4 lĩnh vực ứng dụng của sắc ký lỏng hiệu năng cao

Sắc ký lỏng hiệu năng cao (High Performance Liquid Chromatography: HPLC) được ứng dụng rộng rãi trong đa số các lĩnh vực như phân tích thuốc, kiểm nghiệm thực phẩm, phân tích mẫu sinh học, phân tích môi trường,.... Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc thông tin về ứng dụng của phương pháp sắc ký lỏng hiệu năng cao.
1 Ứng dụng của sắc ký lỏng hiệu năng cao trong kiểm nghiệm thuốc
1.1 Phân tích nguyên liệu thuốc hóa dược
Sự phát triển mạnh mẽ của công nghiệp hóa dược cùng nhu cầu không ngừng tăng của lĩnh vực phòng và điều trị bệnh đã thúc đẩy các nghiên cứu tổng hợp, chiết xuất, vi sinh tạo ra nhiều nguyên liệu hóa dược dùng làm thuốc. Để có thể kiểm soát đúng, chặt chẽ chất lượng hoạt chất tinh khiết sử dụng làm nguyên liệu làm thuốc trong cả giai đoạn phát triển thuốc mới, cũng như kiểm nghiệm trước khi đưa vào sản xuất thì HPLC đóng vai trò quan trọng. Khi phân tích nguyên liệu hóa dược dạng tinh khiết, mẫu thử thường được hòa tan và sắc ký trực tiếp, cũng có thể dẫn xuất hóa với thuốc thử thích hợp để có thể phát hiện bằng detector sử dụng. Phương pháp HPLC sử dụng trong kiểm tra chất lượng thuốc bao gồm nguyên liệu làm thuốc, thuốc thành phẩm đều phải thẩm định đạt yêu cầu theo qui định.
1.1.1 Định tính
Ưu điểm của HPLC là có thể tách riêng chất phân tích ra khỏi các chất khác trong hỗn hợp nên có độ chọn lọc cao. Khi định tính bằng HPLC, mẫu thử được thực hiện song song với mẫu chuẩn và so sánh thời gian lưu píc chất phân tích trên sắc ký đồ của mẫu thử và mẫu chuẩn, nếu trùng nhau thì được coi là dương tính. Đối với phép sác ký thực hiện trên hệ thống HPLC với detector UV - DAD/PDA có thể chồng phổ tại thời gian lưu píc chất phân tích trên sắc ký đồ của mẫu thử và mẫu chuẩn, nếu trùng nhau (hệ số phù hợp match hay similary index xấp xỉ bằng 1000) thì được coi là dương tính. Phép thử định tính bằng HPLC thường được kết hợp trong phép thử định lượng hoặc tạp chất. Tuy nhiên, định tính bằng HPLC không đủ đặc hiệu nên trong kiểm nghiệm nguyên liệu hóa dược, ngoài phép thử định tính bằng HPLC còn có các phép định tính khác như phổ IR, phổ UV - Vis, phản ứng hóa học, đo các hằng số vật lý (nhiệt độ nóng chảy, góc quay cực), ... để khẳng định định tính chất đó.
Trong một số trường hợp có thể sử dụng thông số thời gian lưu tương đối để định tính, ví dụ như định danh tạp chất trong phép thử tạp chất mà không cần chuẩn tạp tương ứng.
Ví dụ 1: Định tính doxycyclin hydroclorid trong nguyên liệu doxycyclin hydroclorid bằng HPLC theo Dược điển Việt Nam V. Tiến hành chuẩn bị dung dịch đối chiếu từ chất đối chiếu doxycyclin hydroclorid và dung dịch thử từ mẫu nguyên liệu cần kiểm tra có nồng độ khoảng 0,8 mg/mL trong dung mỗi pha mẫu. Sắc ký dung dịch thử và dung dịch đối chiếu song song ở điều kiện qui định. Phép thử cho phép kết luận dương tính với chất thử nếu píc chính trên sắc ký đồ của dung dịch thử có thời gian lưu tương ứng với píc chính trên sắc ký đồ của dung dịch đối chiếu.
Ví dụ 2: Định danh các tạp chất trong nguyên liệu levofloxacin hydroclorid bằng HPLC theo Dược điển Việt Nam V. Tiến hành chuẩn bị dung dịch thử có nồng độ khoảng 1 mg/ml trong dung môi pha mẫu. Sắc ký dung dịch thử ở điều kiện qui định. Định tính píc ứng với levofloxacin hydroclorid trên sắc ký đồ mẫu thử dựa vào so sánh với thời gian lưu của píc trên sắc ký đồ mẫu chuẩn levofloxacin hydroclorid nồng độ khoảng 1 mg/ml được tiến hành song song. Định danh các tạp chất trong mẫu thử dựa vào thời gian lưu tương đối của các píc tạp so với píc levofloxacin hydroclorid như sau:
| Tên chất | Thời gian lưu tương đối |
| N-Desmethyl levofloxacin | 0,47 |
| Diamin derivative | 0,52 |
| Levofloxacin N- oxid | 0,63 |
| 9- Desfluoro levofloxacin | 0,73 |
| Levofloxacin | 1,0 |
| D - Isomer | 1,23 |
1.1.2 Định lượng
Nguyên liệu hóa dược có thể định lượng bằng nhiều phương pháp khác nhau từ cổ điển như chuẩn độ, trong đó thường sử dụng phương pháp chuẩn độ trong môi trường khan nước đối với thuốc có bản chất acid yếu hoặc base yếu. Ưu điểm của phương pháp này là rẻ tiền, đơn giản. Phương pháp quang phổ cũng được sử dụng để định lượng một số dược chất có khả năng hấp thụ ánh sáng tử ngoại. Tuy nhiên, cả hai phương pháp này đều là định lượng tổng các chất trong mẫu thử bao gồm chất cần định lượng và cả những chất có đặc tính tương tự chất mục tiêu như tạp chất liên quan, tạp phân hủy nên kết quả không phản ánh đúng lượng hoạt chất có trong mẫu. Đặc biệt, khi các dược chất hóa dược ở dạng đơn đồng phân (có thể có tạp isomer) thì không thể sử dụng hai phương pháp trên mà phải sử dụng phương pháp vừa tách, vừa định tính và định lượng với độ chọn lọc, độ chính xác cao như HPLC hoặc điện di mao quản. Trong định lượng bằng HPLC, phương pháp chuẩn ngoại, chuẩn hóa 1 điểm thường dùng, nồng độ dung dịch cao khoảng 0,1 - 1 mg/ml, cá biệt có trường hợp tới 5 mg/ml, dụng dịch thử và dung dịch chuẩn nồng độ tương đương, yêu cầu nồng độ dung dịch thử nằm trong khoảng ± 20% so với nồng độ chuẩn. Các chất phân tích có thể được định lượng trực tiếp hoặc dẫn chất. Kiểu LC - RP với chế độ đẳng dòng, phát hiện bằng detector UV được sử dụng phổ biến. Kết quả hàm lượng được tính toán dựa trên diện tích píc chất phân tích trên sắc ký đồ của dung dịch chuẩn, dung dịch thử, nồng độ chất phân tích trong dung dịch chuẩn và độ pha loãng của dung dịch thử. Trong các chuyên luận của Dược điển Việt Nam, dược điển các nước (USP - Mỹ, BP - Anh, CP - Trung Quốc,...) dược chất các nhóm thuốc có tác dụng dược lý khác nhau đều có thể được định lượng bằng HPLC.
Trong trường hợp thiết lập chuẩn sơ cấp (chuẩn gốc) sẽ ưu tiên sử dụng HPLC để định lượng, phương pháp chuẩn hóa diện tích được dùng để tính toán hàm lượng hoạt chất trong mẫu phân tích. Kết luận hàm lượng sẽ còn phải tính đến hàm lượng chất vô cơ trong mẫu thử dựa vào giá trị tro và các chất bay hơi.
Bảng 2.11 trình bày một số kháng sinh dạng nguyên liệu được định lượng bằng HPLC theo Dược điển Việt Nam V và Dược điển Mỹ.
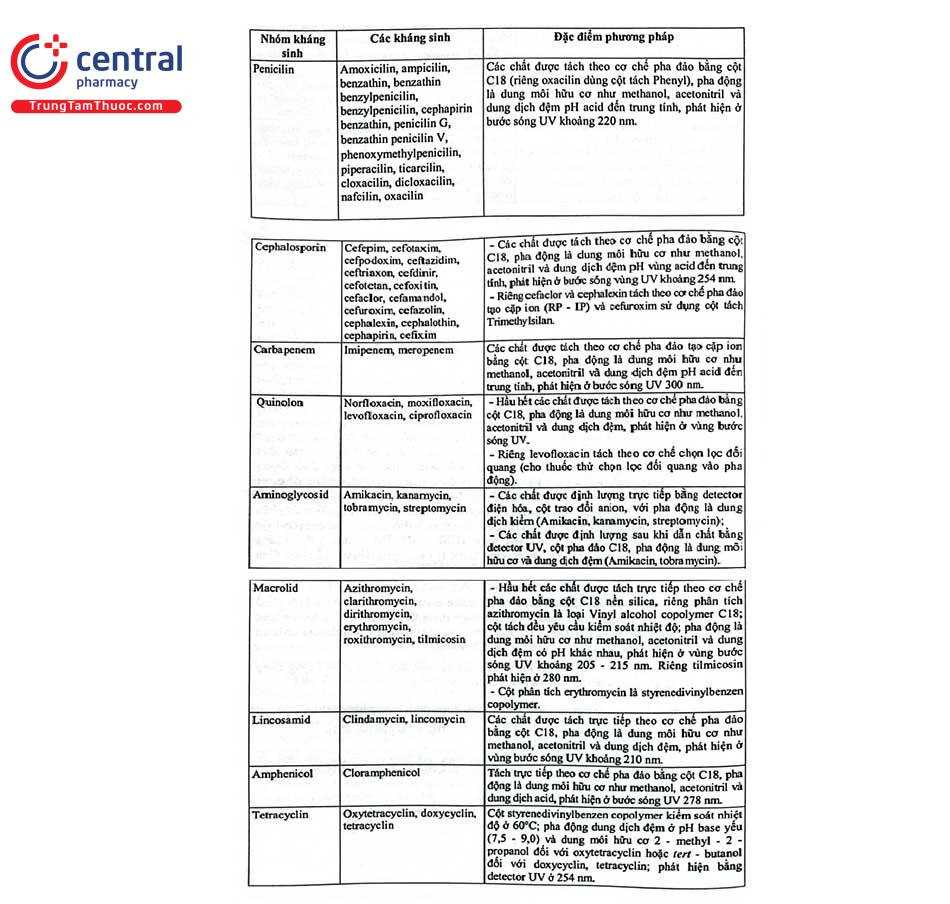
Đa số các nguyên liệu hóa dược định lượng bằng phương pháp HPLC được định lượng trực tiếp với detector UV. Tuy nhiên, có một số chất trong cấu trúc không có nhóm mang màu (vòng thơm, nối đôi liên hợp như các kháng sinh nhóm macrolid). Để phân tích bằng HPLC với detector UV phải dẫn chất với thuốc thử tạo sản phẩm có khả năng hấp thụ UV. Khi tiến hành phân tích cần phải thực hiện một mẫu trắng để loại các píc ứng với thuốc thử trong quá trình xử lý số liệu. Hình 2.29 trình bày sắc ký đồ định lượng tobramycin trong nguyên liệu theo Dược điển Việt Nam V và USP 43. Thí nghiệm được thực hiện như sau: pha dung dịch chuẩn từ chất đối chiếu tobramycin và dung dịch thử từ mẫu nguyên liệu cần kiểm tra có nồng độ chính xác khoảng 0,22 mg/mL trong dung môi pha mẫu. Dẫn chất hóa các dung dịch chuẩn, dung dịch thử và dung dịch mẫu trắng (nước) theo điều kiện qui định thu được dung dịch dẫn xuất chuẩn, dung dịch dẫn xuất thử, dung dịch dẫn xuất mẫu trắng. Sắc ký các mẫu ở điều kiện qui định trong chuyên luận, píc của tobramycin có thời gian lưu khoảng 10,5 phút.
1.1.3 Xác định tạp chất
Hai vấn đề cơ bản nhất của thuốc dùng trong điều trị là an toàn và hiệu quả. Các tạp chất có trong dược chất khi dùng ở liều điều trị của thuốc có thể gây ra tác dụng phụ bởi độc tính của nó, ảnh hưởng đến sự an toàn của thuốc. Các tạp chất được tìm thấy trong một sản phẩm thuốc có thể có tác dụng dược lý và (hoặc) độc tính không mong muốn. Thuốc có chất lượng và đảm bảo về độ an toàn khi các tạp chất được kiểm soát. Do đó, vấn đề quan trọng nhất - theo quan điểm của một nhà hóa học phân tích - trong quá trình phát triển phương pháp phân tích là có thể phát hiện được các tạp chất có thể có.
Các tạp chất liên quan được tìm thấy trong hoạt chất có thể xuất hiện trong các bước tổng hợp, từ nguyên liệu ban đầu, chất trung gian hoặc tạp chất từ nguyên liệu ban đầu phản ứng (tất cả những chất này được gọi là sản phẩm phụ tổng hợp). Khi một hoạt chất được dùng sản xuất thuốc, phải xác định các sản phẩm phân hủy có thể tạo thành (theo hướng dẫn của ỊCH khắc nghiệt hóa điều kiện bảo quản). Lý do quan trọng nhất phải kiểm soát tạp nhằm có một sản phẩm chất lượng đáp ứng yêu cầu an toàn và hiệu quả của thuốc. Mối quan hệ giữa các sản phẩm phụ tổng hợp, các sản phẩm phân hủy và các chất liên quan là các chất liên quan có chứa tổng các sản phẩm phụ tổng hợp (có nguồn gốc từ tổng hợp hóa học khổng thay đổi theo thời gian) và các sản phẩm phân hủy (tăng theo thời gian và thay đổi trong điều kiện bảo quản khác nhau). Tuy nhiên, đôi khi các sản phẩm phụ tổng hợp của hoạt chất cũng có thể là sản phẩm phân hủy của thuốc.
Tạp chất trong nguyên liệu thuốc có thể là tạp chất vô cơ hoặc hữu cơ và được xác định bằng nhiều phương pháp khác nhau như hóa học, quang phổ, sắc ký. Phương pháp chủ yếu dùng để xác định tạp chất liên quan và tạp chất phân hủy của chất hữu cơ là nhóm phương pháp sắc ký, nhiều nhất là HPLC. Xác định tạp của nguyên liệu hóa dược có thể là tạp chất đã biết hoặc tạp chất chưa biết. Tạp định danh được xác định dựa vào so sánh với thời gian lưu của píc tạp chuẩn làm trong cùng điều kiện hoặc dựa vào thời gian lưu tương đối so với chất chính. Khi tính kết quả hàm lượng tạp dựa vào đáp ứng của chất chính có thể dùng hệ số hiệu chỉnh do đáp ứng của chất chính và tạp khác nhau. Xác định tạp chất bằng HPLC có thể thực hiện theo phương pháp chuẩn hóa diện tích, pha loãng hoặc sử dụng chuẩn tạp.
Đối với yêu cầu xác định giới hạn tạp, nếu sử dụng phương pháp pha loãng thì trong đánh giá kết quả sẽ yêu cầu “Bất cứ píc tạp nào trong sắc ký đồ thu được từ dung dịch (1) không được có chiều cao hoặc diện tích píc lớn hơn píc trong sắc ký đồ thu được từ dung dịch (2)”. Dung dịch (1) và dung dịch (2) có chứa chất kiểm tra ở nồng độ cao và thấp tương ứng. Còn nếu sử dụng chuẩn tạp thì phải đáp ứng yêu cầu “Bất cứ píc nào tương đương với [x] trong sắc ký đỗ thu được từ dung dịch (1) không được có chiều cao hoặc diện tích píc lớn hơn píc chính trong sắc ký đồ thu được từ dung dịch (2)”. Dung dịch (1) có chứa chất kiểm tra và dung dịch (2) có chứa tạp chất đã biết.
Khi tiến hành xác định tạp chất, cần chú ý đến độ nhạy của phương pháp và khả năng tách của hệ thống. Khi đánh giá độ phù hợp của hệ thống, trong các chuyên luận thường yêu cầu đánh giá độ phân giải, tỷ lệ đỉnh - hõm và độ nhạy của điều kiện phân tích. Mức độ nhạy yêu cầu có thể chấp nhận là không nhỏ hơn 0,05% hoặc 0,025%.
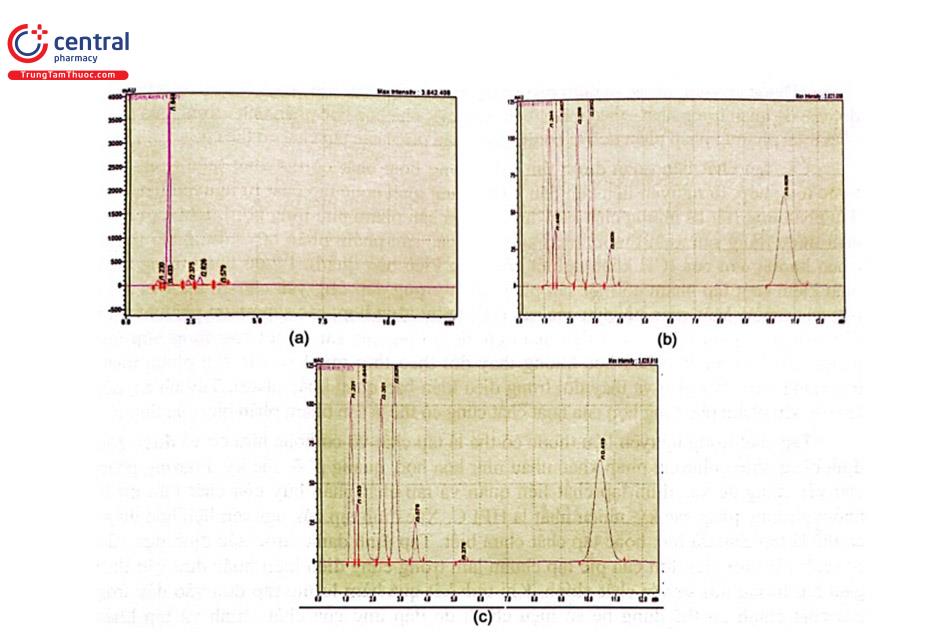
Hình 2.30 trình bày sắc ký đồ của phép thử xác định tạp chất định danh và tạp chất liên quan theo phương pháp sử dụng tạp chất chuẩn và pha loãng của nguyên liệu nifedipin theo DĐVN 5. Dung dịch thử được chuẩn bị bằng cách hòa tan 0,200 g chế phẩm trong vừa đủ 50,0 ml bằng dung môi pha mẫu. Dung dịch đối chiếu là hỗn hợp chứa tạp chất chuẩn A của nifedipin, tạp chất chuẩn B của nifedipin cùng có nồng độ 0,004 mg/mL và nifedipin (pha loãng 1000 lần từ dung dịch thử).
