị tăng tiết chất nhầy. Mapp và cs [96] cho thấy có biểu hiện tăng phản ứng miễn dịch IL-13 trên các tế bào ở thành phế quản của những người hút thuốc bị viêm phế quản mạn tính so với những người hút thuốc không có triệu chứng.
Trên cơ sở một nghiên cứu chứng minh khả năng kiểm soát di truyền đối với tỷ lệ giữa tế bào CD4+ và CD8+, với một tỷ lệ nhỏ (5%) dân số có tỷ lệ tế bào CD4+/CD8+ <1 [97], O'Shaughnessy [98] gợi ý rằng những người có sự gia tăng được xác định về mặt di truyền CD8+ có thể dễ bị tăng thêm tế bào CD8+ khi tiếp xúc với khói thuốc lá. Giả thuyết này có thể giải thích lý do tại sao có sự khác biệt về tế bào CD8+ dưới biểu mô, được phát hiện bởi Lams và cs [85], lại rõ ràng hơn khi so sánh giữa những người hút thuốc bị COPD và những người hút thuốc không có triệu chứng so với giữa những người hút thuốc và những người không hút thuốc. Các dữ liệu này cùng đồng thời cho thấy hai loại tế bào có liên quan nhất quán đến tắc nghẽn luồng khí mạn tính trong COPD, đó là bạch cầu đa nhân trung tính và tế bào CD8+. Bạch cầu trung tính được phân bố ở bề mặt niêm mạc của đường dẫn khí lớn, tức là biểu mô và lòng đường thở, trong khi tế bào CD8+ phân bố rộng rãi hơn dọc theo vùng dưới biểu mô của đường dẫn khí lớn và nhỏ và trong nhu mô phổi, bao gồm cả thành phế nang và động mạch. Kiểu phân bố tế bào viêm này có liên quan đến COPD ở mức độ nhẹ hoặc trung bình. Ngược lại, số lượng bạch cầu trung tính tăng lên ở lớp dưới niêm mạc đã được báo cáo trong đợt cấp của bệnh viêm phế quản mạn tính [78,79] và ở bệnh nhân COPD ổn định với tình trạng tắc nghẽ luồng khí nặng [99]. Khi mức độ tắc nghẽ luồng khí tăng lên, số lượng bạch cầu trung tính ở dưới biểu mô cũng tăng lên, củng cố giả thuyết rằng các tế bào này đóng vai trò trong sự tiến triển của bệnh.
Nhìn nhận về hiểu biết cơ chế sinh bệnh trong COPD Toshinori Yoshida và cs (năm 2007) [100] cho rằng sau nhiều thập kỷ nghiên cứu tập trung vào giả thuyết viêm/protease-antiprotease (hình 5), lĩnh vực COPD đã trải qua sự mở rộng đáng kể các mô hình để giải thích sinh lý học của căn bệnh xuất phát từ các nghiên cứu thực nghiệm cũng như trên con người được thiết kế tốt, kết hợp các kỹ thuật mới, đặc biệt là các quá trình tế bào và phân tử có liên quan tới tín hiệu tế bào và số phận tế bào được phát hiện trong những năm gần đây. Vì không có giả thuyết đơn lẻ nào có thể giải quyết thỏa đáng sự phức tạp của COPD nên các mô hình mới sẽ liên tục viết lại cơ chế bệnh sinh của bệnh. Trong một tổng quan gần đây (năm 2018), Agnieszka Strzelak và cs [101] nhận định rằng sự mất cân bằng giữa chất oxy hóa và chất chống oxy hóa do tiếp xúc với khói thuốc lá dẫn đến stress oxy hóa, tăng viêm niêm mạc và tăng biểu hiện của các cytokine gây viêm (như interleukin (IL)-8, IL-6 và yếu tố hoại tử u α (TNF-α). Tác động trực tiếp của khói thuốc lá lên tế bào biểu mô dẫn đến tăng tính thấm, sản xuất quá mức chất nhầy, suy giảm Độ thanh thải của lông chuyển, tăng giải phóng các cytokine và chemokine tiền viêm, tăng cường huy động các đại thực bào và bạch cầu trung tính và làm rối loạn cân bằng tế bào lympho đối với Th2.
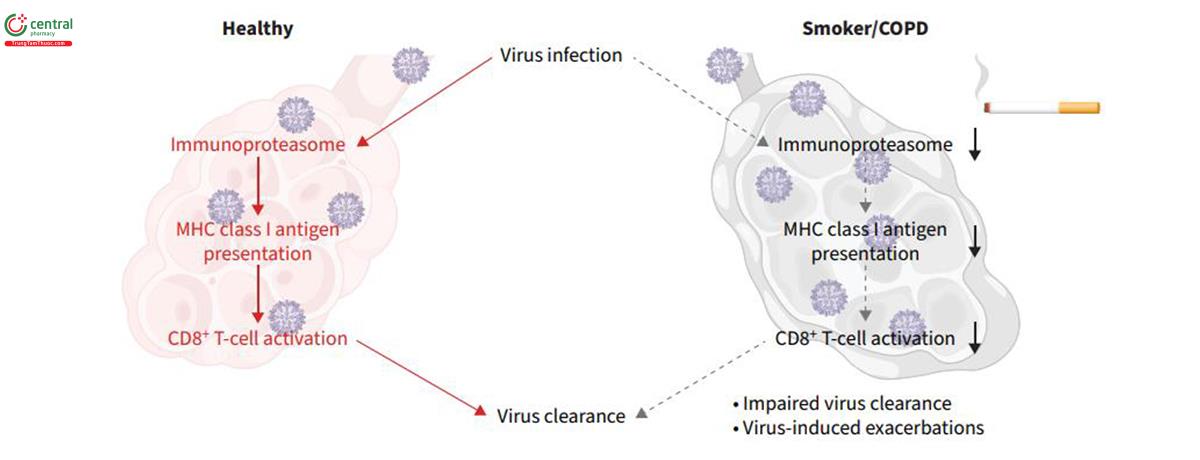
Khói thuốc lá làm giảm chức năng kháng vi-rút. Nhiễm vi-rút làm trầm trọng thêm tiến triển bệnh COPD. Khả năng đáp ứng miễn dịch kháng vi-rút tập trung vào việc kích hoạt các tế bào T CD8+ đặc hiệu với vi-rút bởi các epitope của vi-rút (một epitope cũng được gọi là vùng quyết định kháng nguyên. Đó là một vị trí nhỏ trên kháng nguyên nơi mà kháng thể kết hợp đặc hiệu) được trình bày trên các phân tử phức hợp tương hợp mô chính (major histocompatibility complex, MHC) loại I của các tế bào bị nhiễm bệnh. Một nghiên cứu gần đây [102] đã phân tích tác động của khói thuốc lá đối với việc cảm ứng immunoproteasome (một cấu trúc tế bào có chức năng phân hủy protein chọn lọc. Immunoproteasome tiêu hủy các protein bị tổn thương, bị nhiễm trùng hoặc không còn cần thiết cho tế bào). Các tác giả của nghiên cứu nhận thấy khói thuốc lá làm suy giảm khả năng cảm ứng immunoproteasome bằng tín hiệu cytokine và gây nhiễm vi-rút vào tế bào phổi in vitro, ex vivo và in vivo. Ngoài ra, khói thuốc lá đã làm thay đổi định danh peptide của các kháng nguyên được trình bày trên các phân tử MHC lớp I trong điều kiện viêm. Điều này cho thấy khói thuốc lá cản trở quá trình tạo ra và trình bày kháng nguyên MHC lớp I và do đó góp phần làm suy giảm khả năng kích hoạt tế bào T CD8+ khi bị nhiễm vi-rút. Điều này bổ sung thêm hiểu biết cơ chế quan trọng về cách khói thuốc lá làm trung gian cho việc tăng tính nhạy cảm của người hút thuốc và bệnh nhân COPD đối với nhiễm vi-rút (hình 6).
1.2 Khói sinh khối (biomass)
Sinh khối được định nghĩa là chất hữu cơ có thể được sử dụng làm nhiên liệu như cây lá, gỗ, chất thải động vật (phân), phụ phẩm nông nghiệp, lâm nghiệp. Biomass có chứa nhiều chất có đặc tính hóa học giống với thuốc lá [103]. Tác động của khói sinh khối đối với sức khỏe con người nói chung và COPD nói riêng còn chưa được chú ý và nghiên cứu nhiều [104,105]. Vì phụ nữ có xu hướng ở trong nhà hầu hết thời gian nên việc tiếp xúc với các chất gây ô nhiễm trong nhà chiếm ưu thế trong danh sách tiếp xúc với các chất gây ô nhiễm khác nhau trên toàn thế giới, đặc biệt là ở các nước đang phát triển. Ở hầu hết các nền văn hóa, phụ nữ đóng vai trò chủ đạo trong việc nấu nướng gia đình. Trên toàn cầu, gần 50% số ca tử vong do COPD ở các nước đang phát triển có thể là do phơi nhiễm sinh khối và khoảng 75% trong số này là ở phụ nữ [106]. Báo cáo từ các nghiên cứu khác nhau cho thấy phụ nữ tiếp xúc với khói trong nhà có nguy cơ mắc COPD dưới dạng viêm phế quản mạn tính cao gấp 3 lần so với những phụ nữ nấu ăn bằng điện, gas hoặc nhiên liệu sạch hơn [107]. Ví dụ, ở Colombia, việc sử dụng bếp sinh khối trong 10 năm trở lên có nguy cơ mắc bệnh COPD cao hơn [108]. Hiện nay, COPD liên quan đến ô nhiễm không khí trong nhà do sử dụng nhiên liệu sinh học được coi là một vấn đề sức khỏe cộng đồng với ý nghĩa kép: đây là một bệnh đặc trưng về giới, hầu như chỉ xảy ra ở phụ nữ và mặt khác, ngày càng có nhiều phụ nữ mắc bệnh này trên phạm vi toàn thế giới [107]. Phơi nhiễm với khói sinh khối còn có ý nghĩa khi đó thông thường là phơi nhiễm sớm, liên tục, thậm trí ngay cả khi còn là bào thai. Trẻ tiếp xúc với khói sinh khối có nguy cơ mắc các bệnh nhiễm trùng đường hô hấp cấp tính cao hơn, bao gồm viêm phổi và hen so với trẻ không tiếp xúc [109,110]. Hậu quả là chức năng phổi tăng trưởng chậm hơn, dẫn đến COPD [111].
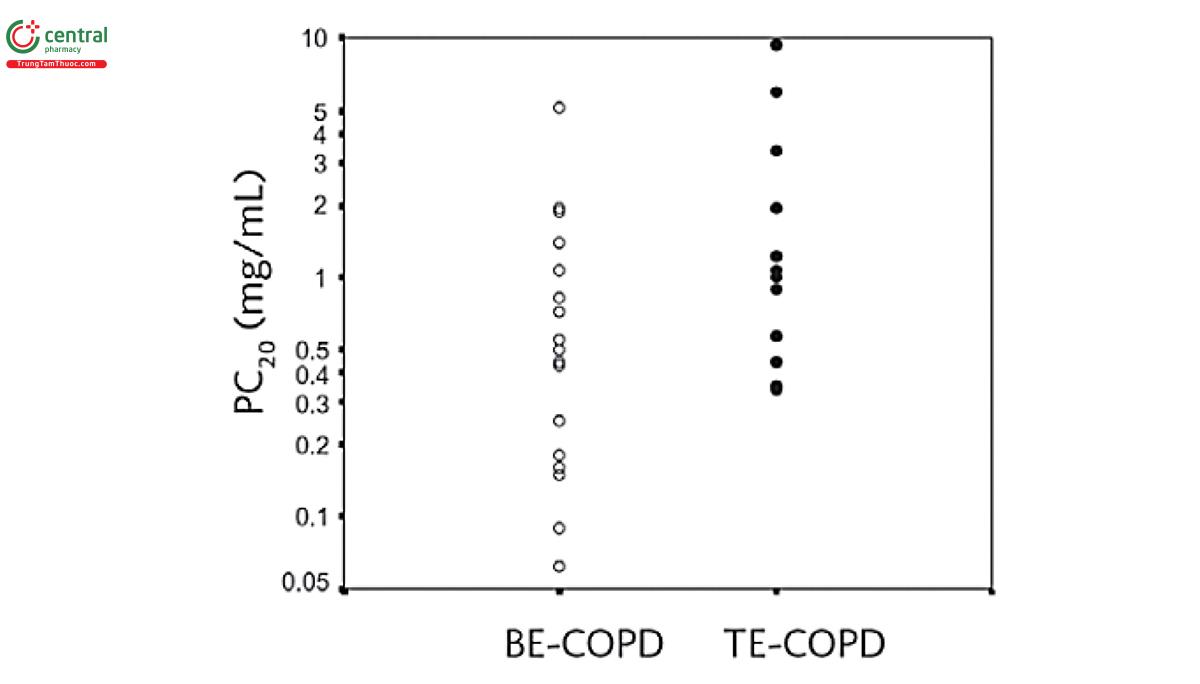
Sự khác biệt giữa kiểu hình COPD do phơi nhiễm khói sinh khối (BE-COPD) và kiểu hình COPD do phơi nhiễm khói thuốc là (TE-COPD) bắt nguồn từ nguồn sinh khói và cách hít khói. Mặc dù khói sinh khối có nhiều thành phần giống như khói thuốc lá nhưng thành phần chính xác lại khác nhau tùy thuộc vào nguồn nhiên liệu, hiệu suất đốt và độ ẩm tương đối. Mặc dù kích thước hạt có thể giống nhau ở cả khói thuốc lá và khói sinh khối [112,113] nhưng sự khác biệt về thành phần hóa học có thể dẫn đến sự khác biệt trong quá trình sinh bệnh (hình 7). Một điểm khác biệt giữa BE-COPD và TE-COPD là kiểu hít khói. Những người hít khói sinh khối sử dụng kiểu thở thông thường và thậm trí thở nông hơn theo phản xạ để giảm hít khói. Kiểu hít này có thể giảm thiểu khói xâm nhập tới đường thở nhỏ và khu vực trao đổi khí, dẫn đến kiểu hình COPD chiếm ưu thế ở đường thở. Ngược lại, người hút thuốc lá thường hút thuốc theo mô hình hai giai đoạn: đầu tiên, khói được hít vào miệng mà không hít trực tiếp vào phổi, sau đó tạm dừng và tiếp theo, khói được hít vào phổi với một lượng không khí bổ sung [113]. Thể tích hít vào trung bình đo được ở mức gần 25% dung tích sống, con số này tương ứng với gần gấp đôi thể tích lưu thông trung bình [113]. Do vậy, thể tích hít vào lớn hơn ở người hút thuốc lá so với những người tiếp xúc với khói sinh khối, có thể cho phép khói đi sâu hơn vào phổi và có thể làm tăng sự lắng đọng khói thuốc lá trong nhu mô phổi, dẫn đến kiểu hình COPD chủ yếu là khu vực đường thở nhỏ và vùng trao đổi khí. Tình huống dịch tễ học tương phản này giữa phụ nữ sử dụng nhiên liệu sinh khối và người hút thuốc lá cho phép hiểu rõ hơn về sự khác biệt trong hình ảnh lâm sàng cũng như các phát hiện về chức năng, mô học và chụp cắt lớp vi tính (CT) của COPD giữa hai kiểu hình này [114]. Sử dụng CT định lượng có độ phân giải cao cho thấy trong BE-COPD, mức tổn thương dạng khí phế thũng không quan trọng bằng trong TE-COPD [115]. Bản thân những người hút thuốc lá, kiểu hít khói tạo nên thể tích khói xâm nhập vào phổi cao hơn có liên quan tới phản ứng pha cấp mạnh hơn trong khi số lượng điếu thuốc và thành phần khói thuốc lại không thấy có liên quan [116].
