3 Ứng dụng của sắc ký khí (CG) trong kiểm nghiệm

Sắc ký khí (Gas Chromatography: GC) có nhiều ứng dụng quan trọng trong kiểm nghiệm thuốc, phân tích chất bảo vệ thực vật hay phân tích chất gây nghiện. Trong bài viết này, Trung Tâm Thuốc Central Pharmacy (trungtamthuoc.com) xin gửi đến bạn đọc thông tin về ứng dụng của kỹ thuật sắc ký khí.
1 Ứng dụng sắc ký khí trong kiểm nghiệm thuốc
Sắc ký khí có thể phân tích được những chất bay hơi được ở điều kiện phân tích, không phân hủy. Có thể đặt nhiệt độ cột thấp hơn nhiệt độ sôi của chất phân tích nhưng nếu áp suất hơi đủ lớn vẫn phân tích được bằng GC. Đa số các chất khí có mùi mà mũi người phát hiện được có thể phân tích bằng GC. Những chất phân cực, ít bay hơi phản ứng với thuốc thử thích hợp thành các dẫn xuất ổn định, dễ bay hơi và ít phân cực và phân tích được bằng sắc ký khí.
1.1 Định tính
Tương tự như đối với HPLC. Mẫu thử được thực hiện song song với mẫu chuẩn và so sánh thời gian lưu píc chất phân tích trên sắc ký đồ của mẫu thử và mẫu chuẩn, nếu trùng nhau thì được coi là dương tính.
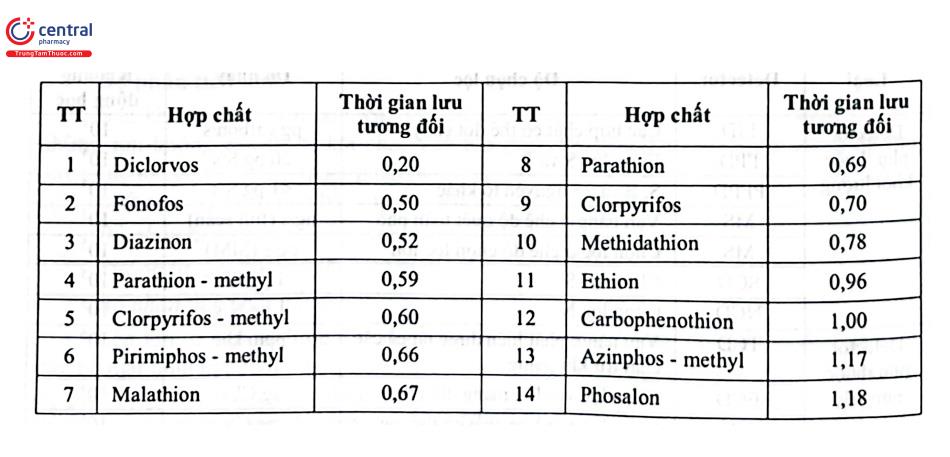
Một cách định tính nữa cũng được sử dụng là dựa vào thời gian lưu tương đối. Trong sắc ký khí hay sử dụng tỷ số thời gian lưu của píc chất phân tích và thời gian lưu pic của một chất đã biết là chất chuẩn nội. Ví dụ, định tính một hóa chất bảo vệ thực vật nhóm phospho hữu cơ trong dược liệu theo Dược điển Việt Nam V, dựa vào thời gian lưu tương đối của chất phân tích với chất chuẩn nội carbophenothion. Điều kiện phân tích gồm cột tách poly(dimethyl)siloxan (30 m × 0,32 mm; 0,25 um); khí mang: H/He/Nz; detector: NPD; chương trình nhiệt độ buồng cột: 80°C trong 1 phút, tăng 30°C/ phút tới 150°C, duy trì trong 3 phút, tăng 4°C/ phút tới 280°C và duy trì trong 1 phút; nhiệt độ bộ phận tiêm mẫu: 250°C; nhiệt độ detector: 275°C; thể tích tiêm mẫu: đảm bảo thời gian lưu tương đối tương đối đạt yêu cầu như Bảng 4.8.