Giới hạn đánh giá:
Tạp chất A: Diện tích píc tạp chất A không được lớn hơn diện tích pic tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (0,1%).
Tạp chất B: Diện tích pic tạp chất B không được lớn hơn diện tích píc tương ứng thu được trên sắc ký đồ của dung dịch đối chiếu (0,1%).
Tạp chất khác: Với mỗi tạp chất, diện tích píc không được lớn hơn diện tích pic nifedipin trên sắc ký đồ của dung dịch đối chiếu (0,1%).
Tổng các tạp chất không được quả 0,3%.
Bỏ qua những píc có diện tích nhỏ hơn 0,1 lần diện tích pic nifedipin thu được trên sắc ký đồ của dung dịch đối chiếu (0,01%)
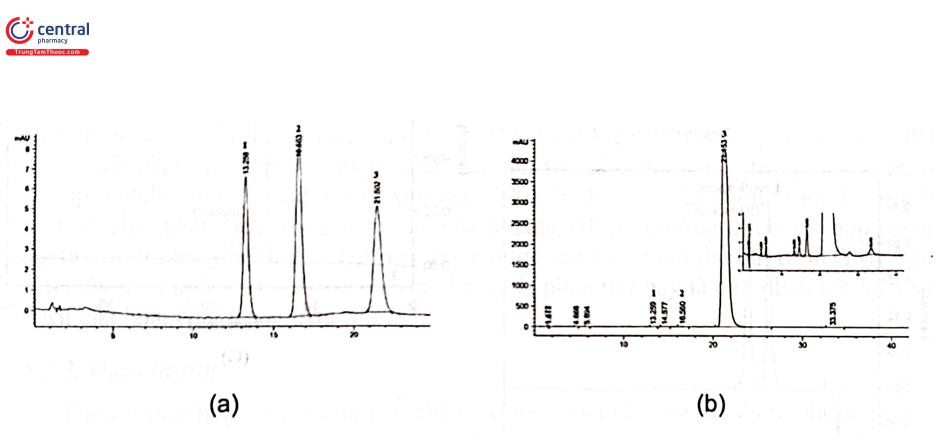
Tách đồng phân đối quang được ứng dụng nhằm kiểm tra đúng dạng đồng phân của chất phân tích khi kiểm tra chất lượng. Ví dụ định lượng và thử tạp chất của levofloxacin trong nguyên liệu theo Dược điển Việt Nam V và USP 43. Thí nghiệm được thực hiện như sau: Pha dung dịch chuẩn từ chất đối chiếu levofloxacin và dung dịch thử từ mẫu nguyên liệu cần kiểm tra có nồng độ chính xác khoảng 1 mg/ml trong dung môi pha mẫu. Song song chuẩn bị dung dịch chuẩn ofloxacin. Các chất được tách bằng cột C18 với pha động gồm amoni acetat, Đồng Sulfat, L - isoleucin và methanol, phát hiện tại UV 360 nm. Kết quả cho thấy 2 đồng phân dạng R - và S - (levofloxacin) của Ofloxacin được tách hoàn toàn và tạp isomer của levofloxacin có thể phát hiện được rõ ràng (Hình 2.31).
Điều kiện sắc ký này cũng được sử dụng để định lượng levofloxacin trong thành phẩm dạng viên nén.
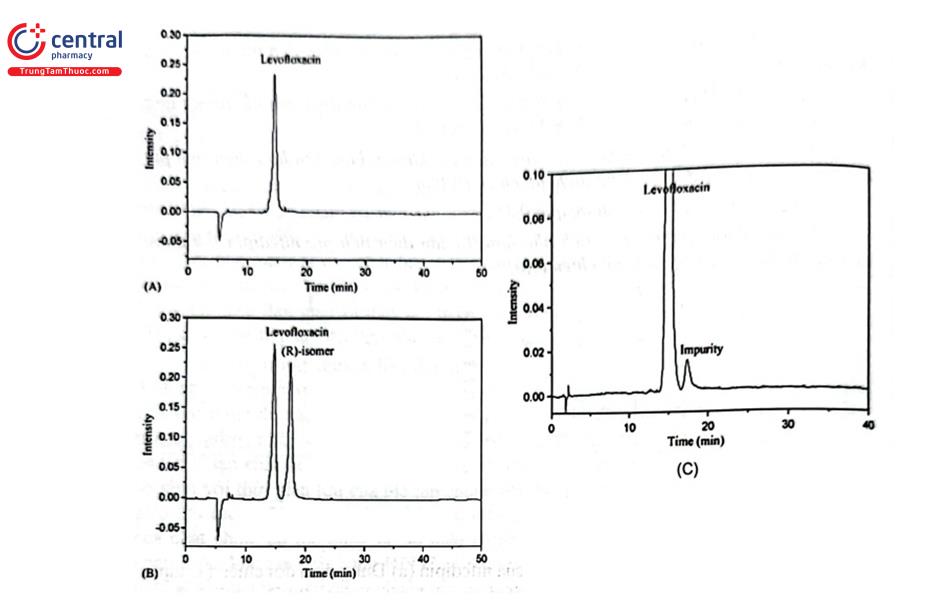
Một khó khăn trong phép thử tạp chất đó là phải có chất chuẩn tạp và/ hoặc các chất chuẩn dùng để đánh giá khả năng tách của hệ thống sắc ký đảm bảo các tạp được tách theo yêu cẫu (chuẩn để chuẩn bị dung dịch đánh giá độ phù hợp hệ thống sắc ký) Hiện nay, số chuyền luận yêu cầu sử dụng các tạp này ngày càng tăng lên, song Số lượng chất chuẩn tạp của 2 đơn vị thiết lập và cung cấp chuẩn phục vụ kiểm nghiệm thuốc ở Việt Nam là Viện Kiểm nghiệm thuốc Trung ương và Viện Kiểm nghiệm thuốc thành phố Hồ Chí Minh rất khiêm tốn. Các chuẩn tạp thường có nguồn gốc của các nhà sản xuất hóa chất lớn như Merck - Đức, Sigma - Aldrich, Mỹ, Hội đồng Dược điển Mỹ, ... Giá chuẩn tạp rát cao, thời gian đặt hàng lâu, có những chuẩn chỉ 1 - 5 mg/ lọ mà giá lên tới hơn 10 triệu đồng và thời gian đặt hàng có thể từ 1 - 3 tháng làm hạn chế khả năng thực hiện chỉ tiêu tạp chất trong kiểm nghiệm thuốc. Do đó, cần đẩy mạnh công tác thiết lập chuẩn tạp để phục vụ kịp thời công tác kiểm tra chất lượng thuốc với giá cả hợp lý.
1.2 Phân tích thành phẩm thuốc hóa dược
Chế phẩm thuốc được bào chế ở nhiều dạng khác nhau như viên nén, viên nang, thuốc mỡ, thuốc tiêm, dung dịch, hỗn dịch, ... Thành phần một đơn vị chế phẩm ngoài dược chất (một hoặc nhiều dược chất) còn có tá dược, chất bảo quản. Do đó, khi sử dụng phương pháp tách như HPLC để phân tích phải lựa chọn được điều kiện mà không có sự cản trở tại thời gian lưu của dược chất từ bất kỳ tá dược, tạp chất, vỏ nang hoặc dung môi. Song song với điều kiện sắc ký thì bước chuẩn bị mẫu cũng vô cùng quan trọng nhằm loại tá dược, chất cản trở. Mẫu thử được đồng nhất, cân hoặc lấy một thể tích chính xác mẫu thử, hòa tan pha loãng tới thể tích thích hợp được qui định trong chuyên luận, lọc hoặc ly tâm thu được mẫu sắc ký. Các chỉ tiêu chất lượng phân tích bằng HPLC là định tính, định lượng hàm lượng hoạt chất hoặc xác định hoạt chất giải phóng trong đánh giá độ hòa tan, độ đồng đều hàm lượng, xác định tạp chất.
1.2.1 Định tính
Tương tự như đối với kiểm nghiệm nguyên liệu. Mẫu thử được thực hiện song song với mẫu chuẩn và so sánh thời gian lưu píc chất phân tích trên sắc ký đồ của mẫu thử và mẫu chuẩn, nếu trùng nhau thì được coi là dương tính. Đối với phép sắc ký thực hiện trên hệ thống HPLC với detector UV - DAD/PDA có thể chồng phố tại thời gian lưu píc chất phân tích trên sắc ký đồ của mẫu thử và mẫu chuẩn, nếu trùng nhau (hệ số phù hợp match/ similary index xấp xỉ bằng 1000) thì được coi là dương tính. Phép thử định tính chất phân tích trong mẫu chế phẩm bằng HPLC thường được kết hợp trong phép thử định lượng hoặc tạp chất. Do thuốc được sản xuất tuân thủ qui trình GMP nên trong một số chuyên luận có thể chỉ cần duy nhất phép thử này là cho phép kết luận sự có mặt của hoạt chất trong mẫu thử.
1.2.2 Định lượng
Định lượng hoạt chất trong chế phẩm có một hoạt chất có thể dùng phương pháp như quang phổ, chuẩn độ thể tích, nhưng HPLC ngày càng được sử dụng rộng rãi trong định lượng hoạt chất trong chế phẩm, đặc biệt đối với chế phẩm có nhiều hoạt chất, nồng độ chất phân tích thấp, nền mẫu phức tạp như viên nang mềm, thuốc mỡ. Trong đánh giá độ hòa tan của các chế phẩm bào chế dạng rắn, phương pháp HPLC được ưu tiên lựa chọn nếu các tá dược gây cản trở khi phát hiện dược chất hoặc khi có nhiều dược chất (chế phẩm đa thành phần) trong một thuốc. Vì quá trình phân tích độ hòa tan có thể yêu cầu thời gian dài do phải thực hiện sáu đơn vị mẫu cho 1 lần thử và có thể có nhiều điểm thời gian lấy mẫu kiểm tra, nên thời gian phân tích ngắn phải được tối nhanh chóng xác định kết quả. Nếu có sẵn phương pháp HPLC thời gian phân tích ngắn khi định lượng thì có thể sử dụng để phân tích mẫu trong phép thử độ hòa tan. Đối với đánh giá độ đồng đều hàm lượng, yêu cầu bắt buộc phải thực hiện chỉ tiêu này trong kiểm nghiệm chế phẩm có hàm lượng hoạt chất thấp, do đó phương pháp HPLC với độ nhạy cao là một lựa chọn phù hợp. Cũng như đối với phép thử độ hòa tan, số lượng mẫu lớn nên cũng cần điều kiện HPLC thực hiện trong thời gian phân tích ngắn. Thông thường điều kiện định lượng của phép thử chỉ tiêu định lượng, độ hòa tan, độ đồng đều hàm lượng của 1 chế phẩm là tương tự nhau.
Sắc ký lỏng pha đảo với detector UV được sử dụng phổ biến, một số trường hợp có thể sử dụng detector huỳnh quang hoặc điện hóa tùy thuộc vào tính chất của chất phân tích và nỗng độ mẫu thử. Phương pháp chuẩn ngoại thường được sử dụng, nhưng có một số trường hợp sử dụng phương pháp chuẩn nội. Dung dịch mẫu thử và dung dịch mẫu chuẩn thường có nồng độ xấp xỉ bằng nhau, trường hợp cá biệt có thể khác nhau nhưng khi sẽ tiêm thể tích với tỷ lệ khác nhau tương ứng để đáp ứng phân tích tích trong mẫu thử và mẫu chuẩn sẽ nằm trong khoảng xác định của phương pháp (đáp ứng tính tuyến tính, độ đúng, độ chính xác). Phép định lượng yêu cầu làm lặp lại tối thiểu 2 lần với mẫu chuẩn và 3 lần với mẫu thử, từ đó xác định hàm lượng trung bình để báo cáo kết quả với RSD thường không được lớn hơn 2,0%.
Ví dụ 1. Định lượng Atropin sulfat trong dung dịch thuốc nhỏ mắt Atropin sulfat 0,01% theo Dược điển Anh bằng HPLC. Sử dụng cột sắc ký C18 (4,6 mm × 15 cm; 5 um), pha động gồm dung dịch natri acetat 0,01 M và natri dioctyl sulfosuccinat 0,005 M trong methanol 60%, điều chỉnh đến pH 5,5 bằng Acid acetic băng, phát hiện tại bước sóng UV 257 nm. Thể tích tiêm mẫu thử (dung dịch chế phẩm) là 100 ul, còn mẫu chuẩn (dung dịch atropin sulfat chuẩn 0,05%) tiêm 20 ul.
Ví dụ 2. Định lượng hoạt chất giải phóng khi thử độ hòa tan viên nén gồm ethinyl Estradiol 30 ug và Levonorgestrel 150 ug theo USP bằng HPLC. Mẫu thử là dung dịch sau khi thử độ hòa tan mỗi viên nén trong 500 ml môi trường hòa tan; mẫu chuẩn là dung dịch ethinyl estradiol và levonorgestrel chuẩn nồng độ chính xác khoảng 0,06 ng/mL và 0,3 uL). Điều kiện sắc ký gồm cột C8 (4 mm × 15 cm; 5 um), pha động acetonitril - nước (6:4), thể tích tiêm mẫu 100 uL, phát hiện tại bước sóng UV 247 nm đối với levonorgestrel và detector huỳnh quang với bước sóng kích thích 285 nm, bước sóng phát xạ 310 nm đối với ethinyl estradiol.
Ví dụ 3. Định lượng Paracetamol và cafein trong viên nén theo Dược điển Việt Nam 5 bằng HPLC được thực hiện theo phương pháp chuẩn nội. Hàm lượng paracetamol và cafein trong chế phẩm có các tỷ lệ 500 mg và 30 mg hoặc 500 mg và 60 mg. Dung dịch thử được chuẩn bị từ chế phẩm viên nén, dung dịch chuẩn từ paracetamol đối chiếu và cafein đối chiếu, mỗi dung dịch này được thêm cùng một lượng chuẩn nội Acid benzoic. Các chất được tách bằng cột C18 (4 mm × 10 cm; 5 um) để ở nhiệt độ 45 ± 1°C, pha động là hỗn hợp nước - methanol - acid acetic băng (69 : 28 : 3). phát hiện tại UV 275 nm. Tính hàm lượng paracetamol (CgH9NO2), cafein (CgHoN,Oz) có trong một đơn vị chế phẩm so với lượng ghi trên nhãn dựa vào tỷ số diện tích pic giữa paracetamol và diện tích píc chuẩn nội, tỷ số diện tích píc giữa cafein và diện tích píc chuẩn nội trên sắc ký đồ của dung dịch thử, dung dịch chuẩn, nồng độ paracetamol, nồng độ cafein trong dung dịch chuẩn và lượng cân mẫu thử.
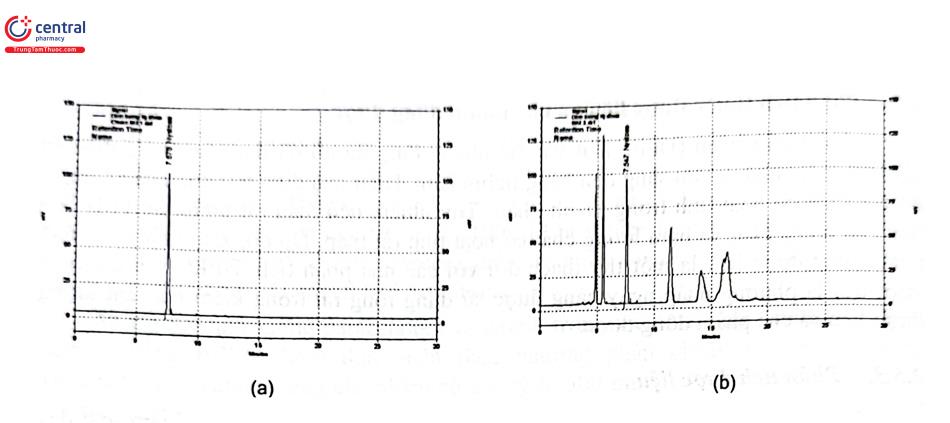
1.2.3 Tạp chất
Tiến hành tương tự như đối với nguyên liệu. Tuy nhiên, khi sử dụng phương pháp chuẩn hóa diện tích cần loại píc của tá dược có thể có bằng cách thực hiện song song với mẫu Placebo. Điều này rất dễ dàng với phòng kiểm nghiệm của đơn vị sản xuất chế phẩm nhưng khó đối với phòng kiểm nghiệm phục vụ quản lý nhà nước như Viện Kiểm nghiệm thuốc, trung tâm kiểm nghiệm thuốc, do đó cần phải lưu ý khi kết luận kết quả kiểm nghiệm. Đối với phương pháp pha loãng có thể loại trừ ảnh hưởng của tá được tới đánh giá kết quả, thường thực hiện pha loãng mẫu chuẩn tới giới hạn yêu cầu và tính kết quả các tạp xuất hiện trên mẫu thử dựa vào píc chất phân tích trong dung dịch chuẩn.
1.3 Phân tích thuốc dược liệu và chế phẩm đông dược
Thuốc cổ truyền (Dược liệu và chế phẩm đông dược) đã được sử dụng điều trị bệnh và kiểm chứng lâm sàng qua hàng nghìn năm. Hiệu quả điều trị bệnh của chúng là do các chất có hoạt tính trong thành phần. Tuy nhiên, nền mẫu rất phức tạp vì là hỗn hợp của nhiều chất và hàm lượng chất có hoạt tính rất thấp. Do đó, xác định các thành phần trong dược liệu là một thử thách đối với các nhà phân tích. HPLC với ưu điểm vượt trội là phương pháp ngày càng được sử dụng rộng rãi trong kiểm tra chất lượng dược liệu và chế phẩm đông dược.