2 PHẢN ỨNG SINH LÝ CỦA ĐƯỜNG THỞ VỚI BỤI - KHÓI, KHÍ ĐỘC HẠI
Như đã trình bày ở phần trên, chất lỏng lót bề mặt đường thở bao gồm lớp chất lỏng ở tầng thấp (pha sol) bao quanh các lông chuyển có hàm lượng nước cao. Bên trên lớp dịch này có một lớp dịch nhầy (pha gel) liên tục lưu chuyển theo hướng từ trong ra ngoài dưới tác động của hoạt động lông chuyển. Ngoài chức năng bắt dính và quét cuốn mang tính cơ học như trên, dịch nhầy còn có các chức năng sinh học. Thành phần của dịch nhầy có chứa peptide kháng khuẩn LL-37, các Lysozyme, β-defensins, lipocalins và chất ức chế protease bạch cầu. Chất nhầy niêm mạc phế quản (mucin, MUC) tăng tiết khi tiếp xúc với kích thích từ bụi-khói, khí. Thí dụ khi phơi nhiễm 2 giờ với khí thải từ động cơ diezel, MUC5B và MUC6 sẽ tăng nồng độ trong dịch tiết phế quản [117].
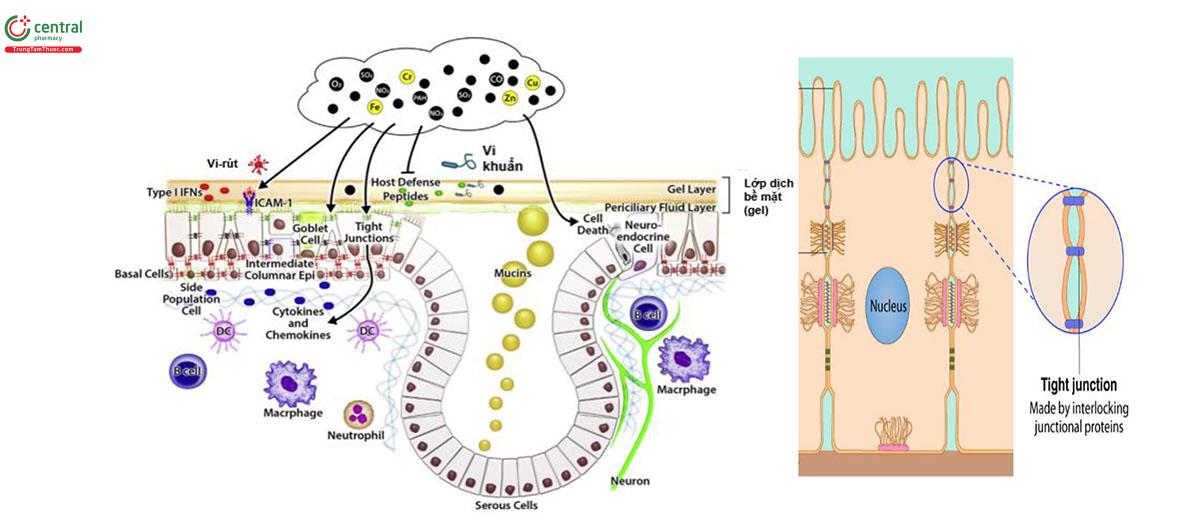
Bên dưới lớp dịch nhầy là hàng rào tế bào biểu mô với đa dạng loại, gắn kết với nhau bằng những kết nối tế bào - tế bào (hình 8). Tế bào biểu mô và cấu trúc kết nối có thể bị phá hủy bởi ô nhiễm khí thở nhưng phải ở tình trạng phơi nhiễm với nồng độ cao [118]. Ô nhiễm khí thở luôn kết hợp với tình trạng tăng dịch tễ nhiễm trùng ở tất cả các lứa tuổi [119-121]. Cơ chế chịu trách nhiệm cho hiện tượng giảm khả năng miễn dịch niêm mạc đường hô hấp và bị tổn hại có thể có nhiều và có thể bao gồm cả những thay đổi trình diện thụ thể với tác nhân gây bệnh, cơ chế kháng vi-rút hoặc thay đổi hoạt tính sinh học các peptide bảo vệ.
Các phân tử dính liên tế bào 1 (Intercellular adhesion molecule-1, ICAM-1) là thụ thể của Human rhinovirus B, và thông qua sự kết dính này gây nhiễm cho các tế bào biểu mô niêm mạc hô hấp và tạo ra bệnh cảnh viêm phế quản cấp [122]. Ô nhiễm không khí làm tăng ICAM-1 [123]. Khi đồng thời phơi nhiễm với vi-rút và nitơ dioxide (NO2) sẽ làm tăng có tính hiệp đồng hoạt động của ICAM-1 [124]. PAHs (Hydrocacbon thơm đa vòng) là sản phẩm phụ sinh ra trong quá trình đốt cháy hóa thạch hoặc sinh khối. PAHs là phối tử (ligand) và đối với thụ thể AhR (The aryl hydrocarbon receptor, một yếu tố phiên mã điều hòa trình diện gen) và gây ra stress oxy hóa, ảnh hưởng tới lộ trình giải độc và sống của tế bào. Các tác động của phơi nhiễm thực nghiệm cho thấy các tế bào biểu mô đường thở của con người suy giảm hoạt động phiên mã sinh tổng hợp interferon và giảm sản xuất các chất trung gian tham gia phản ứng chống vi-rút. Ô nhiễm khí thở cũng làm sản xuất và hoạt tính các peptide bảo vệ có trong dịch nhầy phế quản [125]. Với cơ chế này, khói tro đốt từ than làm giảm khả năng tiêu diệt S. aureus của dịch nhầy phế quản do làm giảm LL-37, nồng độ β-defensin-3 ở người và lysozyme. Tương tự ô nhiễm bụi đô thị làm giảm sinh tổng hợp các protein kháng P. aeruginosa [126].
Tóm lại, mỗi ngày, chúng ta hít thở hơn 10.000L không khí chứa nhiều loại chất gây ô nhiễm có thể gây ra hậu quả tiêu cực cho sức khỏe phổi. Niêm mạc đường hô hấp, là điểm tiếp xúc đầu tiên với tình trạng này, có chức năng như một hàng rào cơ học và miễn dịch. Trong điều kiện bình thường, các tế bào biểu mô đường thở được kết nối bằng các mối nối chặt chẽ, sẽ tiết ra chất nhầy, chất lỏng lót bề mặt đường thở, peptide bảo vệ và chất chống oxy hóa, đồng thời trình diện các thụ thể nhận biết mẫu kháng nguyên để cơ chế miễn dịch bẩm sinh phản ứng với các chất lạ và mầm bệnh bị hít vào. Khi tiếp xúc với ô nhiễm không khí, khả năng phòng vệ của biểu mô đường thở bị tổn hại do giảm chức năng rào cản, suy giảm khả năng phòng vệ của cơ thể đối với mầm bệnh và tạo ra phản ứng viêm quá mức. Trọng tâm của những thay đổi cơ học và miễn dịch do ô nhiễm không khí gây ra là sự kích hoạt các con đường nhạy cảm với oxy hóa khử và vai trò của chất chống oxy hóa trong việc hóa giải những tác động tiêu cực này. Có vai trò của các biến thể di truyền trong các gen quan trọng trong chức năng và kiểu hình của tế bào biểu mô góp phần tạo ra sự đa dạng trong các phản ứng đối với ô nhiễm không khí trong quần thể dân cư ở cấp độ cá nhân và nhóm, đồng thời cho thấy cần có các phương pháp tiếp cận cá thể hóa để làm giảm phản ứng miễn dịch niêm mạc đường hô hấp đối với ô nhiễm không khí.
Giả thuyết y học Dutch được Dick Orie và cs đề xuất năm 1961 từ đại học Groningen (Hà Lan) nên còn gọi là giả thuyết Hà Lan. Giả thuyết cho rằng các dạng bệnh lý tắc nghẽn khác nhau như hen, COPD, viêm phế quản mạn tính hay khí phế thũng không phải là các thực thể bệnh riêng biệt mà nên xem là các biểu hiện khác nhau của một bệnh lý chung gọi là viêm phế quản (Bronchitis) [127,128]. Giả thuyết này nhấn mạnh vai trò của yếu tố cơ địa (nội sinh) bên cạnh yếu tố phơi nhiễm (ngoại sinh) trong sinh bệnh học các bệnh phổi tắc nghẽn. Bên cạnh bụi - khói, khí độc hại, nhiễm trùng thì yếu tố cơ địa cũng là một thành tố quan trọng quyết định tính tăng phản ứng phế quản (Bronchial hyperresponsiveness, BHR) với các kích thích từ bên ngoài để từ đó hình thành nên bệnh hay không [129].
Mặc dù Hen và COPD đều đặc trưng bởi tắc nghẽn luồng khí, thường thay đổi và có thể hồi phục trong bệnh hen nhưng cố định ở COPD nhưng cũng có sự chồng chéo biểu hiện tắc nghẽn thông thường này. Trong cơn hen nặng, có thể có tắc nghẽn luồng khí dai dẳng và tắc nghẽn luồng khí có thể hồi phục một phần trong COPD. Tương tự như vậy, mặc dù một số báo cáo cho rằng có sự khác biệt rõ rệt về mô hình viêm cơ bản, cơ chế tế bào, chất trung gian gây viêm và đáp ứng với điều trị [130] giữa hen và COPD, nhưng cũng có những báo cáo khác cho thấy giữa hen nặng [131-133] và COPD [134-137] có sự không đồng nhất, chồng chéo giữa hai tình trạng bệnh lý này [138-140]. Đã có một cuộc tranh luận đang diễn ra giữa ''giả thuyết Hà Lan'' cho rằng bệnh hen và COPD là những biểu hiện khác nhau của cùng một quá trình bệnh cơ bản, và ''giả thuyết Anh'' (British hypothesis) cho rằng hen và COPD là những thực thể bệnh lý riêng biệt được tạo ra bởi các cơ chế khác nhau [141]. Cần phải tập trung lại nỗ lực để xác định những điểm tương đồng và khác biệt trong bệnh hen, đặc biệt ở những người hen nặng và COPD xét về kiểu (profiles) cytokine [142]. Điều này được nhấn mạnh bởi sự xuất hiện của các chất kháng viêm có hiệu quả đối với các kiểu hình bệnh hơn là đối với tên bệnh [143]. Có lẽ minh họa rõ nhất về hiệu quả kháng viêm là các phương pháp kháng IL-5 đã chứng minh các đáp ứng lâm sàng liên quan đến tình trạng viêm tăng bạch cầu ái toan trong hen [144,145] và các chiến lược tương tự hiện đang được áp dụng thử nghiệm trên COPD. Như vậy, bản chất viêm thông qua kiểu cytokine là đặc điểm mấu chốt phân biệt bản chất viêm trong hen, COPD hay có sự chồng lấp giữa các tên gọi. Xác định kiểu cytokine trong đờm trên người hen và COPD là mục tiêu của một nghiên cứu được thực hiện năm 2014 [146]. Các tác giả phân tích cụm (cluster analysis) trên 18 cytokine các tác giả kết luận việc phân tích cytokine trong đờm có thể xác định các nhóm bệnh nhân khác nhau và các nhóm bệnh nhân chồng chéo nhau giữa hen và COPD. Điều này ủng hộ cả “giả thuyết Hà Lan” và “giả thuyết Anh”.