Đối với GC - MS, định tính còn dựa vào so sánh tỷ lệ m/z của các mảnh phổ (hoặc chồng phổ khối) của mẫu thử với chuẩn làm song song hoặc trong thư viện phổ. Khi định tính dựa vào thư viện phổ, thiết bị GC - MS có độ phân giải càng cao thì độ chính xác của phép thử càng được nâng lên, đặc biệt đối với chất chữa biết. Thông thường yêu cầu độ phân giải của thiết bị ≥ 104
1.2 Định lượng
Nhiều dược chất, tá dược dạng nguyên liệu và chế phẩm được định lượng bằng GC. Detector sử dụng nhiều nhất là FID do chất phân tích chủ yếu là các hydrocarbon. Phương pháp chuẩn nội thường được sử dụng để đảm bảo chính xác lượng mẫu sắc ký vì thể tích mẫu phân tích trong GC rất nhỏ. Phương pháp chuẩn hóa diện tích cũng được sử dụng định lượng trong một số trường hợp. Cột sắc ký chủ yếu là cột mao quản loại WCOT. Bản chất pha tĩnh, cũng như yêu cầu kỹ thuật của cột tách (chiều dài, đường kính trong, bề dày lớp pha tĩnh) được lựa chọn tùy thuộc vào đặc tính chất phân tích. Khi định lượng mẫu đơn giản có thể thực hiện ở điều kiện đẳng nhiệt cột, còn nếu tách mẫu phức tạp chương trình hóa nhiệt độ cột được sử dụng. Chất phân tích có thể được phân tích trực tiếp hoặc dẫn xuất tùy theo đặc tính bay hơi của nó. Dung dịch mẫu thử và dung dịch mẫu chuẩn thường có nồng độ xấp xỉ bằng nhau, một số trường hợp thiết lập đường chuẩn thì nồng độ dung dịch thử phải nằm trong khoảng nồng độ tuyến tính của đường chuẩn. Phép định lượng yêu cầu làm lặp lại tối thiểu 2 lần với mẫu chuẩn và 3 lần với mẫu thử, từ đó xác định hàm lượng trung bình để báo cáo kết quả với RSD không được lớn hơn 2,0%.
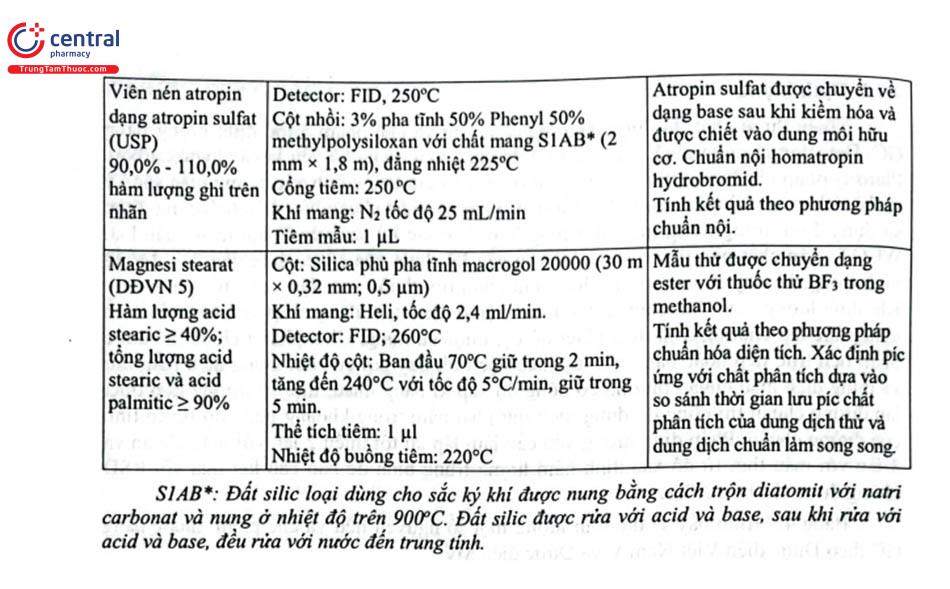
Bảng 4.9 trình bày ví dụ định lượng một số nguyên liệu và chế phẩm thuốc bằng GC theo Dược điển Việt Nam V và Dược điển Mỹ.