1.3.1 Phân tích dược liệu
Nhiều loại thuốc thảo dược được thu hái tự nhiên hoặc chưa được kiểm soát đầy đủ trong quá trình canh tác; ngay cả khi các loại thuốc thảo dược có nguồn gốc canh tác rõ ràng, thì việc chúng trồng ở các vùng khí hậu, thổ nhưỡng đa dạng và được thu hoạch trong điều kiện khác nhau có thể ảnh hưởng tới chất lượng của chúng. Tính xác thực và tính đồng nhất chất lượng thảo dược của một loài thực vật xác định không thể được đảm bảo do đó cần thiết phải kiểm soát chất lượng thông qua chất đánh dấu (fingerprint) trong dược liệu. Chất đánh dấu lý tưởng nhất là chất tạo nên hoạt tính sinh học của thảo dược nhưng cũng có thể là chất xác định được và có hàm lượng lớn trong dược liệu.
Các hoạt chất trong thảo dược có thể chia thành các nhóm alcaloid, Flavonoid, courmarin, Saponin, polysaccharid, phenol, ... Tùy theo cấu trúc của chất phân tích mà sử dụng detector khác nhau trong phát hiện. LC - RP với detector UV được sử dụng khá phổ biến trong phân tích các chất có nối đôi liên hợp, vòng thơm trong cấu trúc như alcaloid, flavonoid; còn detector tán xạ ánh sáng bay hơi hoặc detector khúc xạ được sử dụng đối với chất phân tích không có nhóm mang màu như saponin, polysaccharid, Phương pháp chuẩn ngoại so sánh điểm thường được sử dụng trong định lượng, nhưng có một số trường hợp sử dụng phương pháp đường chuẩn dựa vào mối tương quan giữa diện tích píc và nồng độ chất phân tích hoặc logarith diện tích píc và logarith nồng độ chất phân tích. Do nền mẫu phức tạp là hỗn hợp của nhiều nhóm chất với độ phân cực rất khác nhau nên thời gian phân tích thường dài có thể lên tới 100 phút/ lần sắc ký. Rửa giải gradient thường được áp dụng để tăng tốc độ rửa giải các thành phần không phải đối tượng cần tách trong mẫu và làm sạch cột trước khi thực hiện lần tiêm mẫu tiếp theo.
Dược Điển Việt Nam V có 54 trong 340 chuyên luận dược liệu yêu cầu định lượng chất đánh dấu bằng HPLC. Nếu so sánh với Dược điển Việt Nam IV thì số dược liệu được kiểm nghiệm dựa vào định lượng hoạt chất là chất đánh dấu tăng lên gấp nhiều lần. Tuy nhiên, các chuyên luận về dược liệu của Dược điển Trung Quốc, Hồng Kông, Hàn Quốc, Thái Lan yếu cầu định lượng chất đánh dấu và số chất đánh dấu cần định lượng nhiều hơn so với Dược điển Việt Nam. Ví dụ dược liệu Ba Kích trong Dược điển Việt Nam chưa yêu cầu định lượng chất đánh dấu nhưng Dược điển Trung Quốc 2015 có yêu cầu này. Hình 2.33 trình bẫy sắc ký đồ định lượng nystose trong Ba kích bằng HPLC với detector ELSD, với pha động methanol - nước (3 : 97), nồng độ định lượng khoảng 0,2 - 0,6 mg/mL. Đường chuẩn được xây dựng dựa trên kết quả tiêm mẫu dung dịch chuẩn 0,2 mg/mL thể tích 10 JL và 30 L. Kết quả tính toán dựa trên đường chuẩn logarith diện tích pic và logarith nồng độ chất phân tích của 2 điểm chuẩn.
Một khó khăn trong kiểm nghiệm dược liệu và các chế phẩm đông dược đó là chất chuẩn. Số lượng chất chuẩn dùng trong kiểm nghiệm dược liệu của 2 đơn vị thiết lập và cung cấp chuẩn phục vụ kiểm nghiệm thuốc ở Việt Nam rất ít do chuẩn dược liệu thường không thiết lập bằng con đường tổng hợp mà thông qua con đường chiết xuất và tinh chế từ dược liệu. Quá trình tinh chế thường sử dụng sắc ký cột cổ điển, một số qui trình sử dụng HPLC điều chế để tinh chế nên lượng chuẩn thu được không nhiều, chi phí lớn. Giá chuẩn kiểm nghiệm dược liệu rất cao, có những chuẩn chỉ 5 - 10 mg/ lọ mà giá lên tới hơn 10 triệu đồng. Hầu hết chất chuẩn dùng trong kiểm nghiệm dược liệu thường mua từ Trung Quốc.
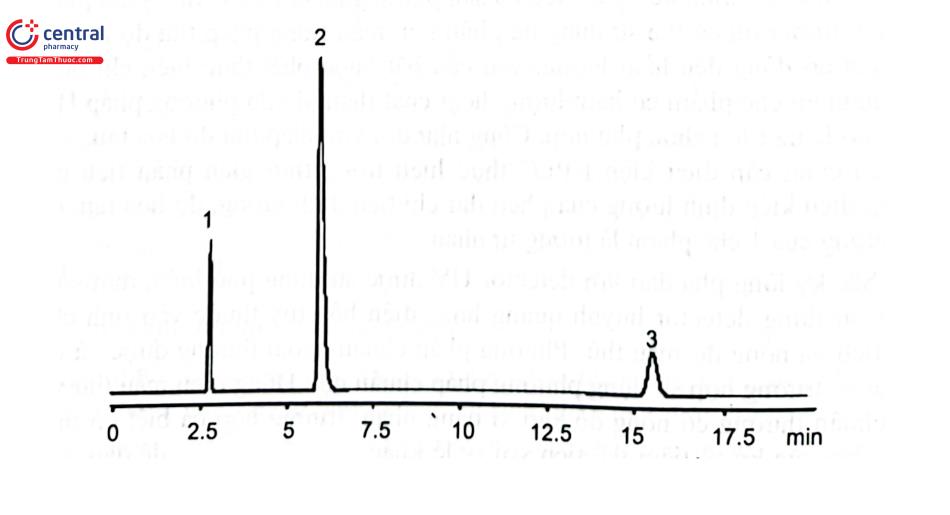
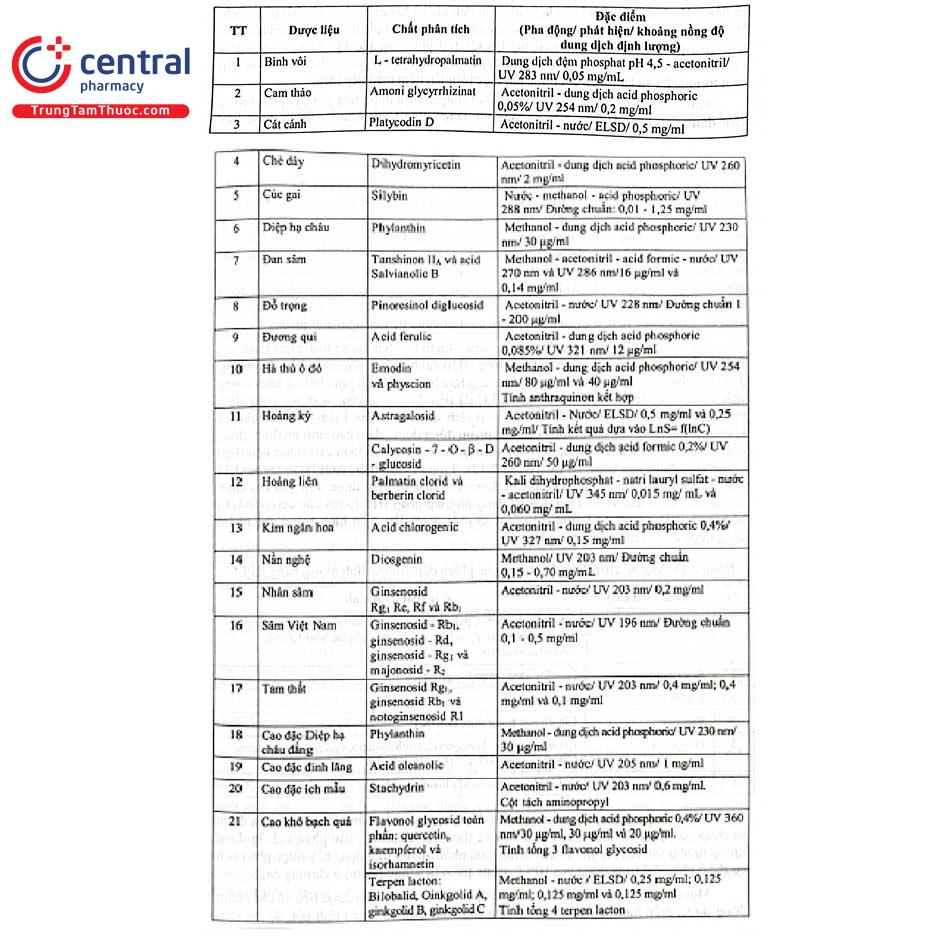
Bảng 2.12 trình bày tóm tắt điều kiện định lượng một số chất đánh dấu trong dược liệu bằng HPLC trong Dược điển Việt Nam V, Dược điển Trung Quốc. Hầu hết các qui trình định lượng đều sử dụng cột sắc ký C18 (25 cm × 4 mm; 5 um) hoặc C18 (25 cm × 4,6 mm; 5 um).
1.3.2 Phân tích chế phẩm đông dược
Chế phẩm đông dược hay thuốc từ dược liệu là sản phẩm bào chế gồm các nguyên liệu từ thiên nhiên được kết hợp dựa trên các nguyên tắc ngũ hành, âm dương, hoạt tính dược lý,... Thuốc đông dược được bào chế theo nguyên lý và lý luận trong Đông y - Y học cổ truyền có tác dụng chữa bệnh, cải thiện sức khỏe, chức năng cơ thể. Các sản phẩm thuốc đông dược hiện nay được sản xuất trong dây chuyền hiện đại, đạt tiêu chuẩn GMP Đông Dược và được kiểm soát nghiêm ngặt từ khâu tuyển chọn nguyên liệu đến nghiên cứu, phân tích và chứng minh lâm sàng hiệu quả của thuốc. Chế phẩm đông dược có dạng bào chế như cao, viên nén, viên nang, hoàn, siro, hầu hết được sản xuất từ nhiều dược liệu, một số ít sản phẩm có một dược liệu trong thành phần.
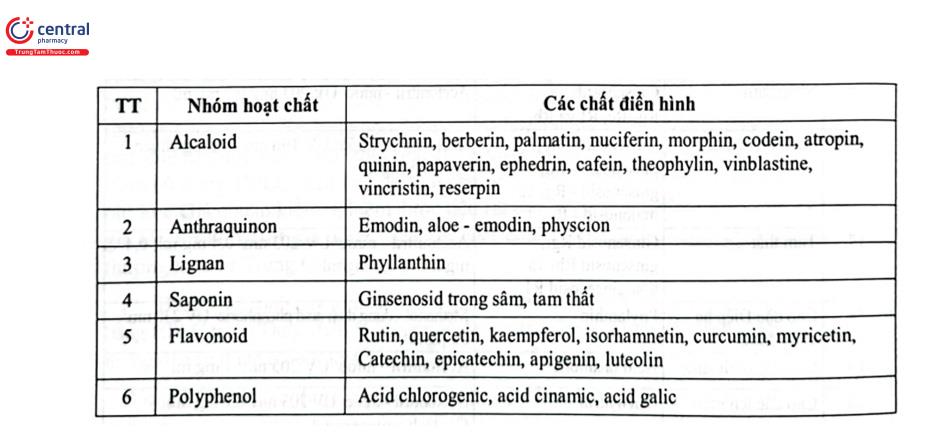
Các chế phẩm đông dược được kiểm soát - kiểm tra chất lượng trong quá trình sản xuất, xuất xưởng và lưu hành trên thị trường. Bên cạnh những phương pháp thường sử dụng như hiển vi, định tính dựa trên phản ứng hóa học, xác định tạp chất và hàm lượng chất chiết bằng phương pháp cân thì HPLC đã dần được yêu cầu sử dụng trong định tính, định lượng các chất đánh dấu có hoạt tính dược lý nhằm kiểm tra chặt chẽ và không ngừng nâng cao chất lượng của chế phẩm đông dược, đảm bảo tính ổn định, đồng nhất về hiệu lực điều trị. Trong DĐVN V có 6 trong 7 chuyên luận cao dược liệu định lượng chất đánh dấu bằng HPLC (Bảng 2.12). Trong Dược điển Anh hoặc Dược điển Mỹ, các cao định chuẩn, các chế phẩm được sản xuất từ thảo dược đều yêu cầu định lượng chất đánh dấu và hầu hết đều sử dụng phương pháp HPLC với các detector UV, ELSD và RID. Bảng 2.13 trình bày một số chất đánh dấu điển hình trong chế phẩm đông dược được định lượng bằng HPLC.
HPLC ứng dụng trong kiểm nghiệm dược liệu và chế phẩm đông dược còn được sử dụng xác định dư lượng hóa chất bảo vệ thực vật. Phương pháp phân tích áp dụng tương tự như với đối tượng rau quả trong thực phẩm do đó ứng dụng này được phân tích cụ thể ở phần 2.7 (ứng dụng của HPLC trong phân tích thực phẩm) ở chương này.
Một ứng dụng nữa của HPLC là xác định độc tố nấm trong dược liệu và chế phẩm đông dược, điển hình là các aflatoxin. Do bản chất mẫu có nhiều tinh bột, độ ẩm cao, ảnh hưởng của điều kiện bảo quản nên có thể mẫu bị nhiễm aflatoxin. Đã có ít nhất 13 loại aflatoxin khác nhau được tạo ra trong thiên nhiên và phần lớn trong số đó được biết là có độc tính cao và gây ung thư, trong đó aflatoxin B1 được xác định là chất độc nhất. Các dược liệu bị nhiễm aflatoxin phải được xác định bằng một phương pháp phân tích đã được thẩm định. Trong Dược điển Anh, Dược điển Việt Nam có trình bày phương pháp xác định aflatoxin B1 bằng HPLC. Aflatoxin B1 trong mẫu thử dược liệu được chiết bằng cột chiết pha rắn ái lực, tách sắc ký bằng cột C18 và được đưa tới buồng phản ứng dẫn chất hóa sau cột với các điều kiện khác nhau như dùng thuốc thử pyridinium hydrobromid perbromid, dựa vào phản ứng quang hóa với đèn UV hơi thủy ngân áp suất thấp 254 nm hoặc brom mới sinh từ phản ứng điện hóa (pin KOBRA). Chất tạo thành sau phản ứng được phát hiện bằng detector huỳnh quang với bước sóng kích thích 366 nm và bước sóng phát xạ 435 nm, thể tích tiêm mẫu 500 L. Nồng độ phân tích của aflatoxin B1 trong dung dịch định lượng nằm trong khoảng 0,05 - 0,4 ng/mL. Giới hạn cho phép dư lượng aflatoxin B1 trong dược liệu không quá 2 ug/kg. Cũng có thể chấp nhận giới hạn cho phép là 4 ug/kg cho tổng lượng các aflatoxin B1, B2, G1 và G2.
2 Ứng dụng của sắc ký lỏng hiệu năng cao phân tích thuốc trong dịch sinh học
2.1 Mục tiêu, tầm quan trọng của phân tích thuốc trong dịch sinh học
Hiện nay, các nhà bào chế không ngừng nghiên cứu cải tiến công thức và phương pháp bào chế để sản xuất thuốc có khả năng phát huy tốt đặc tính dược lý của dược chất, thuận tiện cho người dùng, giảm độc tính. Đa số các thuốc khi vào cơ thể sẽ được hấp thu vào máu, theo máu phân bố đến các cơ quan/ tổ chức trong cơ thể, bị chuyển hóa (chủ yếu ở gan) và thải trừ qua nước tiểu hoặc phân. Thuốc được phân bố tại cơ quan/ tổ chức, mô, đặc biệt là trong dịch sinh học để đi đến nơi phát huy tác dụng. Mặt khác, có một số trường hợp do vô tình hay cố ý thuốc được dùng quá liều gây độc cho cơ thể hoặc có thể dẫn tới tử vong. Nghiên cứu về “cuộc đời” của thuốc trong cơ thể sống được phát triển nhanh chóng và cung cấp các thông tin tương đối toàn diện về hiệu quả điều trị, mức liều, đường dùng cũng như các tác dụng không mong muốn của thuốc. Chuyển hóa thuốc trong cơ thể thường xảy ra ở gan và thận tạo thành các sản phẩm (chất) khác nhau. Có những sản phẩm chuyển hóa của thuốc có hiệu lực tác dụng ngang bằng thậm chí còn mạnh hơn cả thuốc mẹ, nhưng cũng có những sản phẩm chuyển hóa không còn tác dụng, thậm chí có độc tính cao. Các thuốc mới, dạng bào chế mới cần được đánh giá Sinh khả dụng để xác định đường dùng, liều và khoảng cách giữa các lần dùng thuốc thích hợp. Hơn nữa, các thuốc generic còn yêu cầu đánh giá tương đương sinh học để so sánh mức độ và tốc độ hấp thu của thuốc vào hệ tuần hoàn giữa chúng và thuốc phát minh hoặc thế phẩm bào chế giúp cho các bác sĩ lâm sàng có thể thay thế thuốc trong điều trị. Tất cả các yêu cầu này hầu hết đều phải căn cứ vào nồng độ thuốc trong dịch sinh học, thường là máu hoặc huyết tương để thiết lập nên cơ sở dữ liệu làm minh chứng đánh giá mối liên quan giữa nồng độ thuốc trong dịch sinh học với tác dụng/ độc tính của thuốc trong cơ thể.
Trong các nghiên cứu nồng độ thuốc trong dịch sinh học, việc sử dụng các phương pháp phân tích thích hợp để xác định được sự có mặt (định tính) và nồng độ (định lượng) của thuốc và (hoặc) các sản phẩm chuyển hóa trong dịch sinh học (máu, huyết tương, nước tiểu, dịch não tuỷ, ...) là một bước quyết định, là chìa khoá để tính toán các thông số dược động học, sinh khả dụng, giúp các bác sĩ lâm sàng theo dõi nồng độ thuốc trong cơ thể để điều chỉnh hoặc chỉ định cách dùng thuốc hợp lý, cũng như phục vụ cấp cứu hoặc hóa pháp. Phương pháp phân tích để định lượng thuốc và chất chuyển hóa của nó trong dịch sinh học dựa trên kỹ thuật hóa lý và sinh học hiện đại được phát triển và ứng dụng rộng rãi như sắc ký, điện di, phóng xạ miễn dịch, Phương pháp có tính chọn lọc tốt, độ nhạy, độ chính xác cao, có thể tự động hóa được dùng nhiều nhất là HPLC, đặc biệt là LC - MS/MS.