Những người hút thuốc mắc bệnh phổi tắc nghẽn mạn tính và những người hút thuốc không có triệu chứng có chức năng phổi bình thường có phản ứng khác nhau với việc ngưng hút thuốc lá trong một năm, thể hiện trên tình trạng viêm đường hô hấp do khói thuốc gây ra là kết luận của một nghiên cứu mà B.W.M. Willemse và cs thực hiện năm 2025 [147]. Một số khía cạnh của tình trạng viêm đường thở giảm ở những người hút thuốc không có triệu chứng với chức năng phổi bình thường trong khi nó vẫn tồn tại hoặc thậm chí tăng lên ở một số khía cạnh ở những người hút thuốc mắc bệnh phổi tắc nghẽn mạn tính. Hầu hết những thay đổi được thấy ở đờm, đại thực bào, bạch cầu ái toan và nồng độ interleukin-8 giảm đáng kể ở những người hút thuốc không có triệu chứng trong khi bạch cầu đa nhân trung tính, tế bào lympho, interleukin-8 và nồng độ protein cation bạch cầu ái toan (ECP) ở bệnh nhân mắc bệnh phổi tắc nghẽn mạn tính vẫn còn tăng đáng kể. Các tác giả cho rằng tình trạng viêm dai dẳng quan sát được trong bệnh phổi tắc nghẽn mạn tính có thể ít nhất có một phần liên quan đến việc sửa chữa tổn thương mô do khói thuốc gây ra ở đường thở. Hiện tượng này có tác động của yếu tố cơ địa hay không vẫn còn phải có các nghiên cứu làm sáng tỏ. Cũng đã có nghiên cứu xác nhận hút thuốc là và tăng phản ứng phế quản làm tăng triệu chứng hô hấp [148]
3 VIÊM VÀ ĐÁP ỨNG MIỄN DỊCH ĐƯỜNG THỞ
3.1 Khái niệm cơ bản về viêm đường thở
Tạo ra phản ứng viêm là một cơ chế quan trọng mà qua đó các loài động vật có vú phản ứng lại và tự bảo vệ mình đối với các tác động bất lợi nhiễm trùng hay không. Ở phổi, cả cơ chế không đặc hiệu và cơ chế đặc hiệu với kháng nguyên (miễn dịch) đều có thể dẫn đến phản ứng viêm. Mặc dù phản ứng này thường có tác dụng bảo vệ và có lợi nhưng tình trạng viêm cũng có khả năng làm tổn thương các mô, bao gồm cả đường thở.
Biểu hiện cơ bản trong viêm là tăng tưới máu tới vùng tổn thương. Hiện tượng này liên quan tới dãn mạch, tăng lưu lượng máu, tăng tính thấm thành mạch, tăng xâm nhập tế bào viêm, giải phóng các protein huyết tương và cytokines vào tổ chức tổn thương để tạo ra hình ảnh kinh điển của viêm là sưng, nóng, đỏ và đau. Nhìn chung, hiện tương này sẽ tự thoái lui để hồi phục cấu trúc mô tổn thương và trả lại chức năng bình thường của mô tổn thương.
Hình ảnh mô học quan sát được trong phản ứng viêm ở phổi có phần giống nhau và không phụ thuộc vào tổn thương ban đầu như thế nào. Trình tự tăng tính thấm, tích lũy bạch cầu hạt và sau đó là sự xâm nhập của thực bào đơn nhân xảy ra trước khi quá trình viêm kết thúc, đều được thể hiện đối với các kích thích khác nhau, bất kể đó là nhiễm trùng, miễn dịch, hay các thành phần có độ tinh khiết cao của tế bào [149]. Ngoài ra, diễn biến mô học đều có biểu hiện giống nhau ở đường thở lớn cũng như đường thở nhỏ [150]. Hiện tượng dãn mạch, tăng tính thấm xảy ra trong vài phút sau kích thích. Phản ứng tập trung và thoát quản bạch cầu xảy ra trong vòng vài giờ. Thành phần bạch cầu đơn nhân xuất hiện trong 1-2 ngày và tồn tại trong nhiều ngày kể cả khi đã ngưng kích thích [151]. Thoát quản protein huyết tương có thể mang theo các protein ức chế viêm vào khu vực viêm giúp giải quyết làm giảm quá trình viêm bằng các chất ức chế proteinase.
Hình ảnh mô học cụ thể của phản ứng viêm là kết quả hoạt động của một số chất trung gian. Các chất trung gian gây viêm có thể được định nghĩa rộng rãi là các chất truyền tín hiệu hóa học tác động lên mạch máu và/hoặc tế bào để tạo ra hoặc góp phần gây ra phản ứng viêm. Trong số những chất trung gian quan trọng nhất trong giai đoạn đầu của phản ứng viêm, histamine và các eicosanoid khác nhau (sản phẩm của quá trình chuyển hóa a-xít arachidonic) có liên quan đến việc điều hòa lưu lượng máu cục bộ và cũng có thể làm thay đổi tính thấm của mạch máu. Tuy nhiên, mặc dù prostaglandin (cũng là sản phẩm chuyển hóa từ a-xít arachidonic) phát huy một phần tác dụng của chúng bằng cách điều chỉnh lưu lượng máu tới vùng tổn thương, nhưng prostaglandin gây dãn mạch (PGE2 và PGI2) cũng có thêm tác dụng gây viêm trong một số mô hình và có thể làm tăng tác dụng của các chất trung gian khác bao gồm các yếu tố hóa ứng động [152]. Ngoài ra, các prostaglandin dãn mạch như PGE2 cũng có thể có tác dụng chống viêm bằng cách ức chế sự bài tiết từ bạch cầu và dưỡng bào, do đó hạn chế phản ứng viêm. Thật vậy, trong một nghiên cứu gây kích thích thực nghiệm đường thở bằng chất gây dị ứng ở bệnh nhân mắc bệnh hen dị ứng, việc hít PGE2 ngay trước khi tiếp xúc với chất gây dị ứng đã làm giảm sự tích tụ bạch cầu ái toan trong đờm và làm giảm những thay đổi do chất gây dị ứng gây ra trong chức năng đường hô hấp [153]. Do đó, một chất trung gian viêm cũng có thể vừa có cả tác dụng gây viêm và vừa có tác dụng chống viêm, điều này phụ thuộc vào bản chất của kích thích và thời gian được giải phóng của chất trung gian này [151].
Một trong những đặc điểm mô học đáng chú ý nhất của phản ứng viêm là sự tích tụ của các tế bào trong mô phổi. Bạch cầu đa nhân (bạch cầu trung tính, bạch cầu ái toan) thường hiện diện với số lượng nhỏ trong đường dẫn khí và khoang phế nang và chúng được cho là hầu như không có ở các khoảng kẽ của nhu mô phổi. Tuy nhiên, một số lượng lớn bạch cầu đa nhân trung tính nằm rải rác trên giường mạch máu phổi. Nhóm tế bào này, cũng như các tế bào lưu hành trong tuần hoàn, có thể di chuyển vào phổi. Số lượng chất trung gian/chemokine có thể thu hút các bạch cầu này vào phổi và nguồn tế bào có chứa các chất hóa ứng động để thu hút chúng là rất lớn.
Cơ chế phân tử để cho bạch cầu di chuyển ra ngoài mạch máu đã được biết rất chi tiết. Việc thu hút bạch cầu vào phản ứng viêm được thực hiện bởi sự tham gia của các phân tử liên kết tế bào nội mô mạch máu - bạch cầu (là các thụ thể gây dính nằm trong cấu trúc nền ngoại bào, integrins). Các cơ chế tham gia vào phản ứng viêm để bảo vệ phổi bằng cách tiết ra các chất diệt khuẩn và enzyme phá hủy cũng có thể, ngược lại, làm tổn hại cấu trúc phổi. Các tế bào viêm, trong đó có các bạch cầu hạt, tạo ra các men (enzyme) và các chất có gốc oxy tự do (như anion superoxide, hydro peroxide và gốc hydroxyl). Các chất này tạo ra stress oxy hóa và phá hủy cấu trúc, làm thay đổi chức năng của đường thở trong đó có cả khả năng đáp ứng với các kích thích thần kinh [154]. Quá trình hồi phục viêm và lý do dẫn tới viêm kéo dài còn được hiểu biết ít [151]. Đại thực bào có vai trò quan trọng trong quá trình làm sạch môi trường viêm bằng cách thực bào các mảnh vụn, giải phóng các chất kích thích tăng trưởng tế bào. Tuy nhiên, bất chấp các cơ chế sửa chữa, một quá trình phá hủy không hồi phục và tiến triển mạn tính vẫn có thể xảy ra. Có nhiều yếu tố tham gia vào việc quyết định phản ứng viêm sẽ tiến triển theo chiều hướng nào: bảo vệ sự toàn vẹn ban đầu của phổi hay dẫn tới một phản ứng viêm kéo dài, mạn tính với các biến đổi cấu trúc và chức năng. Yếu tố có vai trò quyết định là bản chất của căn nguyên gây tổn thương. Các đặc điểm của căn nguyên sẽ xác định đặc điểm phản ứng viêm sẽ đáp ứng như thế nào (cách thức tham gia của các tế bào viêm, có đáp ứng miễn dịch hay không…). Bên cạnh đó, các đặc điểm cường độ, thời gian, tần suất tác động cũng như thời điểm (tuổi) của cơ thể khi lần đầu tiếp xúc cũng tham gia quyết định chiều hướng diễn biến của phản ứng viêm. Cách phản ứng đã được lập trình về mặt di truyền của cơ thể chủ đối với các yếu tố kích thích từ môi trường cũng có vai trò rất quan trọng.
3.2 Đáp ứng và điều hòa đáp ứng miễn dịch đường thở
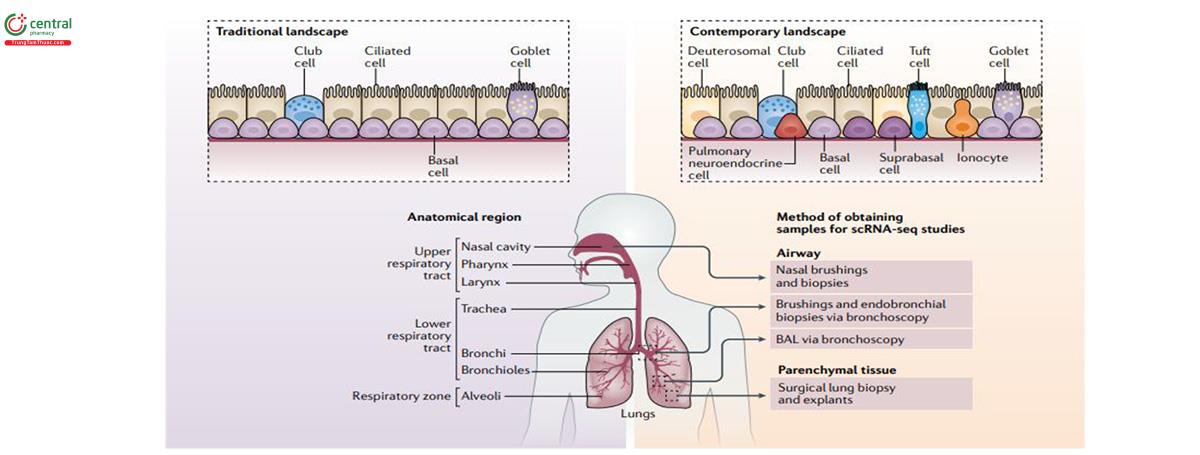
Hiện nay quan niệm đã được chấp nhận rộng rãi cho rằng lớp các tế bào biểu mô đường thở không chỉ tạo thành hàng rào ngăn cách giữa môi trường bên ngoài với lớp trung mô bên dưới. Tập hợp các tế bào trong lớp biểu mô chuyên biệt này phản ứng với các vi khuẩn và các kích thích độc hại vượt qua hàng rào nhầy – lông chuyển và là thành phần quan trọng trong cơ chế bảo vệ của cơ thể chủ, tương tác với các tế bào của hệ thống miễn dịch để duy trì cân bằng nội môi đồng thời tạo điều kiện cho các phản ứng miễn dịch khi cần thiết. Biểu mô hô hấp cũng phải quản lý các phản ứng với các chất độc khác nhau có trong môi trường hít vào và có bằng chứng mới cho thấy rối loạn chức năng biểu mô là nguyên nhân gây ra nhiều bệnh mạn tính ảnh hưởng đến phổi. Quan điểm truyền thống về lớp biểu mô bao gồm một lớp các tế bào đáy ở gần các tế bào tiết và có lông chuyển, tạo thành một đơn vị chặt chẽ duy trì hàng rào vật lý nhưng cũng phản ứng với môi trường hít vào thông qua các tế bào và phân tử từ hệ thống miễn dịch. Tuy nhiên, với sự ra đời của các kỹ thuật giải trình tự gen tiên tiến, quan điểm này đã thay đổi, cho rằng hàng rào biểu mô đường thở là cấu trúc tế bào động, bao gồm nhiều loại tế bào chuyên biệt cao có khả năng đáp ứng với sự thay đổi môi trường, tương tác với các cộng đồng vi sinh vật thường trú và hợp tác với nhiều tế bào chuyên biệt của các hệ thống khác như hệ thống miễn dịch và thần kinh (hình 9).