2.2 Các loại mẫu thử sinh học
Để nghiên cứu đánh giá sinh dược học của thuốc có nhiều loại mẫu sinh học được lấy và phân tích nồng độ thuốc. Phổ biến nhất là máu, huyết tương, huyết thanh, nước tiểu ngoài ra còn có dịch não tủy, nước bọt, sữa, tóc, móng,... Sau đây trình bày đặc điểm của một số loại mẫu sinh học thường dùng trong đánh giá sinh dược học của thuốc.
2.2.1 Máu
Máu được cấu tạo bởi một số loại tế bào gồm thành phần hữu hình (40%) và huyết tương (60%). Độ pH của máu là 7,40 ± 0,05. Tỷ lệ thể tích máu so với trọng lượng cơ thể thay đổi theo lứa tuổi, tình trạng sinh lý bệnh, tỷ lệ này ở trẻ nhỏ cao hơn người trưởng thành, ở phụ nữ có thai cao hơn phụ nữ bình thường.
2.2.2 Huyết tương
Huyết tương là phần lỏng của máu, là dịch trong, hơi vàng. Thành phần của huyết tương có 92% là nước, còn lại là chất hữu cơ, vô cơ. Thành phần chất hữu cơ gồm protein huyết tương như Albumin, fibrinogen và các chất hữu cơ không phải protein. Các chất hữu cơ không phải protein có 2 loại là loại có chứa nitơ như urễ, acid amin tự do, acid uric, creatin, creatinin, bilirubin, amoniac và loại không chứa nitơ như Glucose, lipid, cholesterol, phospholipid, acid lactic. Ngoài ra, còn có hàm lượng rất thấp các chất trung gian hoá học, trung gian chuyển hoá, hormon, vitamin và enzym. Các chất vô cơ trong huyết tương ở dạng ion (C1- Na*, HCO3 - K*, Ca2+, Mg2+...).
2.2.3 Huyết thanh
Huyết thanh có thành phần tương tự như huyết tương nhưng được loại fibrinogen.
2.2.4 Nước tiểu
Là dịch thải ra ngoài cơ thể, gồm những chất không cần thiết, chất khoáng, dịch và một số chất bên trong máu. Thành phần của nước tiểu có 93 - 95% là nước, vật chất khô chiếm 5%. pH nước tiểu của người khỏe mạnh nằm trong khoảng 5 - 6. Trong nước tiểu có các sản phẩm chứa nitơ của quá trình phân hủy protein như ure, acid uric, creatinin, ...; các acid hữu cơ như Acid Lactic, acid béo, enzym, hormon... và các muối vô cơ như HCO3-, CI-, SO42-...
2.3 Các đặc điểm của phân tích thuốc trong dịch sinh học
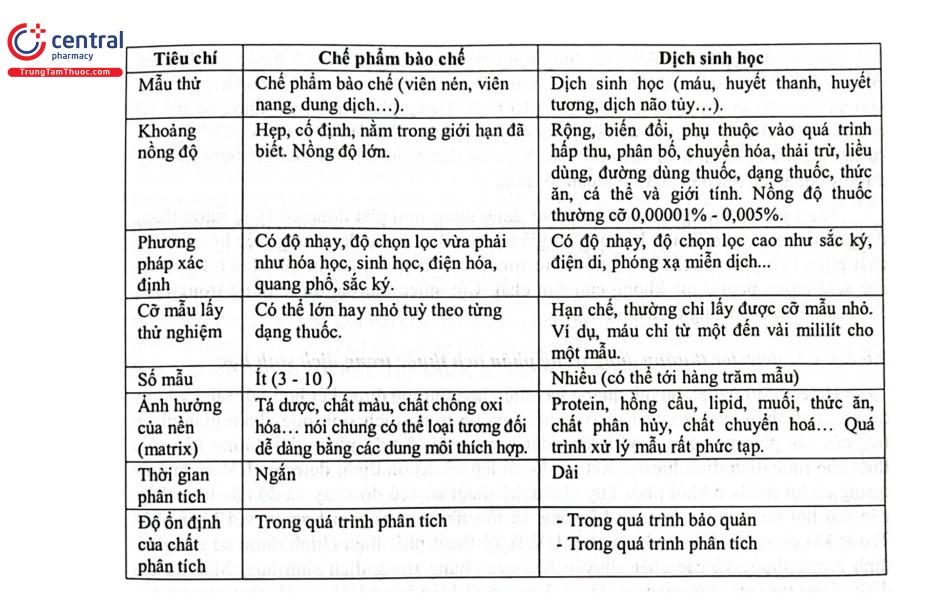
Chế phẩm bào chế và dịch sinh học là hai loại mẫu thử khác hẳn nhau do đó quá trình phân tích thuốc trong chế phẩm bào chế và trong dịch sinh học có nhiều điểm khác nhau, những điểm khác nhau chủ yếu được trình bày ở Bảng 2.14.
2.4 Phân tích mẫu sinh học bằng sắc ký lỏng hiệu năng cao
2.4.1 Xử lý mẫu dịch sinh học
Mẫu thử trước khi đưa vào phân tích sắc ký phải được xử lý để loại các tác nhân cản trở nhưng không được làm mất chất phân tích. Chuẩn bị mẫu một cách thích hợp là một khía cạnh quan trọng của định lượng thuốc trong dịch sinh học, nó chiếm một phần đáng kể trong tổng thời gian phân tích mẫu. Các thành phần trong nền mẫu như protein, chất béo, muối, các hợp chất nội sinh, gây ảnh hưởng tới quá trình phân tích cần được loại bỏ trước khi phân tích. Loại bỏ các thành phần này không chỉ tránh được sự tắc cột phân tích, mà còn cải thiện độ nhạy, độ chọn lọc và độ tin cậy của các phép phân tích. Chất cản trở trong nền mẫu có thể gây nhiễu đối với phát hiện píc chất phân tích khi dùng thiết bị HPLC với detector UV - Vis hoặc huỳnh quang. Tuy nhiên, nếu sử dụng kỹ thuật LC - MS/MS thì ảnh hưởng của chất cản trở trong nền mẫu được giảm thiểu do đó phương pháp này thường ít đòi hỏi khắt khe về làm sạch mẫu. Phương pháp chuẩn bị mẫu thường sử dụng và được ứng dụng rộng rãi đối với các mẫu thử sinh học là kết tủa protein, chiết lỏng - lỏng, chiết pha rắn. Bên cạnh việc xử lý mẫu với các thao tác bằng tay yêu cầu rất nhiều công lao động và tốn thời gian, thì thiết bị chiết tự động có thể chiết đồng thời tới hàng chục mẫu đã được phát triển giúp đẩy nhanh quá trình xử lý mẫu. Ngoài ra, việc tiêm mẫu thử trực tiếp (không qua xử lý) vào hệ thống sắc ký có gắn bộ phận chiết on - line đã tối giản sự phức tạp của quá trình phân tích mẫu sinh học.
2.4.2 Kiểu HPLC trong phân tích dịch sinh học
Sắc ký lỏng pha đảo được sử dụng rộng rãi nhất trong phân tích thuốc và các chất chuyển hóa trong dịch sinh học do phù hợp đối với hầu hết các chất phân tích phân tử nhỏ và mức độ kỵ nước khác nhau của pha tĩnh. Trong phân tích mẫu thực, có thể có mặt các chất chuyển hóa dẫn đến sự phân cực của chất phân tích tăng lên, do đó sẽ giảm sự lưu giữ trên pha tĩnh so với chất mẹ. Việc sử dụng chế độ rửa giải gradient thường được sử dụng để phép phân tích thuận lợi hơn.
Sắc ký lỏng tương tác thân nước sử dụng dung môi pha động có tỷ lệ nước thấp, dung môi hữu cơ cao là một bổ sung có giá trị cho sắc ký pha đảo trong việc lưu giữ các chất phân tích phân cực. HILIC với detector tứ cực chấp ba có thể phân tích trực tiếp các acid amin, peptid mà không cần dẫn chất. Cột silica thường được dùng trong tách các chất phân cực trong dịch sinh học.
2.4.3 Các detector thường dùng trong phân tích thuốc trong dịch sinh học
Detector tử ngoại, huỳnh quang hay điện hóa thường được kết hợp với sắc ký lỏng để xác định thuốc và các chất chuyển hóa của nó trong dịch sinh học. Thời gian phân tích của các phương pháp này thường dài vì yêu cầu đạt độ phân giải đường nền cần thiết cho mục đích định lượng. Xét về độ tái lặp và độ ổn định, detector UV và huỳnh quang có lợi thế hơn khối phổ. Tuy nhiên, kỹ thuật này có độ nhạy và độ đặc hiệu thấp nên đòi hỏi phải chuẩn bị mẫu kỹ càng và tốn nhiều thời gian hơn so với khối phổ. Trước khi có sự ra đời của khối phổ, UV là kỹ thuật phát hiện chính được sử dụng để định lượng thuốc và các chất chuyển hóa của chúng trong dịch sinh học. Mặc dù ổn định, đáng tin cậy, đơn giản và dễ sử dụng, phát hiện bằng UV có độ nhạy tương đối thấp, đặc biệt là đối với các chất không có nhóm mang màu.
Ngược lại với detector UV, detector huỳnh quang có thể cho phép phát hiện chất phân tích rất chọn lọc với độ nhạy cao. Nó được sử dụng phát hiện các chất có khả năng hấp thụ UV và phát ra huỳnh quang ở bước sóng dài hơn. Phát hiện bằng detector này có thể theo phương pháp trực tiếp hoặc gián tiếp. Một số thuốc trong dịch sinh học có thể phát hiện bằng detector này như serotonin, Doxorubicin, Sitagliptin, Tramadol,...
Tương tự như detector huỳnh quang, detector điện hóa loại đo dòng (amperometric detector) cũng cho phép phát hiện chất phân tích với độ chọn lọc và độ nhạy cao. Nó rất phù hợp để xác định chất có khả năng oxy hóa - khử, chẳng hạn như Artemisinin hoặc glutathion trong các mẫu sinh học. Tuy nhiên, nhược điểm của nó là điện cực dễ bị nhiễm bẩn ảnh hưởng tới kết quả phân tích.
LC - MS/MS hiện được coi là phương pháp lựa chọn tối ưu cho phân tích định lượng thuốc và các chất chuyển hóa của nó trong dịch sinh học. Những lợi thế của việc sử dụng kỹ thuật này trong chế độ MRM do độ nhạy, chọn lọc cao, tốc độ nhanh cho phép ứng dụng phương pháp mà yêu cầu rất ít hoặc không cần chuẩn bị mẫu với thời gian phân tích giảm thiểu. Tuy nhiên, hiệu ứng nền mẫu (matrix effect: ME) có thể có ảnh hưởng đáng kể đến quá trình phân tích LC - MS/MS. Vì vậy, đánh giá hiệu ứng nền mẫu cũng như loại bỏ hoặc giảm thiểu ảnh hưởng này cần được xem xét. Hiệu ứng nền mẫu là một thuật ngữ mô tả bất kỳ thay đổi đáp ứng MS của chất phân tích có thể dẫn đến giảm đáp ứng (khử ion: ion suppression) hoặc tăng đáp ứng lên (tăng ion: ion enhancement) của hệ thống LC - MS. ME được gây ra bởi các phân tử có nguồn gốc từ nền mẫu hoặc pha động có khả năng đồng rửa giải với chất phân tích do đó gây cản trở cho quá trình ion hóa trong nguồn ion hóa của MS. Nguồn gốc của ME thường là do phospholipid nội sinh và protein trong huyết tương. Trong sắc ký pha đảo, các lysophospholipid thường rửa giải ra trước và gây ra hiệu ứng nền mẫu nhiều hơn so với các phospholipid rửa giải sau. Phospholipid gẫy khử ion trong cả hai chế độ ESI dương và âm. Có thể loại chúng trong quá trình xử lý mẫu bằng cách chiết pha rắn hoặc chiết lỏng - lỏng thay vì kết tủa protein. Đa số ME xảy ra khi các chất cản trở được rửa giải phía trước chất phân tích nên nếu tăng khả năng lưu giữ chất phân tích có thể giảm thiểu ME. Chuẩn bị mẫu và điều kiện sắc ký phù hợp sẽ tạo cơ sở phát triển một phương pháp LC - MS/MS định lượng thuốc và chất chuyển hóa trong dịch sinh học thành công. Một vấn đề nữa có thể gây ảnh hưởng của ME là lựa chọn giao diện ion hóa. Ion hoá bằng kỹ thuật ESI được sử dụng đối với chất phân tích phân cực mạnh hoặc tồn tại ở dạng ion, còn ion hoá bằng kỹ thuật APCI ứng dụng đối với các chất ít phân cực. APCI thường được coi là ít khử ion hơn so với ESI. Tuy nhiên, độ nhạy và ổn định nhiệt của chất phân tích cần được đánh giá khi sử dụng APCI. Giảm tốc độ dòng pha động (20 pl / phút hoặc thấp nguồn cách chia dòng sau cột cũng có thể giảm bớt hoặc loại bỏ hoàn toàn hiện tượng khử ion. Thêm chất phụ gia vào pha động như triethylamin và acid trifluoroacetic cũng có thể dẫn đến sự khử ion. Việc sử dụng các thuốc thử khắc như acid formic hay acid acetic, acid trifluoroacetic kết hợp với 10 mM amoni acetat hoặc bổ sung 1% acid propionic vào pha động có thể tốt hơn so với sử dụng acid trifluoroacetic để làm giảm ME. Có thể thay thế triethylamin bằng thuốc thử tạo cặp ion khác như hexylamin. Tuy nhiên, cách hiệu quả nhất để loại bỏ ảnh hưởng của ME tới độ chính xác và độ đúng của phương pháp phân tích định lượng thuốc và chất chuyển hóa trong dịch sinh học là sử dụng các đồng vị bền, tương tự làm chất chuẩn nội.
2.5 Các ứng dụng phân tích thuốc trong dịch sinh học của sắc ký lỏng hiệu năng cao
2.5.1 Nghiên cứu dược động học của thuốc
Nghiên cứu số phận chuyển hóa của thuốc là một phần thiết yếu và quan trọng của quá trình phát triển thuốc, nghiên cứu các con đường chuyển hoá thuốc, tương tác thuốc, ảnh hưởng của di truyền (genetic olymorphisms) và các yếu tố khác ảnh hưởng đến chuyển hóa pha I, pha II của thuốc. Các phương pháp in vitro và nghiên cứu in vivo được áp dụng để làm rõ sự chuyển hóa của thuốc. Phân tích các chất chuyển hóa trong mẫu thử sinh học phức tạp là một nhiệm vụ đầy thách thức do đó một số phương pháp phân tích định tính và định lượng các chất chuyển hóa của thuốc được sử dụng. Phương pháp sắc ký lỏng ghép nối với các detector khác nhau như detector UV, huỳnh quang, điện hóa, khối phổ, ... được sử dụng rộng rãi. Trong đó, LC - MS đã trở thành phương pháp phân tích mạnh nhất để sàng lọc và xác định các chất chuyển hóa thuốc trong mẫu thử sinh học. Có nhiều cách tiếp cận khác nhau để phân tích các chất chuyển hóa trong các mẫu sinh học, từ định lượng trực tiếp, gián tiếp thông qua việc định lượng thuốc sau khi thủy phân chất chuyển hóa. Nghiên cứu chuyển hóa thuốc đặc biệt có ý nghĩa khi các chất chuyển hóa có hoạt tính dược lý, độc hoặc khi thuốc có chuyển hóa mạnh. Sự khác biệt về chuyển hoá thuốc giữa các cá thể cũng yêu cầu phải nghiên cứu các yếu tố ảnh hưởng đến chuyển hóa thuốc. Hơn nữa, sự chuyển hóa của các chất có độc tính cũng phải được xem xét. Trong những phát hiện ban đầu, kết quả về chuyển hóa thuốc cung cấp cơ sở để lựa chọn cấu trúc hóa học và các hợp chất tiềm năng với sự chuyển hóa thuốc và dược động học theo mong muốn hoặc có tính an toàn cao. Bên cạnh nghiên cứu chuyển hóa thuốc theo truyền thống, tập trung vào sự hấp thu, phân bố, chuyển hóa và thải trừ in vitro và in vivo của thuốc, các kiến thức trong lĩnh vực dược về di truyền (pharmacogenetics), gen (pharmacogenomics) và vận chuyển mang lại nhiều tiến bộ trong nghiên cứu sự chuyển hóa thuốc.
Yếu tố sinh lý khác nhau như tuổi tác, giới tính, tình trạng bệnh, mang thai, tập thể dục, nhịp sinh học và đói dẫn đến sự trao đổi chất bị suy giảm giữa các cá thể và cần được xem xét khi đánh giá sự chuyển hóa. Liều dùng, khoảng cách giữa các lần dùng, phân bố ở mô và liên kết protein của thuốc ảnh hưởng đến chuyển hóa của nó. Hơn nữa, các yếu tố môi trường như hóa chất môi trường, thuốc điều trị phối hợp, hút thuốc, uống rượu và các thành phần dinh dưỡng có thể thay đổi không chỉ động học của phản ứng enzym mà còn thay đổi toàn bộ mô hình chuyển hóa, dẫn tới biến đổi sinh khả dụng, dược động học, đặc tính dược lý hoặc độc tính của thuốc.
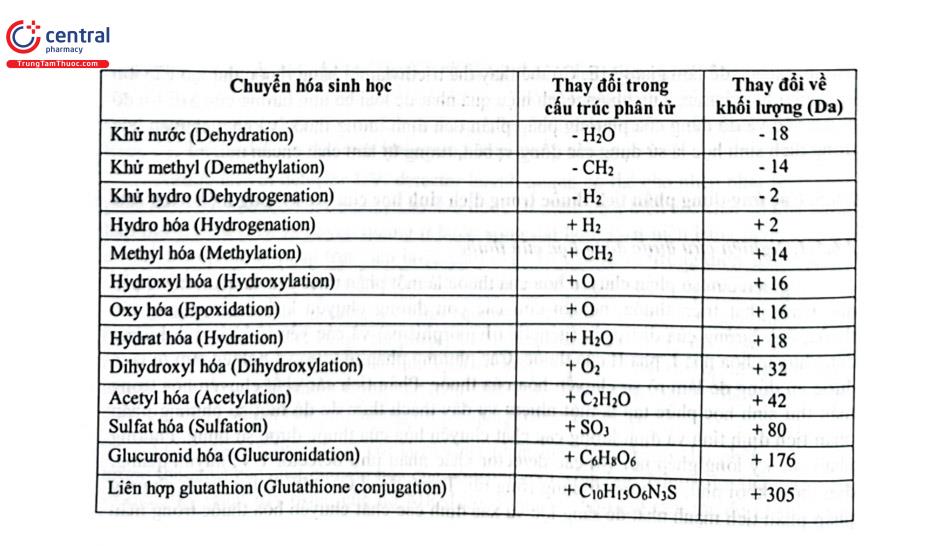
Khi thuốc vào cơ thể và có chuyển hóa thì sẽ thay đổi cấu trúc hóa học và khối lượng chất sẽ thay đổi, Bảng 2.15 trình bày sự thay đổi khối lượng trong quá trình chuyển hóa sinh học của các phản ứng chuyển hóa thông thường.