Đường hô hấp cần được xem là một hệ cơ quan phức tạp, được chia thành đường hô hấp trên, bao gồm: khoang mũi, hầu họng và thanh quản, và đường hô hấp dưới bao gồm: các đường dẫn khí (khí quản, phế quản và tiểu phế quản) và vùng hô hấp (các tiểu phế quản hô hấp và phế nang). Mỗi vùng có một chức năng cụ thể và sự khác biệt về thành phần tế bào theo vùng phản ánh điều này. Các quần thể tế bào biểu mô chuyên biệt trải dọc toàn bộ đường hô hấp từ khoang mũi đến phế nang. Các nghiên cứu bằng kính hiển vi điện tử đã sớm cung cấp cái nhìn sâu sắc về hình thái và cơ sở hạ tầng của các loại tế bào biểu mô chính có trên đường thở của con người. Ngoài việc sử dụng các đặc điểm hình thái được xác định bằng kính hiển vi điện tử cổ điển, nhuộm hóa mô miễn dịch tiêu chuẩn cho các dấu hiệu đặc hiệu của loại tế bào đã được sử dụng để mô tả và định lượng quần thể tế bào biểu mô trên toàn bộ đường hô hấp của con người, xác định ảnh hưởng của vị trí giải phẫu đến thành phần tế bào. Đường dẫn khí được lót bởi các tế bào có lông chuyển và chế tiết, chủ yếu thích nghi để tạo điều kiện thuận lợi cho việc loại bỏ bằng chất nhầy các chất dạng hạt và mầm bệnh trong không khí chúng ta hít thở. Giống như các bề mặt niêm mạc khác, biểu mô đường dẫn khí có bề mặt mở, tiếp xúc với môi trường và do đó có chức năng bảo vệ rất quan trọng. Mặc dù có cùng nguồn gốc phôi thai với niêm mạc ruột, hoạt động miễn dịch ở bề mặt niêm mạc đường thở nhất thiết phải chuyên biệt và được định hình bởi sự khác biệt về điều kiện môi trường (chênh lệch nhiệt độ, luồng khí lưu thông hai chiều), cộng đồng vi khuẩn thường trú và kháng nguyên có trong không khí [155].
Sự ra đời của các kỹ thuật giải trình tự gen hiện đại đã cho phép xác định không chỉ các loại tế bào mới mà còn tiết lộ các chức năng tiềm năng của các loại tế bào được đặt tên trước đó nhưng chưa được biết rõ, ví dụ như tế bào chùm/bàn chải (tuft/brush cell) (hình 9). Ngoài ra còn có dấu hiệu cho thấy tồn tại các loại và trạng thái tế bào không đồng nhất và những thay đổi tinh vi trong chúng dưới tác động của sự thay đổi của môi trường, ví dụ như do hút thuốc lá hoặc tiếp xúc với các chất gây dị ứng hoặc chất ô nhiễm. Thí dụ như trong một nghiên cứu (năm 2019) [156] Jennifer A. Aguiar và cs nhận thấy tác động của khói thuốc lá làm thay đổi trạng thái trình diện gen ABC (gen chịu trách nhiệm sinh tổng hợp protein vận chuyển). Hay trong một nghiên cứu khác, I.H. Heijink và cs (năm 2012) ghi nhận khói thuốc lá làm suy yếu khả năng kết nối giữa các tế bào biểu mô [157]. Các bằng chứng hiện tại cho thấy rõ ràng là thành đường thở đại diện cho một “cộng đồng” năng động các tế bào biểu mô tồn tại trong mối liên hệ chặt chẽ với các tế bào thần kinh và miễn dịch thường trú để tạo ra một đơn vị tích hợp, đóng vai trò quan trọng trong việc duy trì cân bằng nội môi miễn dịch ở niêm mạc cũng như tạo điều kiện cho vật chủ phòng vệ chống lại các tác nhân gây bệnh đường hô hấp [158].
3.3 Các đặc tính miễn dịch của tế bào đường thở
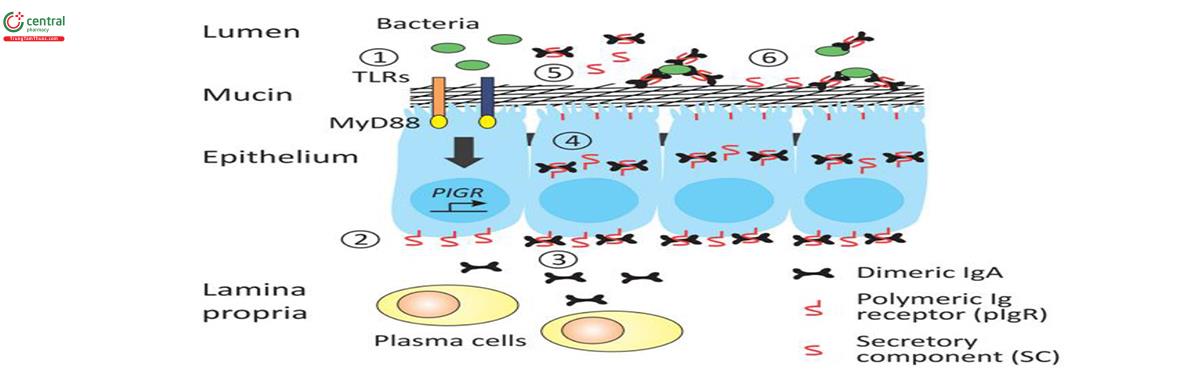
Các chất trung gian hóa học có tác dụng kháng khuẩn và chất nhầy đường thở, bao gồm MUC5AC và MUC5B, như đã nói ở trên, được sản xuất bởi các tế chế tiết hình đài và các tuyến dưới niêm mạc, góp phần tạo nên lớp bảo vệ đường thở đầu tiên ở bề mặt biểu mô. IgA chế tiết (secretory IgA, sIgA) - được sản xuất bởi tương bào nằm dưới lớp biểu mô và được vận chuyển đến bề mặt đỉnh của tế bào biểu mô đường thở thông qua thụ thể globulin miễn dịch đa cực (polymeric immunoglobulin receptor, pIgR) - ngăn chặn sự bám dính của các vi sinh vật có trong không khí lọt vào đường thở trong một quá trình gọi là “loại trừ miễn dịch” [159,160] (hình 10).Các tế bào biểu mô được trang bị các thụ thể nhận dạng mẫu (pattern recognition receptors), thí dụ Toll-like receptor, nhanh chóng nhận dạng và khởi đầu phản ứng miễn dịch trước các kích thích từ vi khuẩn. Các thụ thể cytokine, bao gồm TNFR1 (tumor necrosis factor receptor 1), cho phép chúng phản ứng khi có các tín hiệu do tế bào miễn dịch (như đại thực bào) tạo ra [161]. Các tế bào biểu mô đường thở đứng sát bên nhau, được liên kết bởi các mối nối, tạo ra sự kết dính chúng với nhau nhưng lại có thể điều chỉnh để cho phép có chọn lọc sự khuếch tán của các ion và phân tử đi qua trong khi vẫn duy trì tính toàn vẹn của hàng rào biểu mô [162,163] (hình 8) .
Đường thở là môi trường tiếp xúc liên tục với các chất ô nhiễm, vi sinh gây bệnh và chất gây dị ứng được biết là các tác nhân gây ra hiện tượng chết tế bào theo chương trình [164]. Việc loại bỏ các tế bào chết được thực hiện bởi các thực bào “chuyên trách”, thí dụ như đại thực bào, tế bào đuôi gai [165] hay các tế bào không chuyên trách, bao gồm cả các tế bào có trên biểu mô đường dẫn khí [166]. Hiện tượng này giúp kiểm soát phản ứng viêm đối với các chất gây dị ứng hít vào thông thường [158]. Việc nhận biết các tế bào chết đã được biết là có vai trò của các tế bào đáy nằm trên biểu mô đường thở [167]. Trong tình trạng viêm, cần thay thế các tế bào chết, thí dụ trong COPD, tế bào đáy tăng sinh là hiện tượng xảy ra sớm [168]. Các tế bào đáy không chỉ có khả năng cảm nhận và phản ứng với những thay đổi trong môi trường vi mô gây viêm mà chúng còn có trí nhớ viêm bẩm sinh [169,170]. Tăng sản tế bào đáy là một đặc điểm của việc tái tạo mô trong tình trạng có kích thích viêm mạn tính.
Tế bào biểu mô niêm mạc đường thở có sự tương tác chặt chẽ với các tế bào viêm, miễn dịch. Đại thực bào là tế bào có nhiều nhất trên đường thở. Sự tương tác hai chiều giữa các đại thực bào thường trú trên đường thở và các tế bào biểu mô đảm bảo duy trì trạng thái cân bằng nội môi của khả năng miễn dịch trước những kích thích và phản ứng bảo vệ thích hợp đối với mầm bệnh hít phải với khả năng sửa chữa mô hiệu quả khi được yêu cầu [171]. Khả năng tương tác tế bào - tế bào để tạo thành hệ thống truyền tín hiệu tầm tế bào mà trong y văn thường sử dụng bằng thuật ngữ “trao đổi thông tin chéo” (crosstalk). Hiện tại còn ít nghiên cứu trên người mặc dù đây là lĩnh vực quan trọng mà chúng ta còn chưa biết rõ [158]. Thí dụ người ta đã chứng minh được tế bào biểu mô tăng trình diện kháng nguyên với tế bào đuôi gai để gia tăng đáp ứng dị ứng thu được [172]. Đáp ứng miễn dịch đường thở cũng phụ thuộc nhiều vào các tế bào T nhớ (T cells - tissue-resident memory, TRM) có mặt thường trú tại vùng hay có sự xâm nhập của kháng nguyên [173]. Các tế bào này khi nhận diện được kháng nguyên xâm nhập, chúng sẽ khu trú kháng nguyên tại chỗ và phát động nhanh chóng các đáp ứng miễn dịch bảo vệ [174,175]. Trao đổi thông tin chéo giữa TRM với tế bào biểu mô định hình cách phản ứng với kháng nguyên xâm nhập. Thí dụ viêm phổi do pneumococcal kích hoạt CD4 TRM. CD4 TRM truyền thông tin để tế bào biểu mô tăng cường biểu hiện CXCL5 (cytokine hóa ứng động) gia tăng tập trung bạch cầu đa nhân trung tính nhằm đáp ứng viêm đối với tình trạng nhiễm trùng [176]. Nhưng cũng có thể ngược lại, crosstlak giữa tế bào đáp ứng miễn dịch thường trú trên đường thở làm cản trở quá trình phản ứng của tế bào biểu mô với tác nhân vi sinh gây bệnh. Thí dụ interferons là chất trung gian quan trọng của cơ thể trong sự đáp ứng chống vi-rút lại cản trở quá trình sửa chữa của biểu mô sau nhiễm vi-rút [177,178], tạo điều kiện thuận lợi cho sự xâm nhập của vi khuẩn thứ phát. Có thể nhận định một cách khái quát rằng sự tương tác phức tạp giữa các tế bào biểu mô và các tế bào miễn dịch bẩm sinh và thu được trên đường thở nhằm duy trì khả năng bảo vệ đường thở đối với các kích thích có hại. Tín hiệu từ tế bào biểu mô duy trì sự hiện của các tế bào miễn dịch cư trú tại mô và điều chỉnh phản ứng của chúng với kháng nguyên trong khi ngược lại, các tế bào miễn dịch có thể trực tiếp thay đổi chức năng và kiểu hình của các tế bào biểu mô đường thở.
3.4 Tương tác thần kinh - miễn dịch trên đường thở
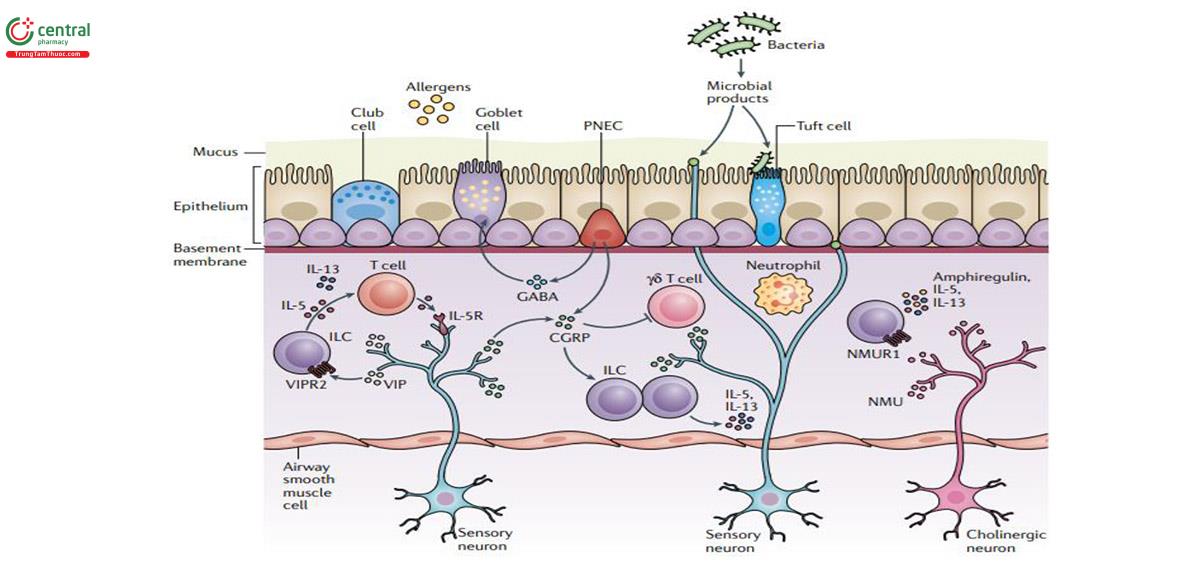
Sự có mặt rộng khắp trên đường dẫn khí cho phép các tế bào biểu mô, giống như một hệ thống cảm biến, cảm nhận và phản ứng với sự thay đổi của môi trường thông qua việc tiết ra một loạt chất trung gian để tạo ra sự tương tác với các tế bào miễn dịch, tế bào mô đệm trong mô liên kết bên dưới niêm mạc. Ngoài các tế bào biểu mô chuyên biệt có chức năng theo dõi đường thở và kích hoạt các phản xạ tránh né (chẳng hạn như ho và hắt hơi), người ta cũng nhận thấy đường hô hấp có rất nhiều tế bào thần kinh cảm giác, tạo điều kiện thuận lợi cho việc thực hiện các phản xạ tránh né này [179]. Đã có bằng chứng gần đây cho thấy rằng có hiện tượng trao đổi thông tin chéo (crosstalk) rộng rãi giữa hệ thống thần kinh và hệ thống miễn dịch, cũng như các tế bào biểu mô phổi có khả năng cảm nhận hóa học (chemosensory) chuyên biệt, rất quan trọng đối với việc điều hòa cân bằng nội mô [180,181] (hình 11). Toàn cảnh hiệu ứng tương tác này có thể tạo ra các phản ứng khi có sự xâm nhập của tác nhân gây bệnh hoặc các hạt độc hại, hoặc để theo dõi những thay đổi từ môi trường bên ngoài như biến động về nhiệt độ, oxy hoặc thậm chí phản ứng với các tác động vật lý như căng cứng hoặc co thắt xảy ra trong quá trình thở nhanh bằng miệng. Ví dụ, PIEZO1, một kênh ion tiếp nhận cảm giác cơ học để chuyển thành tín hiệu sinh hóa hay điện sinh học (PIEZO1 hay PIEZO2 là các kênh có cấu trúc protein được mã hóa bởi gen PIEZO1, PIEZO2), đã được chứng minh là có khả năng cảm nhận được sự thay đổi áp suất thủy tĩnh theo chu kỳ từ môi trường và tạo ra phản ứng tiền viêm [182].
Phổi chứa nhiều loại tế bào thần kinh khác nhau, bao gồm các tế bào thần kinh cảm giác đau, phản ứng với các kích thích độc hại hoặc có khả năng gây hại, trong khi các tế bào thần kinh cholinergic điều chỉnh trương lực đường thở, co cơ trơn đường thở, tiết chất nhầy và dãn mạch thông qua tương tác với các thụ thể muscarinic acetylcholine (mAChRs) trên cơ trơn đường dẫn khí, các tuyến chế tiết và mạch máu phổi. Nếu nhận định rằng nhiều phản ứng hô hấp cơ bản, chẳng hạn như ho, được thực hiện qua trung gian các tế bào thần kinh cảm giác [183], thì không có gì ngạc nhiên khi cho rằng hệ thống thần kinh cảm giác đóng một vai trò quan trọng trong các phản ứng viêm ở phổi. Ngoài việc chuyển tiếp thông tin những thay đổi cảm giác tới não, các tế bào thần kinh phổi khi được kích hoạt cũng có thể tự giải phóng các peptide kích thích, góp phần gây ra phản ứng viêm tiếp theo. Bằng chứng trực tiếp về sự góp phần của các peptide trên vào tình trạng viêm ở phổi bắt nguồn từ các thí nghiệm trong đó sự cắt bỏ hoặc ức chế hóa học Nav1.8+ (Nav1.8+ là một phân nhóm kênh ion ở người được mã hóa bởi gen SCN10A) hoặc kênh cation có khả năng làm thụ thể thoáng qua (transient receptor potential cation channels, TRPV1) trên tế bào thần kinh cảm giác sẽ dẫn đến giảm viêm dị ứng và tăng phản ứng phế quản [184-186]. Tương tự như vậy, hủy bỏ dây thần kinh phó giao cảm phổi có chọn lọc qua nội soi phế quản trên bệnh nhân COPD đã là một liệu pháp được sử dụng với mục đích làm giảm sức cản đường thở, quá tiết chất nhầy và viêm [184,187].
Sự kết hợp của các tế bào cảm nhận các thay đổi hóa học chuyên biệt và tương tác trực tiếp với các tế bào thần kinh cũng có thể là một cách khác để đường hô hấp phản ứng với các tác nhân gây bệnh đường hô hấp hít phải, các chất ô nhiễm trong không khí và các chất gây dị ứng. Việc hoạt hóa TRPV4 trên tế bào biểu mô sẽ dẫn tới kích hoạt phản ứng bảo vệ đối với lipopolysaccharides (LPS) của vi khuẩn, làm tăng tần số rung của lông chuyển và tăng sản xuất oxit nitric diệt khuẩn. Những con chuột thiếu hiển thị TRPV4 sẽ có tình trạng tăng phản ứng của đường thở nặng hơn và tăng cường thu hút bạch cầu đa nhân trung tính vào đường thở khi tiếp xúc với vi khuẩn [188]. Ngược lại, việc loại bỏ đặc hiệu TRPV1+ trên các tế bào thần kinh sẽ dẫn đến tăng cường khả năng bảo vệ miễn dịch đối với bệnh viêm phổi nặng do S. aureus gây ra thông qua việc tăng khả năng cảm ứng cytokine duy trì sự sống tế bào và khả năng thanh thải vi khuẩn [189]. TRPV1 và Nav1.8+ trên các tế bào thần kinh cảm giác đau đã được chứng minh là có ảnh hưởng đến sự phát tán của vi khuẩn thông qua việc ức chế số lượng bạch cầu đa nhân trung tính, các hoạt động giám sát cũng như điều chỉnh số lượng tế bào T (Gamma delta T, γδ T cells) thường trú. Xóa bỏ đặc hiệu các TRPV1 tế bào thần kinh phế vị dẫn đến tăng cường khả năng miễn dịch kháng khuẩn thông qua việc giải phóng neuropeptide thần kinh CGRP (Calcitonin gene-related peptide, neuropeptide gây dãn mạch).
Tương tác biểu mô - thần kinh tạo điều kiện thuận lợi đáp ứng viêm thông qua việc tiết ra các peptide thần kinh và chất dẫn truyền thần kinh. Nhiều tế bào miễn dịch có hiện diện các thụ thể tạo điều kiện giao tiếp với biểu mô cũng như trực tiếp với dây thần kinh [190]. Việc phát hiện ra tế bào ILC2 (tế bào phụ thuộc nang lympho bẩm sinh type 2, type 2 innate lymphoid cells) có một loạt các thụ thể đối với các peptide thần kinh và chất dẫn truyền thần kinh cho thấy cơ chế mà hệ thống thần kinh và miễn dịch phối hợp để cùng tham gia trong việc thúc đẩy một loạt các phản ứng cytokine type 2, tạo điều kiện thuận lợi cho các phản ứng kháng khuẩn, chống viêm, sửa chữa bảo vệ mô type 2 tại các vị trí niêm mạc [191]. Sự hiện diện của các tế bào ILC2 trong đường thở được điều chỉnh bởi các tín hiệu được truyền đi một cách có hệ thống hoặc bởi môi trường mô cục bộ [192] và các tế bào này sẽ tăng sự hiện diện ở những khu vực cụ thể khí có kích thích. Tế bào ILC2 tương tác trực tiếp với tế bào thần kinh thông qua các peptide thần kinh, chất dẫn truyền thần kinh và các yếu tố hỗ trợ thần kinh (Neurotrophic factors, NTFs), và những tương tác như vậy tạo điều kiện thuận lợi cho cả tín hiệu kích thích và ức chế [190]. Những tác dụng khác biệt này của peptide thần kinh đối với việc sản xuất cytokine ILC2 và bệnh lý miễn dịch type 2, đồng thời với các tín hiệu báo động và các peptide thần kinh từ tế bào biểu mô sẽ cung cấp một cơ chế phối hợp trong đó phản ứng của mô với chất gây dị ứng có thể được điều chỉnh tùy theo từng tình huống cụ thể.
Hiểu biết và quan điểm của chúng ta về biểu mô đường thở đã thay đổi đáng kể. Cộng đồng tế bào của biểu mô đường dẫn khí rất đa dạng, năng động và gắn kết về mặt chức năng. Các tế bào biểu mô phối hợp hoạt động với các quần thể tế bào miễn dịch tại chỗ để hỗ trợ hệ thống bảo vệ đa dạng ở vị trí tuyến đầu với các phản ứng sửa chữa vết thương hiệu quả cao. Các tế bào biểu mô, mô đệm và miễn dịch trong phổi có thể được điều hòa bởi nhiều tác động của vi môi trường mô cục bộ, trong đó gồm cả các đặc tính của cấu trúc ECM [193,194], căng thẳng (stress) cơ học [195] và phơi nhiễm với môi trường bụi - khói khí độc hại, thí dụ như khói thuốc lá [196,197].
4 KẾT LUẬN
Với chức năng hô hấp, niêm mạc đường hô hấp, là điểm tiếp xúc đầu tiên với tình trạng ô nhiễm khí thở, có chức năng như một hàng rào cơ học và miễn dịch. Việc tạo ra phản ứng viêm là một cơ chế quan trọng mà qua đó các loài động vật có vú trong đó có con người phản ứng và tự bảo vệ mình khỏi những tác động có hại, lây nhiễm cũng như không lây nhiễm. Ở phổi, cả cơ chế bảo vệ đặc hiệu và không đặc hiệu với kháng nguyên (miễn dịch) đều có thể dẫn đến phản ứng viêm. Mặc dù phản ứng này thường có tác dụng bảo vệ và có lợi nhưng tình trạng viêm cũng có khả năng làm tổn thương các mô, bao gồm cả đường thở, trong phổi.
Các tế bào biểu mô, mô đệm và miễn dịch trong phổi có thể được điều chỉnh bởi nhiều khía cạnh của vi môi trường khu trú của chúng, bao gồm các đặc tính của cấu trúc nền ngoại bào, stress cơ học và sự phơi nhiễm bụi-khói khí độc hại gồm cả thuốc lá. Các kỹ thuật xét nghiệm mới hiện nay đã cho phép lập bản đồ vị trí để hiểu biết tốt hơn kiểu hình và chức năng phân tử của tế bào trong một không gian cụ thể.
Biểu mô đường thở không tĩnh mà rất năng động và được bổ sung liên tục bởi các tế bào tiền thân cơ bản (có thể hiểu đơn giản là tế bào gốc). Công nghệ phiên mã đã nâng cao tầm hiểu biết của chúng ta về bối cảnh tế bào trong phổi con người nhưng vẫn còn những hạn chế cố hữu và nhiều câu hỏi chưa được giải đáp. Các loại tế bào biểu mô hiếm gặp đã thu hút nhiều sự quan tâm nhưng do có số lượng thấp trong phổi nên sẽ là thách thức để xác định sự đóng góp của chúng đối với các bệnh lý khác nhau với hy vọng rằng chúng ta có thể khai thác và điều khiển các tế bào cụ thể trong việc tăng cường sức khỏe phổi trong suốt cuộc đời con người.
5 Tài liệu tham khảo
1. Al-Hegelan, M.; Robert, M.T.; Christian, C.; John, W.H. Ambient ozone and pulmonary innate immunity. Immunol. Res. 2011, 49, 173–191.
2. Michaudel, C.; Mackowiak, C.; Maillet, I.; Fauconnier, L.; Akdis, C.A.; Sokolowska, M. Ozone exposure induces respiratory barrier biphasic injury and inflammation controlled by IL-33. J. Allergy Clin. Immunol. 2018, 142, 942–958.
3. Foster, W.M.; Costa, D.L.; Langenback, E.G. Ozone exposure alters tracheobronchial mucociliary function in humans. J. Appl. Physiol. 1987, 63, 996–1002.
4. Devlin, R.B.; McDonnell, W.F.; Mann, R.; Becker, S.; House, D.E.; Schreinemachers, D.; Koren, H.S. Exposure of humans to ambient levels of ozone for 6.6 h causes cellular and biochemical changes in the lung. Am. J. Respir. Cell Mol. Biol. 1991, 4, 72–81.