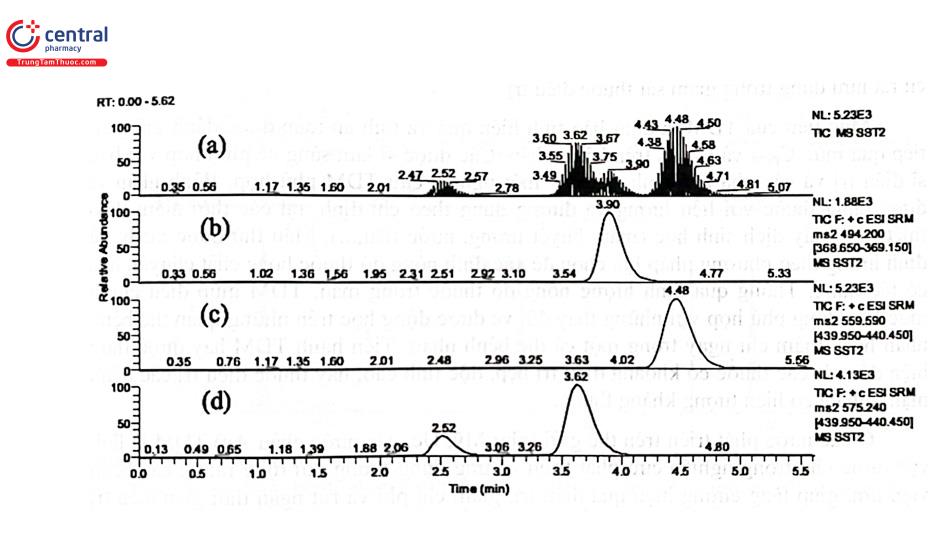
Hình 2.34 trình bày sắc ký đồ thu được khi định lượng huyết tương người tình nguyện khỏe mạnh sau khi uống viên Artovastin 10 mg. Artovastin vào cơ thể sẽ chuyển hóa mạnh ở gan (> 60%) chủ yếu do enzym CYP 3A4 thành các chất chuyển hóa có hoạt tính là ortho - hydroxy atorvasttatin và para - hydroxy atorvastatin. Con đường chuyển hóa là hydroxyl hóa, khối lượng phân tử sẽ tăng lên 16 dalton. Nồng độ artovastatin, 2 chất chuyển hóa trong huyết tương được định lượng bằng LC - MS/MS với chuẩn nội là glibenclamid. Các chất được tách bằng cột Acquity C18 (100 x 2,1 mm; 1,7 cm), ổn định ở 40°C; pha động: methanol - dung dịch acid formic 0,001% tỷ lệ 70 : 30; tốc độ dòng: 0,2 ml/phút; thể tích tiêm mẫu: 20 ul; nhiệt độ buồng tiêm mẫu: 20°C; thời gian phân tích 5,60 phút. Chế độ khối phổ gồm ion hóa chế độ ESI (+), thế phân mảnh của artovastin là 21 V, của ortho - hydroxy atorvasttatin và para - hydroxy Atorvastatin đều là 20 V và của IS là 12 V, chế độ ghi phổ MRM đối với các chất artovastin và IS tại m/z lần lượt là 559,6→ 440,2 dal và 494,2 → 368,9 dal; của ortho - hydroxy atorvasttatin và para - hydroxy atorvastatin đều là 575,2→ 440,2 dal.
2.5.2 Giám sát thuốc trong điều trị
Giám sát thuốc trong điều quan trọng trong cá thể hóa điều trị vì đáp ứng điều trị phụ thuộc chủ yếu vào nồng độ thuốc, thực tế hơn là phụ thuộc vào liều dùng. Trong khi đó, có rất nhiều yếu tố khác nhau ảnh hưởng đến nồng độ thuốc như tuổi, cân nặng bệnh nhân, đường dùng thuốc, các thuốc dùng kèm, tình trạng bệnh lý. Nó bao gồm đánh giá chỉ định lâm sàng đối với biểu hiện giám sát thuốc điều trị, phân tích hóa học mẫu thử sinh học và diễn giải các kết quả can thiệp điều trị nếu có thể xảy ra. Do đó, nồng độ thuốc trong dịch sinh học là cơ sở quan trọng để điều chỉnh chế độ liều dùng phù hợp.
Xác định nồng độ thuốc trong dịch sinh học của từng bệnh nhân là một trong những yêu cầu quan trọng nhất của TDM. Nhiều qui trình phân tích đã được xây dựng để giám sát thuốc trong điều trị. Những kỹ thuật phân tích đã không ngừng được nâng cao, lĩnh vực này của TDM có lẽ được phát triển nhanh hơn nhiều so với các khía cạnh khác. Các kỹ thuật phân tích gồm có định lượng bằng miễn dịch, điện di mao quản, sắc kỷ khí và HPLC. Định lượng bằng miễn dịch ít hoặc không yêu cầu xử lý mẫu, nhanh và dễ tiến hành, nhưng phương pháp này có thể có tính đặc hiệu kém. Điện di mao quản có hiệu lực cao và có thể tách được các chất từ các ion vô cơ có trọng lượng phân tử nhỏ tới các phân tử sinh học lớn nhưng thể tích mẫu nhỏ nên ảnh hưởng tới độ nhạy. Sắc ký khí chỉ ứng dụng đối với dải chất phân tích tương đối hẹp do yêu cầu về tính chất bay hơi và bền với nhiệt, đó chính là hạn chế trong TDM của phương pháp này. HPLC rất đa nặng, đáp ứng được hầu hết các yêu cầu phân tích đã ngày càng khẳng định là công cụ rất hữu dụng trong giám sát thuốc điều trị.
Mục tiêu của TDM là đảm bảo tính hiệu quả và tính an toàn được đánh giá gián tiếp qua mức Cpeak và Ctrough trên bệnh nhân. Các dược sĩ lâm sàng sẽ phối hợp với bác sĩ điều trị và các nhà hóa sinh thiết kế một nghiên cứu TDM phù hợp. Bệnh nhân sẽ được dùng thuốc với liều lượng và đường dùng theo chỉ định, tại các thời điểm theo thiết kế sẽ lấy dịch sinh học (máu, huyết tương, nước tiểu,...). Mẫu thử được xử lý và định lượng theo phương pháp lựa chọn để xác định nồng độ thuốc hoặc chất chuyển hóa có tác dụng. Thông qua định lượng nồng độ thuốc trong máu, TDM giúp điều chỉnh mức liều dùng phù hợp với những thay đổi về dược động học trên những quần thể bệnh nhân hoặc thậm chí ngay trong một cá thể bệnh nhân. Tiến hành TDM hay được thực hiện đối với các thuốc có khoảng điều trị hẹp, độc tính cao, hay thuốc điều trị các bệnh mạn tính dễ có hiện tượng kháng thuốc.
Ở các nước phát triển trên thế giới (như Mỹ, Úc, các nước châu Âu) TDM là lĩnh vực được chú trọng nghiên cứu phát triển và ứng dụng tương đối rộng rãi tại các bệnh viện lớn, giúp tăng cường hiệu quả điều trị, giảm chi phí và rút ngắn thời gian điều trị cho bệnh nhân. Còn tại Việt Nam, giám sát thuốc điều trị chủ yếu thực hiện ở mức độ nghiên cứu. Một số nghiên cứu đã được thực hiện thuộc đề tài nghiên cứu các cấp hoặc của các nghiên cứu sinh thực hiện ở các bệnh viện lớn như Bệnh viện Lao phổi trung ương, Bệnh viện Bạch Mai, Bệnh viện Nhi trung ương, Bệnh viện Tâm thần trung ương 1... Các nghiên cứu đều đưa ra nhận định thực hiện chế độ TDM trong nghiên cứu làm tăng hiệu quả điều trị đồng thời vẫn đảm bảo an toàn trên bệnh nhân. Tuy nhiên, việc triển khai giám sát thuốc điều trị chưa được ứng dụng nhiều trong thực tế do sự hợp tác giữa bác sĩ điều trị và dược sĩ lâm sàng còn hạn chế, hơn nữa trang thiết bị, qui trình phân tích được thẩm định và mức độ sẵn sàng của các nhà phân tích, đặc biệt là kinh phí hạn hẹp là những rào cản để đưa TDM vào thực tiễn.
Hình 2.35 trình bày sắc ký đồ thu được trong nghiên cứu theo dõi và điều trị bệnh nhân tâm thần phân liệt tại bệnh viện Tâm thần trung ương 1 dựa trên nồng độ Haloperidol và clozapin trong huyết tương bệnh nhân dùng thuốc để điều chỉnh liễu sử dụng. Nồng độ haloperidol và clozapin trong huyết tương được định lượng bằng LC - MS/MS với chuẩn nội là carbamazepin. Các chất được tách bằng cột Thermo Hypersil Gold C8 (100 × 2,1 mm; 1,9 um); nhiệt độ cột 40°C; pha động gồm pha động A : pha động B (8:2) với pha động A là hỗn hợp methanol : acetonitril : isopropanol (5:3:1), pha động B là dung dịch amoni acetat 2 mM; tốc độ dòng 200 uL/phút; thể tích tiêm 2 ul; nhiệt độ buồng tiêm mẫu 20°C; thời gian phân tích: 2,5 phút. Chế độ khối phổ gồm ion hóa chế độ ESI (+), thế phân mảnh của clozapin là 17 V, của haloperidol và IS đều là 22 V, chế độ ghi phổ MRM đối với các chất clozapin, haloperidol và IS tại m/z lần lượt là 327,0 → 270,1 dal, 376,0 – 165,1 và 237,1 → 194,0.

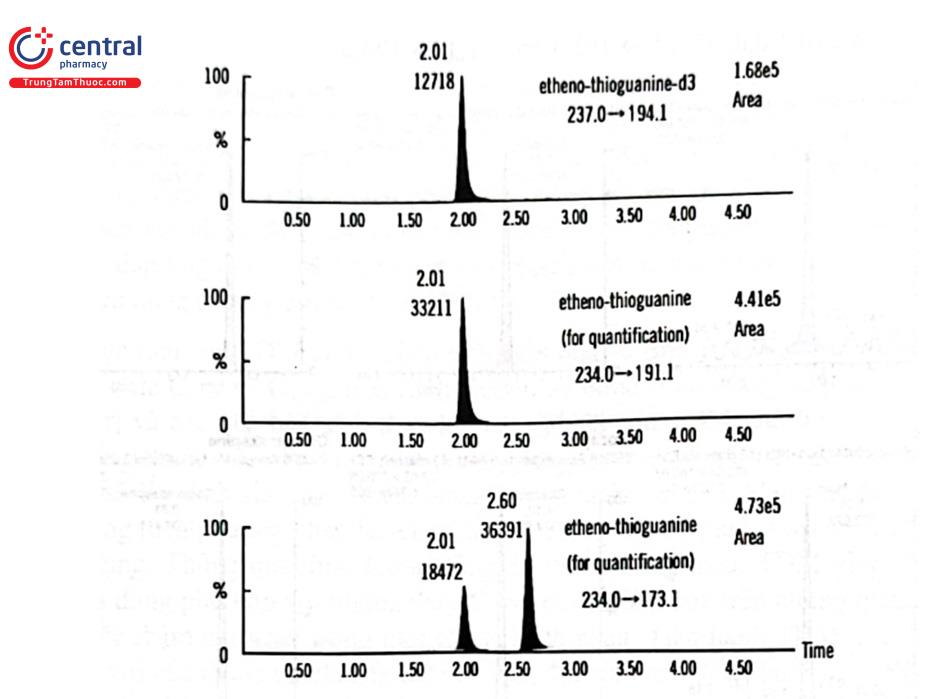
Trong những năm gần đây, LC - MS/MS còn được ứng dụng phân tích nồng độ chất chuyển hóa trong tế bào phục vụ cá thể hóa điều trị. Điển hình là theo dõi nồng độ chất chuyển hóa 6 - thioguanin trong mẫu DNA của trẻ em điều trị Bệnh bạch cầu cấp dòng lympho bằng thuốc Mercaptopurin, từ đó xác định liều dùng phù hợp cho bệnh nhân trong quá trình điều trị. Trong công bố năm 2021 của các nhà khoa học Hàn Quốc, 6 - thioguanin trong DNA được định lượng ở mức nồng độ từ hàng chục đến hàng trăm femtomol trong 1 ũg DNA bằng LC - MS/MS. Chất phân tích trong mẫu thử DNA được dẫn chất hóa với thuốc thử cloroacetaldehyd 1 M trong dung dịch đệm Kali phosphat 90 mM, pH = 5,0 sau đó chiết pha rắn bằng cột chiết C18. Mẫu thử được phân tích trên hệ thống UPLC Aquity kết nối với detector XEVO TQ - S MS/MS (Waters). Điều kiện sắc ký gồm: cột tách HILIC BEH (2,1 mm × 50 mm; 1,7 um), cột bảo vệ cùng loại. Nhiệt độ cột 35°C, rửa giải đẳng dòng bằng pha động gồm 160 mM acid formic và 10 mM amoni format trong acetonitril 95%, tốc độ dòng 0,3 ml/min trong 2,3 min; sau đó 0,8 ml/min trong 0,7 min. Thể tích mẫu tiêm 6 p1. Thời gian phân tích 1 mẫu là 4 phút. Điều kiện khối phổ: chất phân tích etheno - thioguanin và chuẩn nội etheno - thioguanin d3 sau khi dẫn xuất được ion hóa theo chế độ phun điện dương (ESI +), phát hiện bằng chế độ MRM, etheno - thioguanin (thioguanin) m/z 234,0–191,1 và etheno - thioguanin - d3 (thioguanin - d3) m/z 237,0–194,1. Phương pháp đã áp dụng thành công trong định lượng thioguanin trong 257 mẫu DNA của 54 bệnh nhân nhi phục vụ điều trị. Hình 2.36 trình bày sắc ký đồ phân tích thioguanin và chuẩn nội thioguanin d3 sau khi dẫn xuất hóa.
2.5.3 Đánh giá sinh khả dụng và thử tương đương sinh học
Để đánh giá được chính xác chất lượng của thuốc đối với người sử dụng cần phải nghiên cứu sự giải phóng của dược chất ra khỏi dạng thuốc, sự hấp thu dược chất khi vào cơ thể, tức là nghiên cứu sinh khả dụng của thuốc. Mặt khác, khi một chế phẩm generic muốn chứng minh được tính an toàn và hiệu quả phải được so sánh về tương đương sinh học với chế phẩm đối chiếu (thuốc phát minh). Đánh giá sinh khả dụng và tương đương sinh học của thuốc chủ yếu dựa vào nồng độ thuốc trong huyết tương người sử dụng theo những mô hình qui định. Dựa vào nồng độ thuốc trong huyết tương sẽ tính được các thông số dược động học như nồng độ cực đại của thuốc (Cmax), thời gian thuốc đạt cực đại (tmax) trong huyết tương, đường cong dược động học của nồng độ dạng hoạt tính của thuốc trong huyết tương biến đổi theo thời gian (AUC), thời gian bán thải (t12), của thuốc. Từ đó, xác định được sinh khả dụng và so sánh được tương đương sinh học của thuốc.
Các thuốc, chế phẩm cần đánh giá tương đương sinh học đã được qui định cụ thể, đó là các chế phẩm có nguy cơ khác nhau về sinh khả dụng đưa đến không tương đương điều trị như dạng thuốc giải phóng nhanh (viên nén, viên nang, hỗn dịch uống,...) có đặc điểm dược động học của hoạt chất phức tạp (dược động học không tuyến tính, tỷ lệ hoạt chất chuyển hóa cao hoặc đào thải trước khi vào hệ đại tuần hoàn, ...), tính chất lý hoá của hoạt chất không thuận lợi (độ tan thấp, tính thẩm kém, có vấn đề về sinh khả dụng do cấu trúc hóa học của hoạt chất như đồng phân lập thể, ...), tỷ lệ tá dược quá lớn so với hoạt chất, dạng thuốc tác dụng kéo dài, ... Ở Việt Nam hiện nay, khi đăng ký thuốc lưu hành các chế phẩm thuốc bắt buộc phải có nghiên cứu tương đương sinh học là dạng thuốc phóng thích có kiểm soát, dạng thuốc rắn dùng đường uống của 12 dược chất gồm Amlodipin, clarithromycin, glibenclamid, Rifampicin, nifedipin, carbamazepin, azithromycin, cefuroxim, Metformin, cefixim, glyclazid, Metoprolol. Trong thực tế, ngoài các thuốc yêu cầu bắt buộc phải nghiên cứu tương đương sinh học khi đăng ký lưu hành, các nhà sản xuất rất chú trọng nghiên cứu tương đương sinh học của sản phẩm sản xuất nhằm nâng cao chất lượng của thuốc phục vụ điều trị.
Nghiên cứu sinh khả dụng và đánh giá tương đương sinh học là nghiên cứu trên người do đó phải tuân thủ các qui định đã được ban hành. Trong Luật Dược hiện hành (2016) đã có qui định về thử tương đương sinh học trong mục 2 của chương XI. Thử thuốc trên lâm sàng, thử tương đương sinh học của thuốc. Đồng thời Thông tư 10/2020 - BYT của Bộ Y tế có hướng dẫn cụ thể về thử tương đương sinh học của thuốc. Thử tương đương sinh học của thuốc gồm 2 giai đoạn là nghiên cứu lâm sàng và phân tích dịch sinh học. Giai đoạn nghiên cứu lâm sàng là giai đoạn thực hiện thử nghiệm chủ yếu trên người tình nguyện khỏe mạnh do đó cần tuân thủ các yêu cầu về thử thuốc trên lâm sàng, đặc biệt là qui định về đạo đức trong nghiên cứu y sinh học. Giai đoạn phân tích dịch sinh học để xác định nồng độ thuốc trong huyết tương người dùng thuốc đối chứng và thuốc thử nhằm so sánh sinh khả dụng và chứng minh tương đương sinh học của 2 thuốc.