5. Gryparis, A.; Forsberg, B.; Katsouyanni, K.; Analitis, A.; Touloumi, G.; Schwartz, J.; Samoli, E.; Medina, S.; Anderson, H.R.; Niciu, E.M.; et al. Acute effects of ozone on mortality from the “air pollution and health: A European approach” project. Am. J. Respir. Crit. Care Med. 2004, 170, 1080–1087.
6. Foster, W.M.; Stetkiewicz, P.T. Regional clearance of solute from the respiratory epithelia: 18–20 h postexposure to ozone. J. Appl. Physiol. 1996, 81, 1143–1149.
7. Foster, W.M.; Freed, A.N. Regional clearance of solute from peripheral airway epithelia: Recovery after sublobar exposure to ozone. J. Appl. Physiol. 1999, 86, 641–646.
8. Kirkham, P.A.; Barnes, P.J. Oxidative stress in COPD. Chest 2013, 144, 266–273.
9. Halbert, R.J.; Natoli, J.L.; Gano, A.; Badamgarav, E.; Buist, A.S.; Mannino, D.M. Global burden of COPD: Systematic review and meta-analysis. Eur. Respir. J. 2006, 28, 523–532.
10. Park, J.; Kim, H.J.; Lee, C.H.; Lee, C.H.; Lee, H.W. Impact of long-term exposure to ambient air pollution on the incidence of chronic obstructive pulmonary disease: A systematic review and meta-analysis. Environ. Res. 2021, 194, 110703
11. Fletcher, C., and R. Peto. 1977. The natural history of chronic airflow obstruction. B.M.J. 1:1645–1648.
12. Diener, C. F., and B. Burrows. 1975. Further observations on the course and prognosis of chronic obstructive lung disease. Am. Rev. Respir. Dis. 111:719–724.
13. Hogg, J. C., P. T. Macklem, and W. A. Thurlbeck. 1968. Site and nature of airway obstruction in chronic obstructive lung disease. N. Engl. J. Med. 278:1355–1360.
14. Snider, G. L. 1989. Changes in COPD occurrence: chronic obstructive pulmonary disease: a definition and implications of structural determinants of airflow obstruction for epidemiology. Am. Rev. Respir. Dis. 140:S3–S8.
15. Saetta, M., A. Di Stefano, P. Maestrelli, A. Ferraresso, R. Drigo, A. Potena, A. Ciaccia, and L. M. Fabbri. 1993. Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitis. Am. Rev. Respir. Dis. 147:301–306.
16. Di Stefano, A., P. Maestrelli, A. Roggeri, G. Turato, S. Calabro, A. Potena, C. E. Mapp, A. Ciaccia, L. Covacev, L. M. Fabbri, and M. Saetta. 1994. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am. J. Respir. Crit. Care Med. 149:803–810.
17. Turato, G., A. Di Stefano, P. Maestrelli, C. E. Mapp, M. P. Ruggieri, A. Roggeri, L. M. Fabbri, and M. Saetta. 1995. Effect of smoking cessation on airway inflammation in chronic bronchitis. Am. J. Respir. Crit. Care Med. 152:1262–1267.
18. Di Stefano, A., G. Turato, P. Maestrelli, C. E. Mapp, M. Ruggieri, A. Roggeri, P. Boschetto, L. M. Fabbri, and M. Saetta. 1996. Airflow limitation in chronic bronchitis is associated with T-lymphocyte and macrophage infiltration of the bronchial mucosa. Am. J. Respir. Crit. Care Med. 153:629–632.
19. Antonino Di Stefano et al. Severity of Airflow Limitation Is Associated with Severity of Airway Inflammation in Smokers. Am J Respir Crit Care Med Vol 158. pp 1277–1285, 1998
20. Shin S, Bai L, Burnett RT, Kwong JC, Hystad P, van Donkelaar A, et al. Air pollution as a risk factor for incident chronic obstructive pulmonary disease and asthma: 15-year population-based cohort study. Am J Respir Crit Care Med 2021;203:1138–1148.
21. MacNee W. Oxidants/antioxidants and COPD. Chest 2000;117:303s–317s
22. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumour necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996;153:530–534
23. Susan Chinn et al An Increase in Bronchial Responsiveness Is Associated with Continuing or Restarting Smoking. Am J Respir Crit Care Med Vol 172. pp 956–961, 2005
24. Bosse Y et al. Molecular signature of smoking in human lung tissues. Cancer Res 2012; 72: 3753-3763
25. Vogelmeier CF, et al. Global Strategy for the Diagnosis, Management, and Prevention of Chronic Obstructive Lung Disease 2017 Report. GOLD Executive Summary. Am J Respir Crit Care Med 2017; 195: 557-582
26. Agusti A, Faner R. Lung function trajectories in health and disease. Lancet Respir Med 2019; 7: 358-364.
27. Ziller MJ et al. Charting a dynamic DNA methylation landscape of the human genome. Nature 2013; 500: 477-481.
28. Joehanes R et al. Epigenetic Signatures of Cigarette Smoking. Circ Cardiovasc Genet 2016; 9: 436-447.
29. Sundar IK et al. DNA methylation profiling in peripheral lung tissues of smokers and patients with COPD. Clin Epigenetics 2017; 9: 38
30. Morrow JD et al. DNA methylation profiling in human lung tissue identifies genes associated with COPD. Epigenetics 2016: 1-10.
31. Sandra Casas-Recasens et al. Lung DNA Methylation in COPD: Relationship with Smoking Status and Airflow Limitation Severity. AJRCCM Articles in Press. Published August 21, 2020 as 10.1164/rccm.201912-2420LE Copyright © 2020 by the American Thoracic Society
32. Willemse BW, ten Hacken NH, Rutgers B, Lesman-Leegte IG, Postma DS, Timens W. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 2005; 26:835–845
33. MacNee W. Pathogenesis of chronic obstructive pulmonary disease. Clin Chest Med 2007;28:479–513
34. William MacNee et al. New Paradigms in the Pathogenesis of Chronic Obstructive Pulmonary Disease I. Proc Am Thorac Soc Vol 6. pp 527–531, 2009
35. Cosio MG et al. Immunologic aspects of chronic obstructive pulmonary disease. N Engl J Med 2009;360:2445– 2454.
36. MacNee W. Oxidative stress and chronic obstructive pulmonary disease. Eur Respir Monograph 2006;11:100– 129.
37. Rahman I et al. 4-Hydroxy-2-nonenal, a specific lipid peroxidation product, is elevated in lungs of patients with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 2002;166: 490–495.
38. Schaur RJ et al. The lipid peroxidation product 4-hydroxynonenal is formed by–and is able to attract–rat neutrophils in vivo. Free Radic Res 1994; 20:365–373. 8
39. Liu W et al. 4-hydroxynonenal triggers an epidermal growth factor receptor-linked signal pathway for growth inhibition. J Cell Sci 1999;112:2409–2417.
40. Liu W et al. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J Cell Sci 2000;113:635–641
41. Parola M et al. 4-Hydroxynonenal as a biological signal: molecular basis and pathophysiological implications. Antioxid Redox Signal 1999;1:255–284
42. Cavarra E et al. Human SLPI inactivation after cigarette smoke exposure in a new in vivo model of pulmonary oxidative stress. Am J Physiol Lung Cell Mol Physiol 2001;281:L412–L417.
43. Shapiro SD. Proteolysis in the lung. Eur Respir J Suppl 2003;44:30s–32s.
44. Shapiro SD. Proteinases in chronic obstructive pulmonary disease. Biochem Soc Trans 2002;30:98–102
45. Rahman I, Adcock IM. Oxidative stress and redox regulation of lung inflammation in COPD. Eur Respir J 2006;28:219–242
46. Cosio BG et al. Theophylline restores histone deacetylase activity and steroid responses in COPD macrophages. J Exp Med 2004;200:689–695
47. Kazuhiro Ito et al. Decreased Histone Deacetylase Activity in Chronic Obstructive Pulmonary Disease. N Engl J Med 2005;352:1967-76
48. Segura-Valdez L, Pardo A, Gaxiola M, Uhal BD, Becerril C, Selman M. Upregulation of gelatinases A and B, collagenases 1 and 2, and increased parenchymal cell death in COPD. Chest 2000;117:684–694.
49. Kasahara Y et al. Endothelial cell death and decreased expression of vascular endothelial growth factor and vascular endothelial growth factor receptor 2 in emphysema. Am J Respir Crit Care Med 2001;163:737–744.
50. Majo J et al. Lymphocyte population and apoptosis in the lungs of smokers and their relation to emphysema. Eur Respir J 2001;17:946–953
51. Yokohori N et al. Increased levels of cell death and proliferation in alveolar wall cells in patients with pulmonary emphysema. Chest 2004;125:626–632
52. Tuder RM. Aging and cigarette smoke: fueling the fire. Am J Respir Crit Care Med 2006;174:490–491.
53. Tang K, et al. Lung-targeted VEGF inactivation leads to an emphysema phenotype in mice. J Appl Physiol 2004;97:1559–1566.
54. Marwick JA et al. Cigarette smoke disrupts VEGF165-VEGFR-2 receptor signaling complex in rat lungs and patients with COPD: morphological impact of VEGFR-2 inhibition. Am J Physiol Lung Cell Mol Physiol 2006;290:L897–L908.
55. Tuder RM et al. Apoptosis and emphysema: the missing link. Am J Respir Cell Mol Biol 2003;28: 551–554.
56. Tuder RM et al. Oxidative stress and apoptosis interact and cause emphysema due to vascular endothelial growth factor receptor blockade. Am J Respir Cell Mol Biol 2003;29: 88–97.
57. Zheng T et al. Role of cathepsin S-dependent epithelial cell apoptosis in IFN-g-induced alveolar remodeling and pulmonary emphysema. J Immunol 2005;174:8106–8115
58. Kang M-J et al. Cigarette smoke selectively enhances viral PAMP and virusinduced pulmonary innate immunity and remodeling responses. J Clin Invest 2008;118:2771–2784
59. Tuder RM, Yoshida T, Arap W, Pasqualini R, Petrache I. Cellular and Molecular Mechanisms of Alveolar Destruction in Emphysema: An Evolutionary Perspective. Proc Am Thorac Soc 2006;3:503–510.
60. MacNee W. Accelerated lung aging: a novel pathogenic mechanism of chronic obstructive pulmonary disease (COPD). Biochem Soc Trans 2009;37:819–823.
61. Saretzki G, von Zglinicki T. Replicative aging, telomeres, and oxidative stress. Ann N Y Acad Sci 2002;959:24– 29.
62. Morla M, Busquets X, Pons J, Sauleda J, MacNee W, Agusti AG. Telomere shortening in smokers with and without COPD. Eur Respir J 2006;27:525–528
63. Abed DA, Goldstein M, Albanyan H, et al. Discovery of direct inhibitors of Keap1-Nrf2 protein-protein
64. Liu Q, Gao Y, Ci X. Role of Nrf2 and Its Activators in Respiratory Diseases. Oxid Med Cell Longev 2019;2019:7090534.
65. Rojo AI, Sagarra MR, Cuadrado A. GSK-3β downregulates the transcription factor Nrf2 after oxidant damage: relevance to exposure of neuronal cells to oxidative stress. J Neurochem 2008;105:192-202
66. Mari Hikichi et al. Pathogenesis of chronic obstructive pulmonary disease (COPD) induced by cigarette smoke. J Thorac Dis 2019;11 (Suppl 17): S2129-S2140 | http://dx.doi.org/10.21037/jtd.2019.10.43
67. Stanescu D, Sanna A, Veriter C, Kostianev S, Calcagni PG, Fabbri LM, Maestrelli P. Airways obstruction, chronic expectoration, and rapid decline of FEV1 in smokers are associated with increased levels of sputum neutrophils. Thorax 1996;51:267–271.
68. Maestrelli P, Calcagni PG, Saetta M, Bertin T, Mapp CE, Sanna A, Veriter C, Fabbri LM, Stanescu D. Integrin upregulation on sputum neutrophils in smokers with chronic airway obstruction. Am J Respir Crit Care Med 1996;154:1296–1300
69. Keatings VM, Collins PD, Scott DM, Barnes PJ. Differences in interleukin-8 and tumour necrosis factor-alpha in induced sputum from patients with chronic obstructive pulmonary disease or asthma. Am J Respir Crit Care Med 1996;153:530–534
70. Rutgers SR, Postma DS, ten Hacken NH, Kauffman HF, van Der Mark TW, Koeter GH, Timens W. Ongoing airway inflammation in patients with COPD who do not currently smoke. Thorax 2000;55:12–18
71. Balzano G, Stefanelle F, Iorio C, De Felice A, Melillo EM, Martucci M, Melillo G. Eosinophilic inflammation in stable chronic obstructive pulmonary disease. Relationship with neutrophils and airway function. Am J Respir Crit Care Med 1999;160:1486–1492.
72. Thompson AB, Daughton D, Robbins RA, Ghafouri MA, Oehlerkin M, Rennard SI. Intraluminal airway inflammation in chronic bronchitis: characterization and correlation with clinical parameters. Am Rev Respir Dis 1989;140:1527–1537.