Nồng độ thuốc trong dịch sinh học được định lượng chủ yếu bằng phương pháp HPLC với các detector khác nhau, đặc biệt UPLC - MS/MS được dùng rộng rãi để đẩy nhanh quá trình phân tích và tăng khi nồng độ chất phân tích rất thấp (ng/ml). Phương pháp phân tích phải được thẩm định đạt yêu cầu qui định. Định lượng tất cả các mẫu thử phải hoàn thành trong khoảng thời gian đã khảo sát độ ổn định. Các mẫu của mỗi cá thể tốt nhất nên phân tích trong một lần tiến hành. Mỗi mẫu thử có thể phân tích một lần hoặc lặp lại nếu cần. Với mỗi lô mẫu phân tích sinh học (các mẫu phân tích cùng một thời gian trong một buổi hoặc một ngày), cần thiết lập một đường chuẩn mới để phân tích và dùng các mẫu chuẩn đối chiếu ở 3 mức nồng độ (thấp, trung bình và cao) để định lượng đồng thời trong mỗi lô phân tích. Mẫu chuẩn đối chiếu (Quality Control samples: QCs) được dùng để so sánh khi định lượng là những mẫu tự tạo biết trước nồng độ, chuẩn bị bằng cách pha chất chuẩn của chất cần phân tích trong mẫu sinh học trắng tương tự như các mẫu dùng để thẩm định.
Ví dụ: Một nghiên cứu tương đương sinh học của chế phẩm generic viên nén Azithromycin 500 mg của một công ty sản xuất được so sánh với chế phẩm đối chiếu là viên nén Zithromax 500 mg của Pfizer Australia Pty Ltd. sản xuất. Nghiên cứu được thực hiện theo mô hình chéo đôi 2 giai đoạn ở trạng thái đói trên 24 người tình nguyện khỏe mạnh đáp ứng yêu cầu sức khỏe, chế độ ăn uống. Mẫu máu được lấy tại 16 thời điểm gồm ngay trước khi uống thuốc và từ 0,5 đến 120 giờ sau khi uống thuốc. Huyết tương được tách ra và azithromycin trong mẫu được phân tích bằng LC - MS/MS với chuẩn nội là Propranolol. Mẫu thử được xử lý theo phương pháp kết tủa protein bằng acetonitril. Các chất được tách bằng cột Synergi 4 | POLAR - RP - 80A (50 × 2,0 mm; 4 um) nối tiếp với cột bảo vệ AQ C18, (4 × 2,0 mm; 4 pm) nhiệt độ cột 40°C; pha động chạy chế độ gradient gồm acid formic 0,1% trong acetonitril và acid formic 0,1% trong nước; tốc độ dòng 0,7 mL/phút; thời gian phân tích 4 phút. Chế độ khối phổ gồm ion hóa ESI (+), chế độ ghi phổ MRM đối với azithromycin và IS với m/z lần lượt là 750,1 158,3 và 260,0 → 116,1. Đường chuẩn được xây dựng trong khoảng nồng độ từ 2 - 500 ng/mL, hồi qui tuyến tính với trọng số 1/x2 có tương quan tuyến tính chặt chẽ. Phương pháp đáp ứng yêu cầu các chỉ tiêu thẩm định theo qui định đối với phân tích dịch sinh học. Kết quả nghiên cứu của chế phẩm thử so với chế phẩm đối chiếu được trình bày ở Hình 2.37, Bảng 2.16 và Bảng 2.17 cho thấy chế phẩm thử tương đương sinh học với chế phẩm đối chiếu.
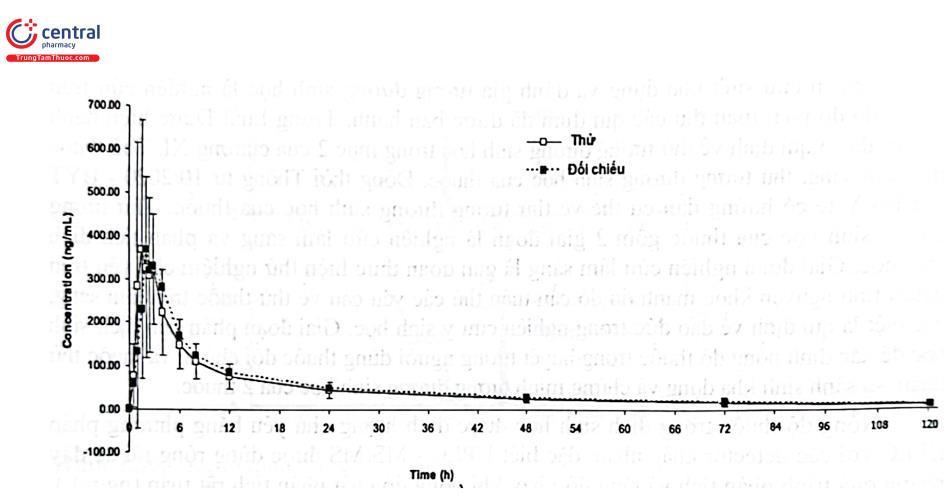
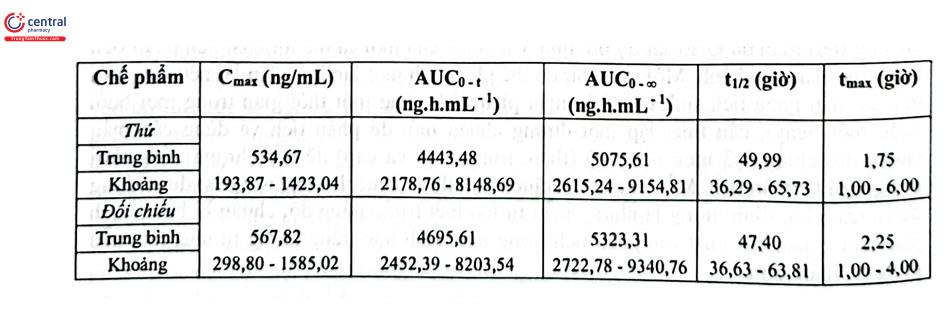
Bảng 2.17. Khoảng tin cậy 90% của các thông số dược động học trung bình(N=24)
| Thống kê | Thử/đối chiếu (%) | Giới hạn tin cậy (%) | Power (%) |
| Cmax (ng/mL) | 94,16 | 80,31-110,41 | 92,51 |
| AUC0-t (ng.h.mL-1) | 94,63 | 86,27 - 103,81 | 90,25 |
| AUC0-∞ (ng.h.mL-1) | 95,35 | 87,15-104,31 | 91,08 |
2.5.4 Độc chất - Pháp y
Ngộ độc bệnh viện thường đề cập đến các trường hợp người bệnh nhập viện do nghi ngờ bị ngộ độc hoặc để được hỗ trợ điều trị. Phạm vi các chất có thể gặp khá lớn, có thể là các tác nhân được học, thuốc bất hợp pháp, khí, dung môi, thuốc trừ sâu, kim loại độc hại và chất độc môi trường trong dịch sinh học. Các phòng xét nghiệm lý tưởng ở bệnh viện là có khả năng định tính và định lượng được các chất trên khi cần thiết, nhưng thực tế có ít phòng thí nghiệm có thể kiểm tra các chất một cách toàn diện.
Ở các nước phát triển, các phòng thí nghiệm hóa học lâm sàng trong bệnh viện được thiết kế để hướng đến cung cấp các dịch vụ cơ bản và khi gặp các trường hợp đặc biệt thì dựa vào hỗ trợ từ các phòng thí nghiệm độc chất chuyên biệt trung ương. May mắn là phần lớn các trường hợp có thể chẩn đoán gián tiếp dựa trên các bằng chứng lâm sàng và dựa vào kinh nghiệm mà không cần thiết phải phân tích ngay. Tuy nhiên, khi tình trạng bệnh nhân nặng và chẩn đoán không rõ ràng thì việc kiểm tra độc tính có thể rất quan trọng và kết quả phân tích cần được đưa ra nhanh chóng (thường là trong vòng 1 - 2 giờ kể từ khi nhập viện). Lý tưởng nhất là trong khoảng thời gian đó có thể vừa định tính vừa định lượng được chất độc, nhưng nếu chỉ kịp định tính thì vẫn cần thông báo kết quả ngay cho bác sĩ điều trị vì nó có giá trị lớn nếu các triệu chứng phù hợp với chất độc được xác định. Sự hạn chế về mặt thời gian đòi hỏi nhà phân tích cần phải cân nhắc giữa tốc độ và độ chính xác phân tích. Để làm được điều đó, các phương pháp định lượng sử dụng có thể được lược bớt so với qui chuẩn, nhưng vẫn cần phải có đủ các đặc điểm cần thiết để có thể đưa ra một quyết định lâm sàng thích hợp.
Trong phòng thí nghiệm lâm sàng lớn, việc sử dụng các kỹ thuật sắc ký lỏng khối phổ khác nhau cho các việc giám sát điều trị thuốc và phân tích chất độc đã tăng lên đáng kể. Phương pháp LC - MS/MS giúp tăng độ nhạy và độ tin cậy của các ứng dụng này trong độc chất pháp y. Mặc dù LC - MS/MS không phải là một phương pháp kiểm tra toàn diện, nhưng nếu có đầy đủ các thông tin về nguyên nhân gây ra ngộ độc trong các trường hợp quá liều thuốc, nó có thể sử dụng dữ liệu về định tính, định lượng của các loại thuốc và các chất chuyển hóa của chúng trong thời gian khoảng 1 giờ.
Sàng lọc bằng HPLC sử dụng qui trình định tính độc chất hệ thống (the systematic toxicological identification procedure: STIP) đối với thuốc đã được triển khai và thu được hiệu quả tích cực. Qui trình dựa trên phương pháp xử lý mẫu nhanh và đơn giản, sau đó phân tích bằng LC - RP và phát hiện bằng detector mảng diod. Thư viện thời gian lưu và phổ UV có sẵn cho khoảng 400 loại thuốc thông thường. Qui trình này được trình bày trong tài liệu Clarke’s Analysis of Drugs and Poisons in pharmaceuticals, body fluids and postmortem material năm 2011 của nhà xuất bản Pharmaceutical Press. Một nhược điểm của qui trình là có số lượng lớn thuốc rửa giải từ 1 đến 3 phút và vấn đề này trở nên khó khăn hơn với các chất không có phổ UV đặc trưng (cực đại<210 nm). Trong trường hợp này, cần phân tích tiếp để có được kết quả chính xác. Qui trình xử lý mẫu sau được ứng dụng đối với thuốc có tính acid và base trong phân tích sàng lọc được tiến hành như sau: Mẫu thử được xử lý bằng cách cho 1 mL acetonitril vào 1,0 mL mẫu thử (huyết thanh hoặc huyết tương). Lắc xoáy 2 giây. Thêm 6 mL dicloromethan. Lắc xoáy 2 giây. Thêm 100 uL dung dịch HCl 2 mol/L hoặc dung dịch NaOH 2 mol/L. Lắc nhẹ trong 2 phút và ly tâm. Chuyển lớp dung môi sang ống khác. Làm bay hơi dung môi tới khô ở 40°C dưới luồng khí nitơ. Hòa cắn trong 100 uL pha động. Tiêm 40 uL vào hệ thống sắc ký với điều kiện cột tách Lichrospher RP - 18e; (125 mm× 4,0 mm, Sum). Pha động được chuẩn bị như sau: trộn 530 mL nước siêu tinh khiết với 146 ut triethylamin và khoảng 750 uL acid phosphoric 85%; điều chỉnh tới pH 3,3 bằng dung dịch KOH 10%; thêm 470 mL acetonitril (pH – 4,0). Tốc độ dòng 0,6 mL/min. Thời gian lưu trong thư viện và trên sắc ký đồ phải giống nhau (cửa sổ <10%), nếu chưa đạt, phải điều chỉnh nồng độ phosphat hoặc tốc độ dòng của pha động. Phổ của píc chất phân tích được chồng với phổ tham chiếu có trong cơ sở dữ liệu.
Một lĩnh vực xét nghiệm nữa mà HPLC cũng được ứng dụng rộng rãi đó là phân tích ma túy trong dịch sinh học. Phân tích ma túy đóng vai trò vô cùng quan trọng trong lâm sàng và trong độc chất pháp y. Mẫu để xác định ma túy trong dịch sinh học thường là nước tiểu, máu. Tuy nhiên, các chất ma túy được phát hiện trong dịch sinh học có cửa sổ phát hiện hẹp, chỉ vài ngày sau khi sử dụng, nên có thể bổ sung thêm phân tích mẫu thử là tóc vì mẫu này có thể phát hiện được người sử dụng ma túy trước đó vài tuần đến vài tháng. Đã có qui trình phát hiện sàng lọc các chất gây nghiện bằng HPLC chế độ rửa giải gradient với detector mảng diod, huỳnh quang hoặc điện hóa được công bố (cũng ở tài liệu Clarke’s Analysis of Drugs and Poisons ... đề cập ở trên). Các chất được tách bằng cột Lichrospher RP - 18e (125 × 4,0 mm, 5 um); pha động gồm pha động A là dung dịch đệm triethylamoni phosphat 0,025 mol/L trong nước (pH 3,0); pha động B là acetonitril, chế độ gradient với tốc độ dòng 1 mL/min.
| Thời gian (phút) | %A | %B | Thời gian (phút) | %A | %B |
| 0 | 100 | 0 | 40 | 20 | 80 |
| 30 | 30 | 70 | 45 | 100 | 0 |
| 35 | 30 | 70 | 60 | 100 | 0 |
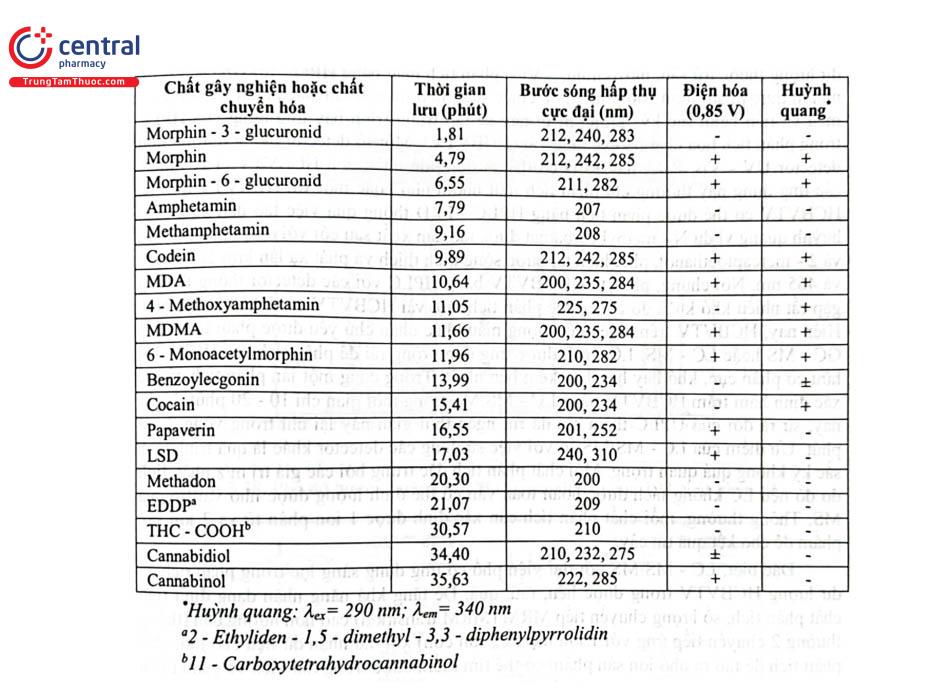
Mẫu thử là 1,0 mL huyết thanh hoặc hoặc nước tiểu được thêm 50 uL dung dịch chuẩn nội (nalorphin 25 ug/L trong nước) và 3,0 mL dung dịch đệm phosphat 0,1 mol/L pH 6. Lắc xoáy trong vài giây, ly tâm nếu dung dịch không trong. Chuyển hỗn hợp lên cột (cột SPE loại Certify solid - phase extraction của Varian đã được hoạt hóa lần lượt bằng 2 mL methanol và 2 mL dung dịch đệm pH 6,0) không dùng chân không. Rửa cột với 1 mL dung dịch đệm pH 6,0 không dùng chân không. Rửa tiếp với 1 mL hỗn hợp dung dịch đệm phosphat pH 6,0 - methanol (80 ; 20), sau đó với 1,0 mL dung dịch acid acetic 1 moVL. Làm khô cột SPE trong 2 phút bằng chân không ở mức tối đa. Làm khô thành cột SPE. Rửa cột bằng 1,0 mL n - hexan với chân không nhẹ và làm khô cột SPE trong 5 phút bằng chân không ở mức tối đa. Rửa giải bằng 1,2 mL hỗn hợp n - hexan - ethyl acetat (80 : 20) không dùng chân không. Phân đoạn này là các thành phần dạng acid. Rửa giải bằng 2,4 mL hỗn hợp dicloromethan - propan - 2 - ol - amoni hydroxyd 25% (80 : 20 : 2) không dùng chân không vào một ống nghiệm khác. Phân đoạn này là các thành phần dạng base. Làm bay hơi cả 2 dịch chiết dưới luồng khí nitơ. Cho vào mỗi ống nghiệm chứa dịch chiết các chất dạng acid 100 FL acetonitril 33% trong nước và lắc xoáy 30 giây. Cho vào mỗi ống nghiệm chứa dịch chiết các chất dạng base 100 UL dung dịch đệm triethylamoni phosphat 0,025 mol/L pH 3 và lắc xoáy 30 giây. Ly tâm 1500 g trong 5 phút. Tiêm 60 UL mẫu thử vào hệ thống sắc ký. LOQ của mỗi chất khoảng 50 kg/L đối với mẫu thử nước tiểu khi phát hiện bằng detector UV và thấp hơn nhiều khi phát hiện bằng detector huỳnh quang hoặc điện hóa. Trong thời gian phân tích khoảng 36 phút đã tách và phát hiện được 41 chất gồm morphin, dẫn chất của Morphin và chất chuyển hóa của chúng; các amphethamin, dẫn chất của amphethamin và chất chuyển hóa của chúng; các chất từ cần sa và chất chuyển hóa của chúng; các chất từ cỏ cựa gà và chất chuyển hóa; methadon và chất chuyển hóa; Bảng 2.18 trình bày các ma túy, dẫn chất và chất chuyển hóa phân tích bằng HPLC theo điều kiện trên.
3 Ứng dụng sắc ký lỏng hiệu năng cao trong phân tích thực phẩm
Hóa học phân tích áp dụng trong phân tích thực phẩm sử dụng các kỹ thuật tách để xác định đặc tính của thực phẩm trên cơ sở các hợp chất cấu thành chính và phụ của chúng. Các hợp chất phụ (hàm lượng nhỏ) thường làm nổi bật những điểm khác biệt quan trọng hơn là các hợp chất chính. Kỹ thuật sắc ký đã được áp dụng rộng rãi; sắc ký khí bị hạn chế sử dụng do độ bền nhiệt của chất phân tích. Dẫn chất hóa có thể sử dụng do các chất tạo thành sau phản ứng bền, nhưng nhìn chung các qui trình này khá tốn thời gian, phức tạp và có thể tạo các sản phẩm phụ khác. HPLC cho phép giải quyết những vấn đề này, đồng thời với nhiều loại detector cho phép phát hiện được hầu hết các chất trong phân tích thực phẩm với độ nhạy, độ chọn lọc đáp ứng yêu cầu. HPLC có thể phân tích các thành phần dinh dưỡng trong thực phẩm như lipid, carbohydrat và các chất liên quan, protein, peptit, acid amin, phenol, vitamin. Kết quả định lượng có thể thu được bằng cách sử dụng chuẩn ngoại hoặc chuẩn nội.
Song song với việc xác định các chất dinh dưỡng trong thực phẩm thì xác định những chất ô nhiễm được đặc biệt coi trọng. Một số chất gây ô nhiễm thực phẩm chính như độc tố nấm mốc, dư lượng chất kháng khuẩn, dư lượng chất kích thích tăng trưởng, dư lượng thuốc trừ sâu, nitrosamin,... Việc phân tích mẫu bằng HPLC đối với các chất ở nhiễm thực phẩm luôn bao gồm bước chiết xuất, tiếp theo là làm sạch mẫu và làm giàu mẫu và tiến hành sắc kỷ. Trong phần này sẽ tập trung trình bày ứng dụng của HPLC trong phân tích hóa chất bảo vệ thực vật (HCBVTV). Một số detector thông thường như detector UV - Vis, PDA hay FLD có thể sử dụng để phân tích HCBVTV. Tuy nhiên, các ứng dụng này thường chỉ phân tích một nhóm nhỏ hoặc một vài HCBVTV đơn lẻ. HCBVTV có thể được phân tích bằng HPLC - FLD thông qua việc tạo dẫn xuất phát huỳnh quang ví dụ N - methyl carbamat được tạo dẫn xuất sau cột với o - phthalaldehyd và 2 - mercaptoethanol, phát hiện tại bước sóng kích thích và phát xạ lần lượt là 330 nm và 465 nm. Nói chung, phân tích HCBVTV bằng HPLC với các detector thông thường gặp rất nhiều khó khăn do chỉ có thể phân tích một vài HCBVTV có tính chất đặc biệt. Hiện nay, HCBVTV trên nhiều đối tượng mẫu khác nhau chủ yếu được phân tích bằng GC - MS hoặc LC - MS. LC - MS được ứng dụng rộng rãi để phân tích các HCBVTV hữu cơ phân cực, khó bay hơi, hay kém bền nhiệt. Trong cùng một lần phân tích có thể xác định hàm trăm HCBVTV bằng LC - MS/MS trong thời gian chỉ 10 - 20 phút. Ngày nay, sự ra đời của UPLC thậm chí đã rút ngắn thời gian này lại chỉ trong vòng 2 - 4 phút. Ưu điểm của LC - MS/MS so với việc sử dụng các detector khác là quá trình tách sắc ký không quá quan trọng. Mỗi chất phẩn tích đặc trưng bởi các giá trị m/z nhất định do đó nếu LC không tách được hoàn toàn vẫn có thể định lượng được nhờ vai trò của MS. Thông thường, mỗi chất phân tích cần xác định được 1 ion phân tử và 2 ion sản phẩm để cho kết quả tin cậy.
Đặc biệt, LC - MS/MS với thư viện phổ có ứng dụng sàng lọc trong phân tích đa dư lượng HCBVTV trong dược liệu, rau, quả. Để tăng khả năng nhận dạng định tính chất phân tích, số lượng chuyển tiếp MRM (MRM transition) cao hơn hướng dẫn (thông thường 2 chuyển tiếp ứng với 1 ion mẹ và 2 ion con) khi thu nhận dữ liệu cho mỗi chất phân tích để tạo ra phổ ion sản phẩm có thể tìm kiếm được trong thư viện và giảm thiểu nguy cơ âm tính hoặc LC - MS/MS, mẫu thử được xử lý theo phương pháp QuEChERS, 193 HCBVTV được cứu phân tích HCBVTV bằng phân tích đồng thời với điều kiện sắc ký: Cột HSS T3 (100 × 2,1 mm, 1,7 pm) của Waters; nhiệt độ 40°C; lưu lượng dòng 0,4 mL/min; pha động A) dung dịch amoni format 5 mM trong nước; pha động B) amoni format 5 mM trong methanol; gradient: 0 - 1,50 phút 35% B, 1,50 - 11,50 phút tăng tuyến tính tới 100% B, 11,50 - 13,00 phút 100% B, 13,01 - 15,00 phút 3% Bị tiêm mẫu 0,1 –L mẫu thử + 30 uL nước (water co - injection). Chế độ MS: số lượng chuyển tiếp MRM kiểu phổ 1291; chế độ ion hóa ESI +/ - ; chuyển cực 5 ms; nhiệt độ interface 350°C; nhiệt độ đường chuyển solvat hóa 150°C; khí phun sương 3 L/phút,
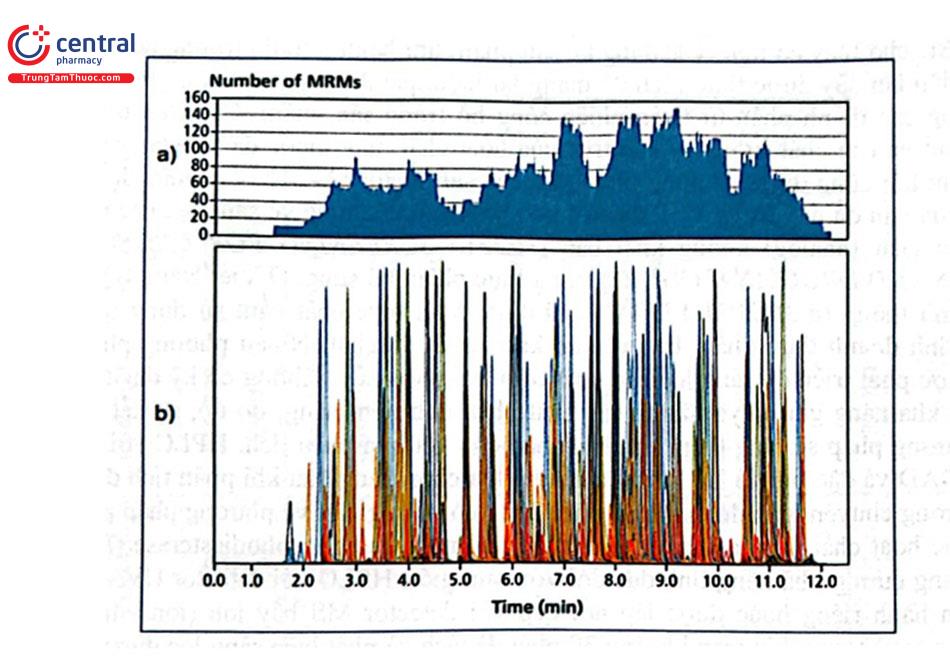
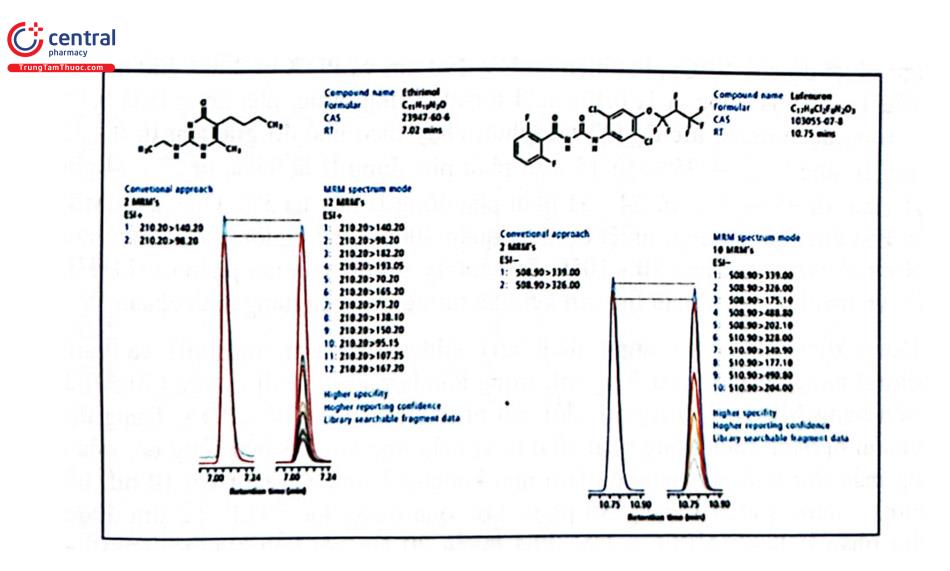
Hình 2.38 là phổ đồ MRM ghi 1291 chuyển tiếp MRM đo 193 loại HCBVTV (0,010 mg/kg). Hiệu năng của phương pháp này được so sánh với phương pháp MRM thông thường theo dõi hai quá trình chuyển tiếp đối với mỗi loại thuốc trừ sâu. Hình 2.39 cho thấy sự so sánh các phương pháp tiếp cận khác nhau trên hai chất ethirimol và lufenuron thêm vào quả bơ với lượng 0,1 mg/kg. Dữ liệu thu được cho thấy bất kể số lượng ion mảnh được giám sát nhiều như thế nào, cường độ tín hiệu tuyệt đối đối với cả hai phương pháp gần như giống hệt nhau ở chế độ ion dương và âm.
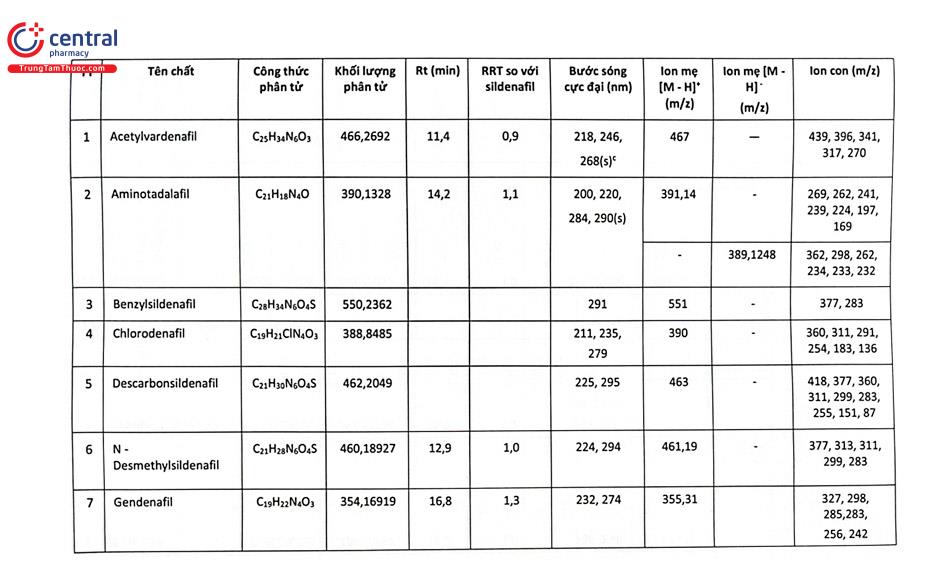
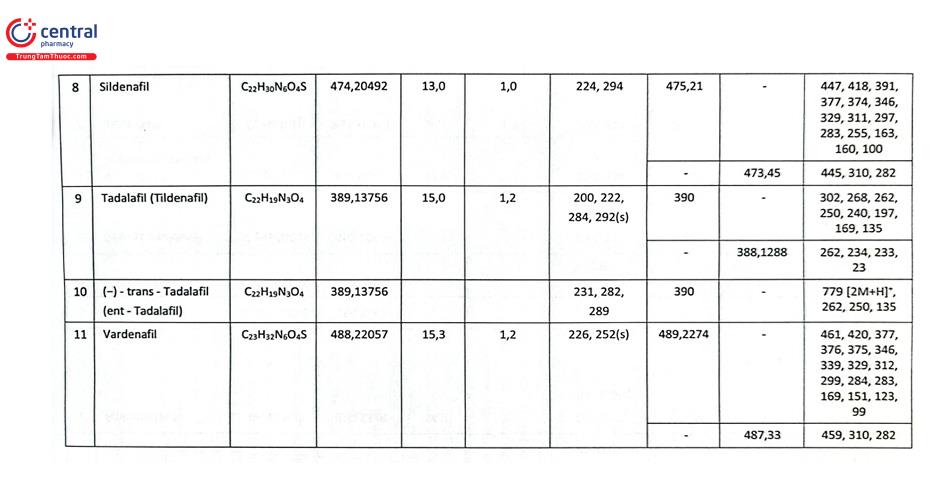
Gần với lĩnh vực dược phẩm là thực phẩm bảo vệ sức khỏe (TPBVSK) xuất hiện tình trạng trộn trái phép các thuốc hóa dược ngày càng nghiêm trọng cả ở trong nước và trên thế giới. Các thuốc hóa dược ở hầu hết các nhóm tác dụng dược lý đã phát hiện có trong các sản phẩm này. Các nhóm thuốc hóa dược điển hình trộn vào chế phẩm đông dược phải kể đến tăng cường khả năng tình dục, giảm cân, tăng cường hiệu suất thể thao, điều trị xương khớp, hạ đường huyết, kháng histamin,... Đã có nhiều nghiên cứu được công bố ở Việt Nam và trên thế giới phát hiện các thuốc hóa dược trong sản phẩm TPBVSK cho thấy có một tỷ lệ đáng kể sản phẩm lưu hành trên thị trường có tình trạng này. Gian lận này được thực hiện để mang lại hiệu quả điều trị mà không thể đạt được chỉ bằng các thành phần từ thiên nhiên công bố trong sản phẩm. Các chất trung gian tổng hợp và các chất tương tự cấu trúc của hoạt chất, hay thuốc đã bị ngừng sản xuất hoặc thu hồi cũng được sử dụng thêm vào các sản phẩm này. Thấy rõ mức độ nghiêm trọng của vấn đề này trong USP đã có một chuyên luận chung về sàng lọc các thuốc và các dẫn chất (analog) không khai báo (<2251> SCREENING FOR UNDECLARED DRUGS AND DRUG ANALOGUES) trong thực phẩm bổ sung. Ở Việt Nam, Bộ Y tế đã ban hành thông tư 10/2021/TT-BYT qui định danh mục chất cấm sử dụng trong sản xuất, kinh doanh thực phẩm bảo vệ sức khoẻ gồm 88 chất. Nhiều phương pháp phân tích được phát triển để sàng lọc các chất trộn trái phép này. Không có kỹ thuật riêng lẻ nào có khả năng giải quyết tất cả các chất phân tích tiềm năng; do đó, sự kết hợp của các phương pháp sẽ tăng thêm sự chắc chắn cho kết quả phân tích. HPLC với detector UV - DAD và đặc biệt là LC - MS là những lựa chọn hàng đầu khi phân tích đối tượng này. Trong chuyên luận đó của USP năm 2021 có hướng dẫn về phương pháp phân tích sàng lọc hoạt chất và các analog của thuốc nhóm ức chế phosphodiesterase (PDE - 5) dùng tăng cường khả năng tình dục đối với nam giới. HPLC với detector UV - PDA có thể tiến hành riêng hoặc được lắp nối tiếp với detector MS bẫy ion (ion - trap mass spectrometer) trong thời gian khoảng 30 phút đã tách và phát hiện sàng lọc được 64 chất. Điều kiện sắc ký như sau: Cột tách C18 (2,1 mm × 15 cm; 5 um) cột Zorbax SB - C18 là phù hợp; nhiệt độ cột 40°C; phát hiện tại UV 290 nm và PDA từ 200 - 400 nm, thể tích tiêm mẫu 1 "L; pha động A là 0,1% acid formic trong nước, pha động B là 0,1% acid formic trong acetonitril, tốc độ 0,2 mL/phút, chạy theo chế độ gradient từ 0 - 15 phút pha động B tăng từ 5 → 95%; từ 15 - 23 phút pha động B là 95%; từ 23 - 24 phút pha động B giảm từ 95 → 5%; từ 24 - 31 phút pha động B duy trì 5%. Điều kiện MS: Kiểu ion hóa ESI âm hoặc dương; nhiệt độ mao quản 300°C; thế nguồn 5 kV; năng lượng và chạm 45 meV; dải quét m/z 90 - 1050. So sánh tỷ số m/z của ion phân tử [M+H]* hoặc [M-HJ và mảnh ion của mẫu thử với kết quả tương ứng của dung dịch chuẩn.
Dung dịch chuẩn là dung dịch của Sildenafil citrat, Tadalafil và Vardenafil hydroclorid nồng độ mỗi chất 5 ng/mL trong hỗn hợp acetonitril - nước (50:50) đối với phát hiện bằng MS và 100 kg/mL đối với phát hiện bằng UV - PDA. Dung dịch thử được chuẩn bị bằng cách đồng nhất số đơn vị mẫu ứng với 1/5 liều công bố, siêu âm 10 - 20 mg mẫu thử hoặc vỏ nang đã làm nhỏ khoảng 3 mm × 3 mm với 10 mL hỗn hợp acetonitril - nước (50:50) trong 30 phút. Lọc qua màng lọc PTFE 0,2 um được dung dịch thử phân tích với HPLC - UV. Pha loãng 20 lần với hỗn hợp acetonitril - nước (50:50) trước khi phân tích bằng LC - MS.
Bảng 2.19 minh họa điều kiện phát hiện một số chất nhóm ức chế PDE - 5 bằng HPLC và LC - MS/MS.