73. Di Stefano A, Maestrelli P, Roggeri A, Turato G, Calabro S, Potena A, Mapp CE, Ciaccia A, Covacev L, Fabbri LM, Saetta M. Upregulation of adhesion molecules in the bronchial mucosa of subjects with chronic obstructive bronchitis. Am J Respir Crit Care Med 1994;149:803–810
74. Saetta M et al. Inflammatory cells in the bronchial glands of smokers with chronic bronchitis. Am J Respir Crit Care Med 1997;156:1633–1639.
75. Saetta M et al. Goblet cell hyperplasia and epithelial inflammation in peripheral airways of smokers with both symptoms of chronic bronchitis and airflow limitation. Am J Respir Crit Care Med 2000;161:1016–1021
76. Takanashi S, Hasegawa Y, Kanehira Y, Yamamoto K, Fujimoto K, Satoh K, Okamura K. Interleukin-10 level in sputum is reduced in bronchial asthma, COPD and in smokers. Eur Respir J 1999;14:309–314.
77. Chanez P, Vignola AM, O’Shaugnessy T, Enander I, Li D, Jeffery PK, Bousquet J. Corticosteroid reversibility in COPD is related to features of asthma. Am J Respir Crit Care Med 1997;155:1529–1534.
78. Saetta M et al. Airway eosinophilia in chronic bronchitis during exacerbations. Am J Respir Crit Care Med 1994;150: 1646–1652
79. Zhu J, Qiu YS, Majumdar S, Gamble E, Matin D, Turato G, Fabbri LM, Barnes N, Saetta M, Jeffery PK. Exacerbation of bronchitis. Bronchial eosinophilia and gene expression for interleukin-4, interleukin-5 and eosinophil chemoattractants. Am J Respir Crit Care Med 2001;164: 109–116.
80. Gibson PG, Wooley KL, Carty K, Murree-Allen K, Saltos N. Induced sputum eosinophil cationic protein (ECP) measurement in asthma and chronic obstructive airway disease (COAD). Clin Exp Allergy 1998;28: 1081–1088.
81. Saetta M et al. CD8+ T-lymphocytes in the peripheral airways of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1998;157:822–826.
82. Di Stefano A et al. Airflow limitation in chronic bronchitis is associated with T-lymphocyte and macrophage infiltration in the bronchial mucosa. Am J Respir Crit Care Med 1996;153:629–632.
83. O’Shaughnessy TC et al. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med 1997;155:852–857
84. Saetta M et al. Activated T-lymphocytes and macrophages in bronchial mucosa of subjects with chronic bronchitis. Am Rev Respir Dis 1993;147:301–306.
85. Lams BEA, Sousa AR, Rees PJ, Lee TH. Subepithelial immunopathology of the large airways in smokers with and without chronic obstructive pulmonary disease. Eur Respir J 2000;15:5
86. Saetta M, Baraldo S, Corbino L, Turato G, Braccioni F, Rea F, Cavallesco G, Tropeano G, Mapp CE, Maestrelli P, Ciaccia A, Fabbri LM. CD8+ve cells in the lungs of smokers with chronic obstructive pulmonary disease. Am J Respir Crit Care Med 1999;160:711–717.
87. Cannon MJ et al. Cytotoxic T-cells clear virus but augment lung pathology in mice infected with respiratory syncytial virus. J Exp Med 1988;168:1163–116
88. Salgame P et al. Differing lymphokine profiles of functional subsets of human CD4 and CD8 T cell clones. Science 1991;254:279–282.
89. Del Prete GF et al. Allergen exposure induce activation of allergen-specific Th2 cells in the airway mucosa of patients with allergic respiratory disorders. Eur J Immunol 1993;23:1445–1449.
90. Maestrelli P et al. CD8 T cell clones producing interleukin-5 and interferon-gamma in bronchial mucosa in toluene diisocyanate asthma. Scand J Work Environ Health 1994;20: 376–381.
91. Coyle AJ, Erard F, Bertrand C, Walti S, Pircher H, Le Gros G. Virus-specific CD8+ cells can switch to interleukin-5 production and induce airway eosinophilia. J Exp Med 1995;181:1229–1233.
92. Maestrelli P, Tsai J-J, Cromwell O, Kay AB. The identification and partial characterization of a human mononuclear cell-derived neutrophil chemotactic factor apparently distinct from IL-1, IL-2, GM-CSF, TNF and IFN-gamma. Immunology 1988;64:219–225.
93. Maestrelli P, O’Hehir RE, Tsai J-J, Lamb JR, Cromwell O, Kay AB. Antigen-induced neutrophil chemotactic factor derived from cloned human T lymphocytes. Immunology 1988;65:605–609.
94. Zheng T, Zhu Z, Wang Z, Homer RJ, Ma B, Riese RY Jr, Chapman HA Jr, Shapiro SD, Elias JA. Inducible targeting of IL-13 to the adult lung causes matrix metalloproteinase- and cathepsin-dependent emphysema. J Clin Invest 2000;106:1081–1093.
95. Shim JJ, Dabbagh K, Ueki IF, Dao-pick T, Burgel PR, Takeyama K, Tam DC, Nadel JA. IL-13 induces mucin production by stimulating epidermal growth factor receptors and by activating neutrophils. Am J Physiol Lung Cell Mol Physiol 2001;280:L134–L140.
96. Mapp CE, Miotto D, Buzzacaro E, Maestrelli P, Saetta M, Braccioni F, Cavallesco G, Papi A, Fabbri LM. Increased expression of IL-13 in the central airways of smokers with chronic bronchitis. Am J Respir Crit Care Med 2000;161:A571.
97. Amadori A, Zamarchi R, De Silvestro G, Forza G, Cavatton G, Danieli GA, Clementi M, Chieco-Bianchi L. Genetic control of the CD4/CD8 T-cell ratio in humans. Nat Med 1995;1:1279–1283
98. O’Shaughnessy TC, Ansari TW, Barnes NC, Jeffery PK. Inflammation in bronchial biopsies of subjects with chronic bronchitis: inverse relationship of CD8+ T lymphocytes with FEV1. Am J Respir Crit Care Med 1997;155:852– 857.
99. Di Stefano A, Capelli A, Lusuardi M, Balbo P, Vecchio C, Maestrelli P, Mapp CE, Fabbri LM, Donner CF, Saetta M. Severity of airflow limitation is associated with severity of airway inflammation in smokers. Am J Respir Crit Care Med 1998;158:1277–1285
100. Toshinori Yoshida et al. Pathobiology of Cigarette Smoke-Induced Chronic Obstructive Pulmonary Disease. Physiol Rev 87: 1047–1082, 2007;
101. Agnieszka Strzelak et al. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033; doi:10.3390/ijerph15051033
102. Chen J, Wang X, Schmalen A, et al. Antiviral CD8+ T-cell immune responses are impaired by cigarette smoke and in COPD. Eur Respir J 2023; 62: 2201374 [DOI: 10.1183/ 13993003.01374-2022]
103. Salvi SS, Barnes PJ. Chronic obstructive pulmonary disease in non-smokers. Lancet. 2009;374:733-43
104. Kodgule R, Salvi S. Exposure to biomass smoke as a cause for airway disease in women and children. Curr Opin Allergy Clin Immunol. 2012;12:82-90
105. Biomass.net. Following the Energy Trail With Biomass History. Available from: http://www.biomass.net/Biomass-History.html. [Last accessed on 2018 Apr 10th].
106. Eisner MD, Anthonisen N, Coultas D, et al. An official American thoracic society public policy statement: novel risk factors and the global burden of chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2010;182:693-18.
107. Ramírez-Venegas A, Sansores RH, Velázquez-Uncal M, Pérez-Bautista O. Nonsmokers and biomass exposure. ERS Monogr. 2015;69:35-46.
108. Caballero A, Torres-Duque CA, Jaramillo C, et al. Prevalence of COPD in five Colombian cities situated at low, medium, and high altitude (PREPOCOL study). Chest. 2008;133:343-9.
109. Mishra V, Retherford RD. Cooking smoke increases the risk of acute respiratory infection in children. Natl Fam Health Surv Bull. 1997;8:1-4.
110. Po JYT, FitzGerald JM, Carlsten C. Respiratory disease associated with solid biomass fuel exposure in rural women and children: systematic review and meta-analysis. Thorax. 2011;66:232-9.
111. Heinzerling AP, Guarnieri MJ, Mann JK, et al. Lung function in woodsmoke-exposed Guatemalan children following a chimney stove intervention. Thorax. 2016;71:421-8.
112. Naeher LP, Brauer M, Lipsett M, et al. Woodsmoke health effects: a review. Inhal Toxicol. 2007;19:67-6.
113. Bernstein DM. A review of the influence of particle size, puff volume, and inhalation pattern on the deposition of cigarette smoke particles in the respiratory tract. Inhal Toxico. 2004; 16:675-89
114. Rivera RM, Cosío MG, Ghezzo H, Salazar M, Pérez-Padilla R. Comparison of lung morphology in COPD secondary to cigarette and biomass smoke. Int J Tuberc Lung Dis. 2008;12:972-7
115. Fernandes L, Gulati N, Fernandes Y, et al. Small airway imaging phenotypes in biomass- and tobacco smoke-exposed patients with COPD. ERJ Open Res 2017; 3: 00124-2016.
116. Tim Higenbottam et al. Cigarette smoke inhalation and the acute airway response. Thorax, 1980, 35, 246-254
117. Mookherjee N, Piyadasa H, Ryu MH, Rider CF, Ezzati P, Spicer V, et al. Inhaled diesel exhaust alters the allergen-induced bronchial secretome in humans. Eur Respir J 2018;51.
118. Zarcone MC, Duistermaat E, van Schadewijk A, Jedynska A, Hiemstra PS, Kooter IM. Cellular response of mucociliary differentiated primary bronchial epithelial cells to diesel exhaust. Am J Physiol Lung Cell Mol Physiol 2016; 311:L111-23.
119. MacIntyre EA, Gehring U, Molter A, Fuertes E, Klumper C, Kramer U, et al. Air pollution and respiratory infections during early childhood: an analysis of 10 European birth cohorts within the ESCAPE Project. Environ Health Perspect 2014;122:107-13.
120. Wong CM, Yang L, Thach TQ, Chau PY, Chan KP, Thomas GN, et al. Modification by influenza on health effects of air pollution in Hong Kong. Environ Health Perspect 2009;117:248-53.
121. Pirozzi CS, Jones BE, VanDerslice JA, Zhang Y, Paine R 3rd, Dean NC. Short-term air pollution and incident pneumonia. A case-crossover study. Ann Am Thorac Soc 2018;15:449-59.
122. Jartti T, Gern JE. Role of viral infections in the development and exacerbation of asthma in children. J Allergy Clin Immunol 2017;140:895-906.
123. Liu CW, Lee TL, Chen YC, Liang CJ, Wang SH, Lue JH, et al. PM2.5-induced oxidative stress increases intercellular adhesion molecule-1 expression in lung epithelial cells through the IL-6/AKT/STAT3/NF-kappaB-dependent pathway. Part Fibre Toxicol 2018;15:4.
124. Spannhake EW, Reddy SP, Jacoby DB, Yu XY, Saatian B, Tian J. Synergism between rhinovirus infection and oxidant pollutant exposure enhances airway epithelial cell cytokine production. Environ Health Perspect 2002;110: 665-70
125. Lakey PS, Berkemeier T, Tong H, Arangio AM, Lucas K, Poschl U, et al. Chemical exposure-response relationship between air pollutants and reactive oxygen species in the human respiratory tract. Sci Rep 2016;6:32916.
126. Chen X, Liu J, Zhou J, Wang J, Chen C, Song Y, et al. Urban particulate matter (PM) suppresses airway antibacterial defence. Respir Res 2018;19:5.
127. Orie NGM, Sluiter HJ, de Vries K, et al. The host factor in bronchitis. function. Am Rev Respir Dis 1988;138:26– 30. In: Orie NGM, Sluiter HJ, eds. Bronchitis: an international symposium Assen, Netherlands: Royal van Gorcum, 1961
128. Woolcock AJ, Peat JK, Salome CM, et al. Prevalence of bronchial hyperresponsiveness and asthma in a rural adult population. Thorax 1987;42:361–8.
129. M H Brutsche et al. Bronchial hyperresponsiveness and the development of asthma and COPD in asymptomatic individuals: SAPALDIA Cohort Study. Thorax 2006;61:671–677. doi: 10.1136/thx.2005.052241
130. Barnes PJ. Mechanisms in COPD: differences from asthma. Chest 2000;117: 10S-4S.
131. Haldar P, Pavord ID, Shaw DE, Berry MA, Thomas M, Brightling CE, et al. Cluster analysis and clinical asthma phenotypes. Am J Respir Crit Care Med 2008;178: 218-24.
132. Moore WC, Meyers DA, Wenzel SE, Teague WG, Li HS, Li XN, et al. Identification of asthma phenotypes using cluster analysis in the Severe Asthma Research Program. Am J Respir Crit Care Med 2010;181:315-23.
133. Brightling CE, Gupta S, Gonem S, Siddiqui S. Lung damage and airway remodelling in severe asthma. Clin Exp Allergy 2012;42:638-49.
134. Burgel PR, Paillasseur JL, Caillaud D, Tillie-Leblond I, Chanez P, Escamilla R, et al. Clinical COPD phenotypes: a novel approach using principal component and cluster analyses. Eur Respir J 2010;36:531-9.
135. Garcia-Aymerich J, Gomez FP, Benet M, Farrero E, Basagana X, Gayete A, et al. Identification and prospective validation of clinically relevant chronic obstructive pulmonary disease (COPD) subtypes. Thorax 2011;66:430-7.
136. Weatherall M, Travers J, Shirtcliffe PM, Marsh SE, Williams MV, Nowitz MR, et al. Distinct clinical phenotypes of airways disease defined by cluster analysis. Eur Respir J 2009;34:812-8.
137. Barker BL, Brightling CE. Phenotyping the heterogeneity of chronic obstructive pulmonary disease. Clin Sci 2013;124:371-87.
138. Gibson PG, Simpson JL. The overlap syndrome of asthma and COPD: what are its features and how important is it? Thorax 2009;64:728-35.
139. Vanfleteren LE, Kocks JW, Stone IS, Breyer-Kohansal R, Greulich T, Lacedonia D, et al. Moving from the Oslerian paradigm to the post-genomic era: are asthma and COPD outdated terms? Thorax 2014;69:72-9.
140. Iwamoto H, Gao J, Kosela J, Kinnula V, Kobayashi H, Laitinen T, et al. Differences in plasma and sputum biomarkers between COPD and COPD-asthma overlap. Eur Respir J 2014;43:421-9.
141. Barnes PJ. Against the Dutch hypothesis: asthma and chronic obstructive pulmonary disease are distinct diseases. Am J Respir Crit Care Med 2006;174:240-3.
142. Bafadhel M, McCormick M, Saha S, McKenna S, Shelley M, Hargadon B, et al. Profiling of sputum inflammatory mediators in asthma and chronic obstructive pulmonary disease. Respiration 2012;83:36-44.
143. Desai D, Brightling C. Cytokines and cytokine-specific therapy in asthma. Adv Clin Chem 2012;57:57-97.
144. Haldar P, Brightling CE, Hargadon B, Gupta S, Monteiro W, Sousa A, et al. Mepolizumab and exacerbations of refractory eosinophilic asthma. New Engl J Med 2009;360:973-84.
145. Pavord ID, Korn S, Howarth P, Bleecker ER, Buhl R, Keene ON, et al. Mepolizumab for severe eosinophilic asthma (DREAM): a multicentre, double-blind, placebo-controlled trial. Lancet 2012;380:651-9.
146. Michael A. Ghebre et al. Biological clustering supports both ‘‘Dutch’’ and ‘‘British’’ hypotheses of asthma and chronic obstructive pulmonary disease. J Allergy Clin Immunol 2015;135:63-72
147. B.W.M. Willemse et al. Effect of 1-year smoking cessation on airway inflammation in COPD and asymptomatic smokers. Eur Respir J 2005; 26: 835–845 DOI: 10.1183/09031936.05.00108904
148. Désirée F. Jansen et al. Smoking and Airway Hyperresponsiveness Especially in the Presence of Blood Eosinophilia Increase the Risk to Develop Respiratory Symptoms A 25-year Follow-up Study in the General Adult Population. Am J Respir Crit Care Med Vol 160. pp 259–264, 1999
149. Larsen, G. L. 1999. Host defense systems of the lung. In L. M. Taussig, L. I. Landau, P. N. Le Souëf, W. H. Morgan, F. D. Martinez, and P. D. Sly, editors. Pediatric Pulmonary Medicine. C. V. Mosby, St. Louis, MO. 57–75.
150. Behrens, B. L., R. A. F. Clark, D. M. Presley, J. P. Graves, D. C. Feldsien, and G. L. Larsen. 1987. Comparison of the evolving histopathology of early and late cutaneous and asthmatic responses in rabbits after a single antigen challenge. Lab. Invest. 56:101–113.
151. Gary L. Larsen et al. The Concept of Airway Inflammation. Am J Respir Crit Care Med Vol 162. pp S2–S6, 2000
152. Larsen, G. L., R. O. Webster, G. S. Worthen, R. S. Gumbay, and P. M. Henson. 1985. The additive effect of intravascular complement activation and brief episodes of hypoxia in producing increased permeability in the rabbit lung. J. Clin. Invest. 75:902–910.
153. Gauvreau, G. M., R. M. Watson, and P. M. O’Byrne. 1999. Protective effects of inhaled PGE2 on allergen-induced airway responses and airway inflammation. Am. J. Respir. Crit. Care Med. 159:31–36
154. Larsen, G. L et al. 1998. Transgenic mice that overexpress pulmonary copper-zinc superoxide dismutase are resistant to changes in neural control of airways following allergen sensitization and challenge (abstract). Am. J. Respir. Crit. Care Med. 157:A231
155. Huffnagle, G. B., Dickson, R. P. & Lukacs, N. W. The respiratory tract microbiome and lung inflammation: a two-way street. Mucosal Immunol. 10, 299–306 (2017).
156. Jennifer A. Aguiar et al. The impact of cigarette smoke exposure, COPD, or asthma status on ABC transporter gene expression in human airway epithelial cells. Scientific Reports | (2019) 9:153 | DOI:10.1038/s41598-018-36248-9
157. I.H. Heijink et al. Cigarette smoke impairs airway epithelial barrier function and cell–cell contact recovery. Eur Respir J 2012; 39: 419–428
158. Richard J. Hewitt et al. Regulation of immune responses by the airway epithelial cell landscape. Nature reviews | immunology reviews volume 21 | june 2021. 347-362 )
159. Johansen, F. E. & Kaetzel, C. S. Regulation of the polymeric immunoglobulin receptor and IgA transport: new advances in environmental factors that stimulate pIgR expression and its role in mucosal immunity. Mucosal Immunol. 4, 598–602 (2011).
160. Richmond, B. W. et al. Airway bacteria drive a progressive COPD-like phenotype in mice with polymeric immunoglobulin receptor deficiency. Nat. Commun. 7, 11240 (2016).
161. Lehmann, R. et al. Differential regulation of the transcriptomic and secretomic landscape of sensor and effector functions of human airway epithelial cells. Mucosal Immunol. 11, 627–642 (2018).
162. Brune, K., Frank, J., Schwingshackl, A., Finigan, J. & Sidhaye, V. K. Pulmonary epithelial barrier function: some new players and mechanisms. Am. J. Physiol. Lung Cell Mol. Physiol. 308, L731–L745 (2015).
163. Vermeer, P. D. et al. Segregation of receptor and ligand regulates activation of epithelial growth factor receptor. Nature 422, 322–326 (2003)
164. White, S. R. Apoptosis and the airway epithelium. J. Allergy 2011, 948406 (2011).
165. Henson, P. M. & Hume, D. A. Apoptotic cell removal in development and tissue homeostasis. Trends Immunol. 27, 244–250 (2006).
166. Juncadella, I. J. et al. Apoptotic cell clearance by bronchial epithelial cells critically influences airway inflammation. Nature 493, 547–551 (2013).
167. Fujino, N. et al. Sensing of apoptotic cells through Axl causes lung basal cell proliferation in inflammatory diseases. J. Exp. Med. 216, 2184–2201 (2019).
168. Shaykhiev, R. & Crystal, R. G. Early events in the pathogenesis of chronic obstructive pulmonary disease. Smoking-induced reprogramming of airway epithelial basal progenitor cells. Ann. Am. Thorac. Soc. 11 (Suppl. 5), S252–S258 (2014).
169. Natoli, G. & Ostuni, R. Adaptation and memory in immune responses. Nat. Immunol. 20, 783–792 (2019).
170. Ordovas-Montanes, J., Beyaz, S., Rakoff-Nahoum, S. & Shalek, A. K. Distribution and storage of inflammatory memory in barrier tissues. Nat. Rev. Immunol. 20, 308–320 (2020).
171. Puttur, F., Gregory, L. G. & Lloyd, C. M. Airway macrophages as the guardians of tissue repair in the lung. Immunol. Cell Biol. 97, 246–257 (2019).
172. Moon, H. G. et al. Airway epithelial cell-derived colony stimulating factor-1 promotes allergen sensitization. Immunity 49, 275–287.e5 (2018).
173. Masopust, D. & Soerens, A. G. Tissue-resident T cells and other resident leukocytes. Annu. Rev. Immunol. 37, 521–546 (2019).
174. Turner, D. L. et al. Lung niches for the generation and maintenance of tissue-resident memory T cells. Mucosal Immunol. 7, 501–510 (2014).
175. Oja, A. E. et al. Trigger-happy resident memory CD4+ T cells inhabit the human lungs. Mucosal Immunol. 11, 654–667 (2018).
176. Shenoy, A. T. et al. Lung CD4+ resident memory T cells remodel epithelial responses to accelerate neutrophil recruitment during pneumonia. Mucosal Immunol. 13, 334–343 (2020).
177. Broggi, A. et al. Type III interferons disrupt the lung epithelial barrier upon viral recognition. Science 369, 706– 712 (2020).
178. Major, J. et al. Type I and III interferons disrupt lung epithelial repair during recovery from viral infection. Science 369, 712–717 (2020)
179. 112. Prescott, S. L., Umans, B. D., Williams, E. K., Brust, R. D. & Liberles, S. D. An airway protection program revealed by sweeping genetic control of vagal afferents. Cell 181, 574–589.e14 (2020).
180. Ordovas-Montanes, J. et al. The regulation of immunological processes by peripheral neurons in homeostasis and disease. Trends Immunol. 36, 578–604 (2015).
181. Godinho-Silva, C., Cardoso, F. & Veiga-Fernandes, H. Neuro-immune cell units: a new paradigm in physiology. Annu. Rev. Immunol. 37, 19–46 (2019).
182. Solis, A. G. et al. Mechanosensation of cyclical force by PIEZO1 is essential for innate immunity. Nature 573, 69–74 (2019). This article describes how mechanical forces influence innate immune pathways in the lungs via the expression of PIEZO1, a mechanically activated ion channel
183. Coleridge, H. M., Coleridge, J. C. & Schultz, H. D. Afferent pathways involved in reflex regulation of airway smooth muscle. Pharmacol. Ther. 42, 1–63 (1989).
184. Slebos, D. J. et al. Safety and adverse events after targeted lung denervation for symptomatic moderate to severe chronic obstructive pulmonary disease (AIRFLOW). A multicenter randomized controlled clinical trial. Am. J. Respir. Crit. Care Med. 200, 1477–1486 (2019).
185. Talbot, S. et al. Silencing nociceptor neurons reduces allergic airway inflammation. Neuron 87, 341–354 (2015).
186. Trankner, D., Hahne, N., Sugino, K., Hoon, M. A. & Zuker, C. Population of sensory neurons essential for asthmatic hyperreactivity of inflamed airways. Proc. Natl Acad. Sci. USA 111, 11515–11520 (2014)
187. Kistemaker, L. E., Slebos, D. J., Meurs, H., Kerstjens, H. A. & Gosens, R. Anti-inflammatory effects of targeted lung denervation in patients with COPD. Eur. Respir. J. 46, 1489–1492 (2015).
188. Alpizar, Y. A. et al. TRPV4 activation triggers protective responses to bacterial lipopolysaccharides in airway epithelial cells. Nat. Commun. 8, 1059 (2017).
189. Baral, P. et al. Nociceptor sensory neurons suppress neutrophil and γδ T cell responses in bacterial lung infections and lethal pneumonia. Nat. Med. 24, 417–426 (2018). This study shows how lung sensory neurons contribute to innate host defence mechanisms against lethal bacterial infections.
190. Klose, C. S. & Artis, D. Neuronal regulation of innate lymphoid cells. Curr. Opin. Immunol. 56, 94–99 (2019).
191. Huh, J. R. & Veiga-Fernandes, H. Neuroimmune circuits in inter-organ communication. Nat. Rev. Immunol. 20, 217–228 (2020).
192. Germain, R. N. & Huang, Y. ILC2s - resident lymphocytes pre-adapted to a specific tissue or migratory effectors that adapt to where they move? Curr. Opin. Immunol. 56, 76–81 (2019).
193. Puttur, F. et al. Pulmonary environmental cues drive group 2 innate lymphoid cell dynamics in mice and humans. Sci. Immunol. 4, eaav7638 (2019). This study shows that extracellular matrix influences cell dynamics in vivo and in vitro, changing the course of inflammation.
194. Chen, H. et al. Mechanosensing by the alpha6-integrin confers an invasive fibroblast phenotype and mediates lung fibrosis. Nat. Commun. 7, 12564 (2016).
195. Wu, H. et al. Progressive pulmonary fibrosis is caused by elevated mechanical tension on alveolar stem cells.Cell 180, 107–121.e17 (2020).
196. Goldfarbmuren, K. C. et al. Dissecting the cellular specificity of smoking effects and reconstructing lineages in the human airway epithelium. Nat. Commun. 11, 2485 (2020)
197. Duclos, G. E. et al. Characterizing smoking-induced transcriptional heterogeneity in the human bronchial epithelium at single-cell resolution. Sci. Adv. 5, eaaw3413 (2019)
198. Tiến sĩ, Bác sĩ Nguyễn Văn Thành - Phó chủ tịch Hội Hô hấp Việt Nam, Phó chủ tịch Hội Phổi Việt Nam, "Ảnh hưởng của bụi - khói, khí độc hại và phản ứng của phế quản - phổi". Tải bản PDF TẠI ĐÂY