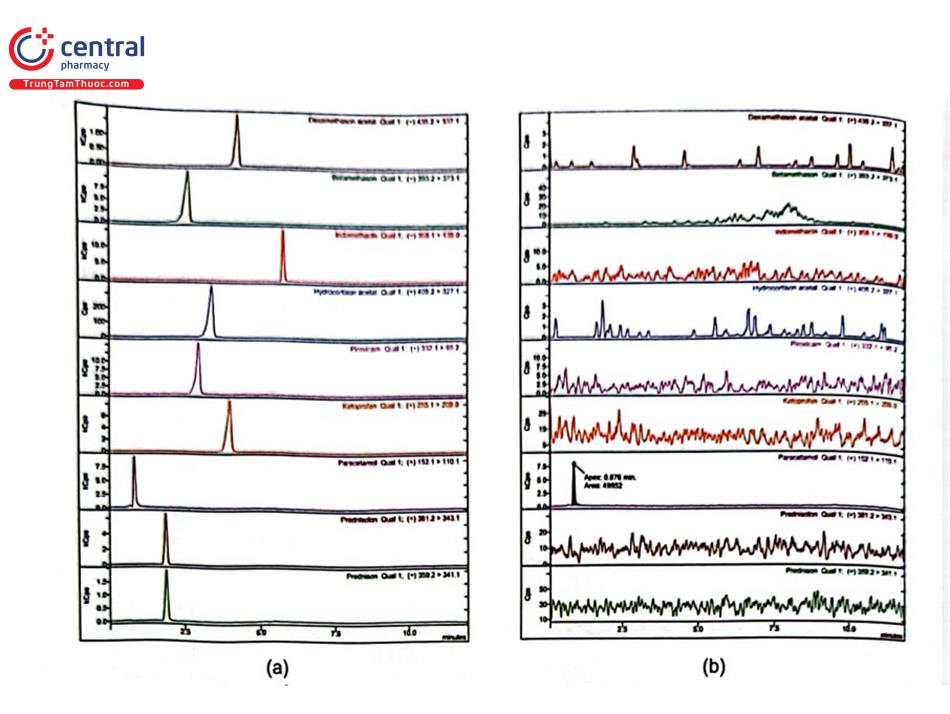
Một nghiên cứu tại Trường Đại học Dược Hà Nội đã xây dựng phương pháp xác định 9 thuốc hóa dược nhóm glucocorticoid và NSAIDs trộn trái phép trong chế phẩm đông dược bằng LC - MS/MS. Các chất được tách bằng cột Restek Ultra II C18 (100 mm × 2,1 mm; 1,9 um), pha động gồm dung dịch acid formic 0,1% trong nước và dung dịch acid formic 0,1% trong acetonitril rửa giải theo chế độ gradient, chế độ ion hóa ESI (+), xử lý số liệu theo chế độ MRM. Các chất được định tính dựa vào ion mẹ và 2 mảnh ion con (4 điểm IP), định lượng dựa vào đáp ứng của mảnh ion con có cường độ lớn và ổn định nhất (mảnh ion con có m/z in đậm).
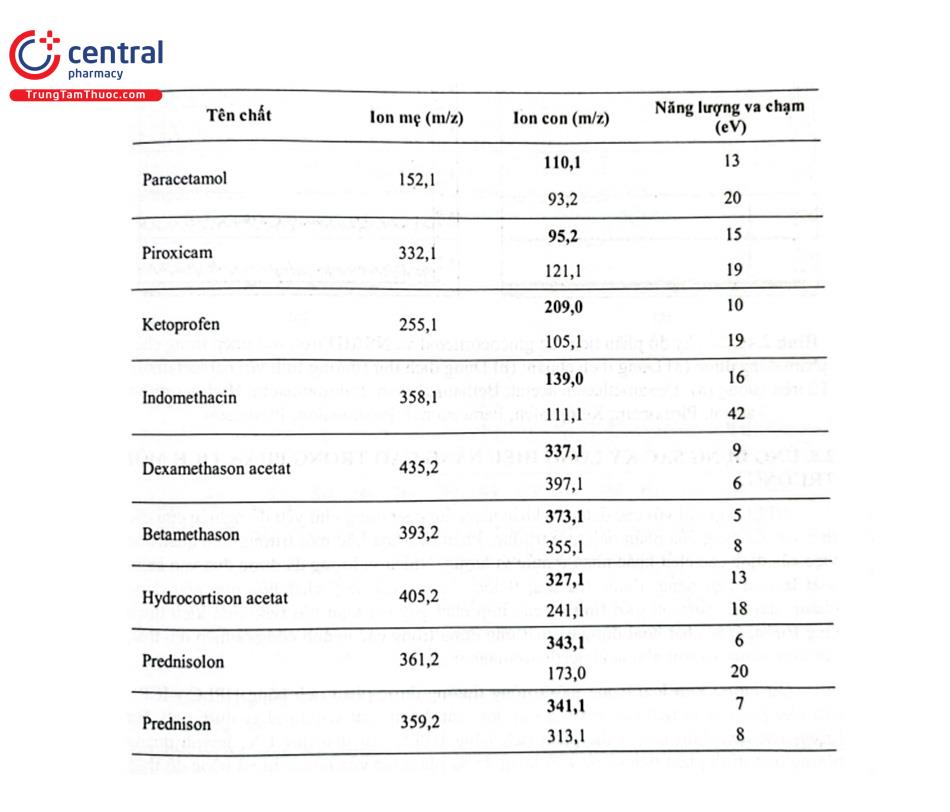
Nghiên cứu cũng đã phát hiện có 11 mẫu dương tính với paracetamol, dexamethason acetat và Indomethacin trong 119 mẫu chế phẩm đông dược dùng trong điều trị hoặc hỗ trợ điều trị cơ xương khớp thu thập được. Kết quả được minh họa ở Hình 2.40.
4 Ứng dụng sắc ký lỏng hiệu năng cao trong phân tích môi trường
HPLC kết nối với các detector khác nhau được sử dụng chủ yếu để nghiên cứu các lĩnh vực đa dạng của phân tích môi trường. Phân tích hóa học môi trường liên quan đến việc xác định các chất khác nhau ở mức vi lượng. Nhiều vi lượng đã được đưa vào kiểm soát là kim loại nặng, thuốc trừ sâu, thuốc nhuộm, các hợp chất hóa học như được phẩm, steroid, hormon giới tính và các hợp chất gây rối loạn nội tiết, chất kích thích tăng trưởng, các chất hoạt động bề mặt ứng dụng trong các ngành công nghiệp nội thất, dệt may, nhựa và sơn như acid perfluorooctanoic,...
Dư lượng kim loại trong môi trường thường được phân tích bằng HPLC - ICP - MS cho phép phân tích hầu hết các kim loại cần kiểm soát với độ nhạy dưới ppb, Dư lượng các chất hữu cơ có thể phân tích bằng HPLC với detector UV, huỳnh quang nhưng quá trình phân tích sẽ rất khó khăn do sự phức tạp của nền mẫu và nồng độ thấp của chất phân tích có trong mẫu. LC - MS/MS luôn là lựa chọn đầu tiên khi phân tích dư lượng các chất hữu cơ trong mẫu thử môi trường, trong trường hợp cần xác định các sản phẩm phân hủy thì LC - MS với kiểu bẫy ion ở chế độ MS" sẽ nâng cao độ nhạy cũng như định danh chính xác chất phân tích. Trong phân tích môi trường, giai đoạn xử lý mẫu rất quan trọng cho phép loại tạp và làm giàu mẫu. Chiết pha rắn với các loại pha tĩnh khác nhau tùy vào bản chất chất phân tích được sử dụng, đồng thời sử dụng chuẩn nội đồng vị cũng tăng độ chính xác của kết quả thử nghiệm.
Một ví dụ về phân tích môi trường là xác định dư lượng hóa chất bảo vệ thực vật trong nước. Lĩnh vực này rất được quan tâm và có nhiều phương pháp được thiết lập cho phép áp dụng ở các phòng thí nghiệm khác nhau trên phạm vi toàn thế giới. Phương pháp US EPA 531.2 của Bộ môi trường Mỹ (United States Environmental Protection Agency: EPA) cung cấp hướng dẫn giám sát 11 hóa chất nhóm carbamat trong nước ngầm, nước bề mặt và nước uống. Mẫu thử được thêm chuẩn nội (4 - bromo - 3,5 dimethylphenyl N - methylcarbamat: BDMC), lọc và tiêm vào hệ thống sắc ký với thể tích có thể từ 200 - 1000 uL. Các chất phân tích được tách bằng cột C18 dùng cho phân tích carbamat (ví dụ Acclaim Carbamate 4,6 × 250 mm; 5 um) với pha động là methanol - nước rửa giải theo chế độ gradient. Sau khi rửa giải khỏi cột, chất phân tích được thủy phân trong phản ứng sau cột với natri hydroxyd 0,075 N ở 80 - 100°C để tạo thành methyl amin. Methyl amin sẽ phản ứng với o - phthalaldehyd và 2 - mercaptoethanol (hoặc N, N dimethyl - 2 - mercaptoetylamin) để tạo thành isoindol, phát hiện bằng detector huỳnh quang tại bước sóng kích thích 330 nm và bước sóng phát xạ 465 nm. (Hình 2.41).
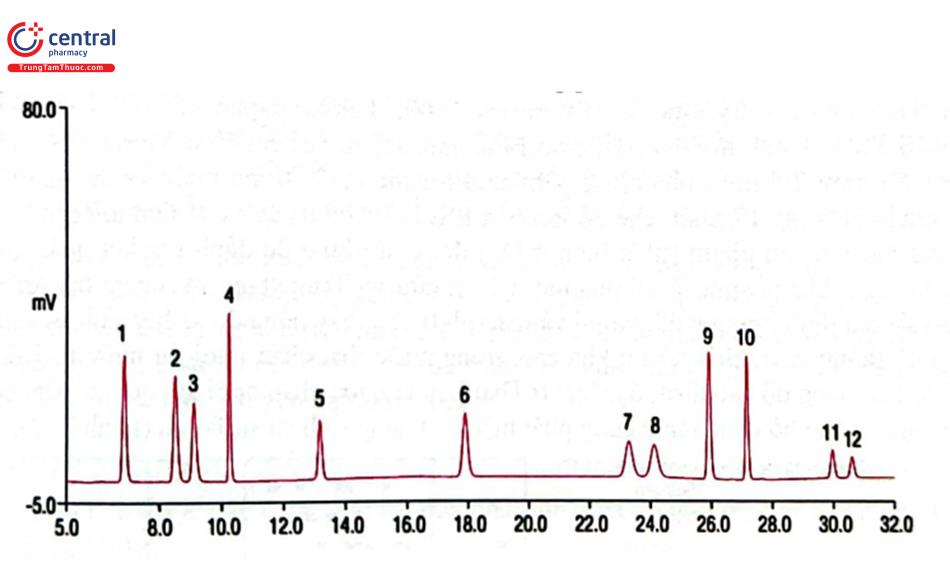
1. Aldicarb sulfoxid, 2. Aldicarb sulfon, 3. Oxamyl, 4. Methomyl, 5. 5-Hydroxycarbofuran, 6. Aldicarb, 7. Propoxur, 8. Carbofuran, 9. Carbaryl, 10. 1 - Naphthol, 11. Methiocarb, 12. BDMC (surrogate)
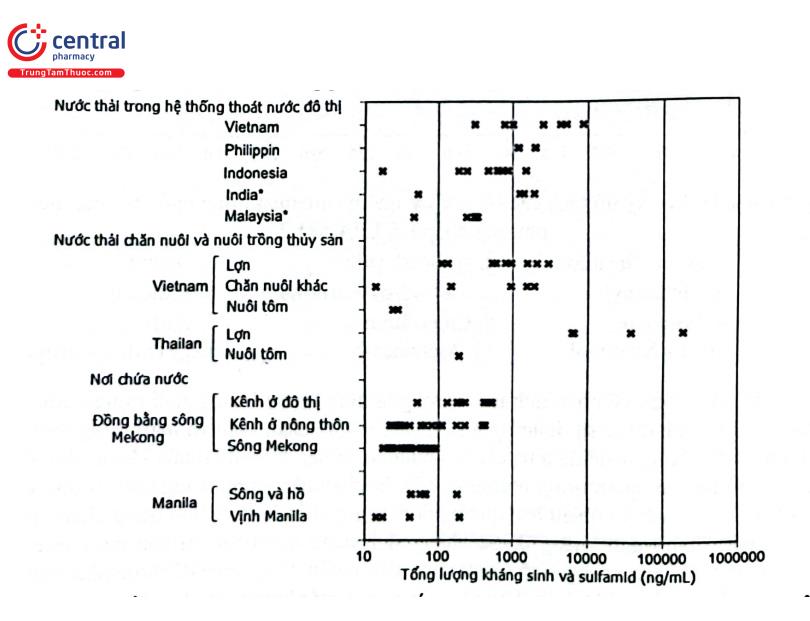
Một ví dụ nữa về phân tích môi trường là định lượng kháng sinh trong nước thải. Thuốc kháng sinh được sử dụng rộng rãi trong y học, chăn nuôi và nuôi trồng thủy sản với mục đích phòng hoặc điều trị các bệnh nhiễm trùng. Mặc dù thuốc kháng sinh đóng một vai trò hết sức quan trọng nhưng sự tồn dư của kháng sinh trong môi trường nước nói chung có thể gây ra nhiều hậu quả nghiêm trọng, điển hình là tình trạng kháng thuốc kháng sinh. Tình trạng kháng thuốc kháng sinh đang là một thảm họa mà con người phải đối mặt. Để loại kháng sinh trong nước thải nhiều công nghệ đã được phát triển và thu được kết quả đáng khích lệ. Tuy nhiên, vẫn còn một lượng rất nhỏ kháng sinh tồn tại trong nước thải có thể dẫn đến hậu quả làm biến đổi gen của hệ sinh thái vi sinh vật trong nước dẫn tới tình trạng kháng kháng sinh. Do yêu cầu về độ nhạy nên có thể phân tích một số kháng sinh có khả năng phát huỳnh quang bằng HPLC với detector huỳnh quang như các kháng sinh họ floquinolon, nhưng định lượng dư lượng đa kháng sinh trong nước thải chủ yếu vẫn sử dụng LC - MS/MS. Trong một nghiên cứu hợp tác giữa các nhà khoa học Nhật Bản, Việt Nam, Philippin và Malaysia đã đánh giá mức độ tồn dư kháng sinh và sulfamid trong hệ thống thoát nước đô thị, kênh rạch và các sông bị ảnh hưởng nặng nề bởi nước thải ở Việt Nam (Hà Nội, Thành phố Hồ Chí Minh, Cần Thơ), Philippin (Manila), Indonesia (Jakarta), An Độ (Kolkata) và Malaysia (Kuala Lumpur) từ năm 2006 đến năm 2010. Nhóm nghiên cứu đã sử dụng LC - MS/MS để phân tích. Mẫu thử được xử lý sơ bộ và thêm chuẩn nội gồm sulfamethoxazol - d, Clarithromycin - d, Roxithromycin - d và oxytetracyclin - i3C, - d3, sau đó tiếp tục chiết pha rắn bằng cột 200 mg Oasis HLB resin, Waters. Các chất phân tích gồm 7 sulfonamid (sulfapyridin, sulfamethoxazol, sulfathiazol, sulfamerazin, sulfamethizol, sulfamethazin, sulfadimethoxin), 9 kháng sinh (azithromycin, Erythromycin, roxithromycin, clarithromycin, tylosin, Lincomycin, tetracyclin, doxycyclin, oxytetracyclin) và Trimethoprim được phân tích bằng hệ thống sắc ký của Agilent series 1100, Tokyo, Japan kết nối với hệ thống MS/MS TSQ Quantum 7000, Thermo Finnigan, Japan với cột tách Xterra MS C18 (2,1 mm × 50 mm; 2,5 um), pha động gồm acid formic 0,1% trong nước và acetonitril, rửa giải gradient trong 17 phút, chế độ ion hóa ESI (+). Giá trị m/z của tiền ion (phát hiện ở Q1) và hai ion sản phẩm (phát hiện ở Q3) được sử dụng để đánh giá kết quả. Kết quả tổng lượng 9 kháng sinh, 7 sulfonamid nghiên cứu và Trimethoprim (ngoại trừ Ấn Độ và Malaysia chỉ định lượng sulfonamid và macrolid) cho thấy nồng độ kháng sinh và sulfamid trong hệ thống nước thải đô thị khá cao, trong nước thải chăn nuôi thì nước thải từ chăn nuôi lợn có nồng độ cao nhất, đặc biệt ở Thái Lan là nước chăn nuôi lợn qui mô lớn, nước ở kênh, rạch, sông hồ chứa nước cũng phát hiện có kháng sinh và sulfamid (Hình 2.42).
5 Kết luận
Hệ thống HPLC rất đa dạng với nhiều loại pha tĩnh, nhiều loại detector khác nhau và sự linh hoạt khi sử dụng pha động cho phép tách và phân tích các chất khó bay hơi và các chất dễ bị phân hủy nhiệt có cấu trúc rất khác nhau, từ những chất có khối lượng phân tử nhỏ đến các chất có khối lượng phân lớn, các kim loại, anion vô cơ, các chất hữu cơ, bao gồm cả những chất có nguồn gốc thiên nhiên, sản phẩm sinh học và tổng hợp. Phương pháp dễ dàng đáp ứng yêu cầu về độ đúng, độ chính xác, độ chọn lọc cũng như độ nhạy ứng với các qui trình phân tích tương ứng. Nhiệm vụ của nhà phân tích là phải lựa chọn, đánh giá điều kiện phân tích để phù hợp với đối tượng mẫu và yêu cầu qui định.
Phạm vi ứng dụng của phương pháp rất rộng, với khoảng nồng độ có thể phân tích từ cỡ hàng trăm ppm đến dưới ppb, HPLC là phương pháp rất phổ biến và được ứng dụng trong nhiều ngành khác nhau như kiểm nghiệm thuốc, kiểm nghiệm thực phẩm, phân tích môi trường, phân tích dịch sinh học, ...Trong quá trình phát triển thuốc và lưu hành thuốc, HPLC càng quan trọng hơn khi nó được ứng dụng trong hầu hết các giai đoạn nghiên cứu từ phân tích cấu trúc, xác định độ tinh khiết của chất tổng hợp được, phân tích dược động học, sinh khả dụng của thuốc, tiêu chuẩn hóa chất lượng thuốc và kiểm tra, giám sát chất lượng thuốc lưu hành, cũng như phục vụ giám sát điều trị.
Tuy nhiên, giá thành thiết bị, hóa chất, vật tư tiêu hao cao là một điểm hạn chế khi ứng dụng vào thực tiễn. Đặc biệt, yêu cầu về chất chuẩn trong qui trình phân tích cũng là một khó khăn mà nhà phân tích phải đối diện. Do đó, cần nhận thức rõ khả năng ứng dụng của phương pháp cũng như năng lực thực tiễn của phòng thí nghiệm để trang bị thiết bị HPLC phù hợp với yêu cầu phân tích.
6 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Ứng dụng của sắc ký lỏng hiệu năng cao trong kiểm nghiệm thuốc", Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 67 - 105. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