Trong bào chế thuốc tiêm, thuốc nhỏ mắt, nhỏ tai, một thành phần quan trọng trong đó là các chất làm giảm thiểu nguy cơ xâm nhập của vi khuẩn cho phần còn lại sau khi đã dùng một phần chế phẩm, đó là các chất bảo quản kháng khuẩn. Các ester của acid p - hydroxybenzoic, phenol, benzyl alcol và clorobutanol là các chất bảo quản kháng khuẩn hay được sử dụng trong thành phần của những dạng thuốc trên. Có thể xác định các chất này bằng phương pháp HPLC và GC, trong đó GC dùng cột nhồi được sử dụng nhiều với điều kiện sắc ký khá đơn giản. (Bảng 4.10).
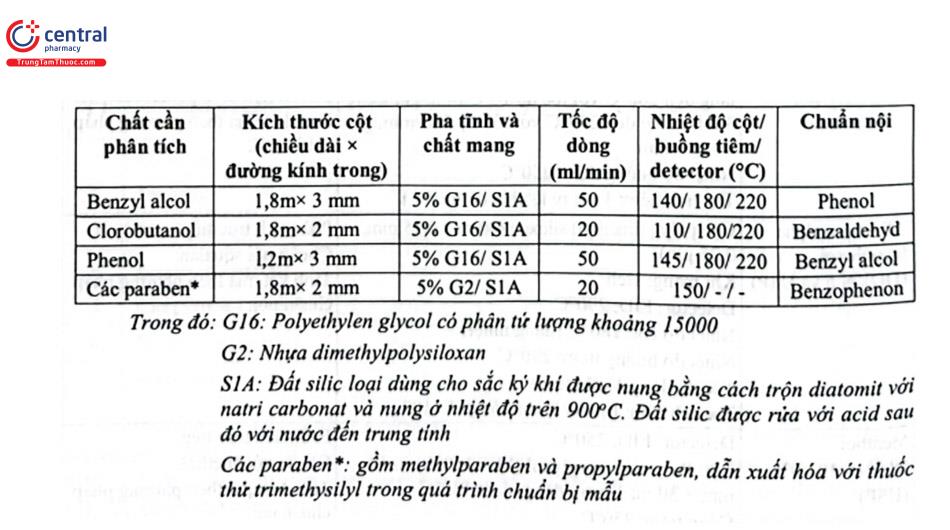
Các chất đều được phát hiện bằng detector FID với khí mang heli hoặc nitơ. Ngoài điều kiện sử dụng cột nhồi như trên, các chất benzyl alcol, clorobutanol, phenol có thể được tách bằng sắc ký khí sử dụng cột mao quản với các điều kiện cụ thể như sau: Cột DB1 (30 m × 0,25 mm ID × 0,25 um), pha tĩnh 100% dimethylpolysiloxan, tốc độ dòng 1,2 mL/min, nhiệt độ cột là 320°C.
1.3 Xác định tạp chất
Tạp chất trong thuốc phân tích bằng GC gồm sản phẩm phân hủy, tạp chất liên quan, tạp chất bay hơi và dung môi tồn dư. Xác định tạp chất liên quan, tạp phân hủy có thể thực hiện theo 3 cách tương tự như đối với HPLC, bao gồm chuẩn hóa diện tích, sử dụng chất chuẩn và pha loãng. Còn xác định tạp chất bay hơi và dung môi tồn dư được thực hiện bằng phương pháp so sánh với chuẩn.
1.3.1 Tạp chất liên quan, tạp chất định danh
Đa số qui trình xác định tạp chất trong thuốc bằng GC được thực hiện với detector FID, nhiệt độ cột tách được chương trình hóa để đạt được hiệu quả tách tốt.
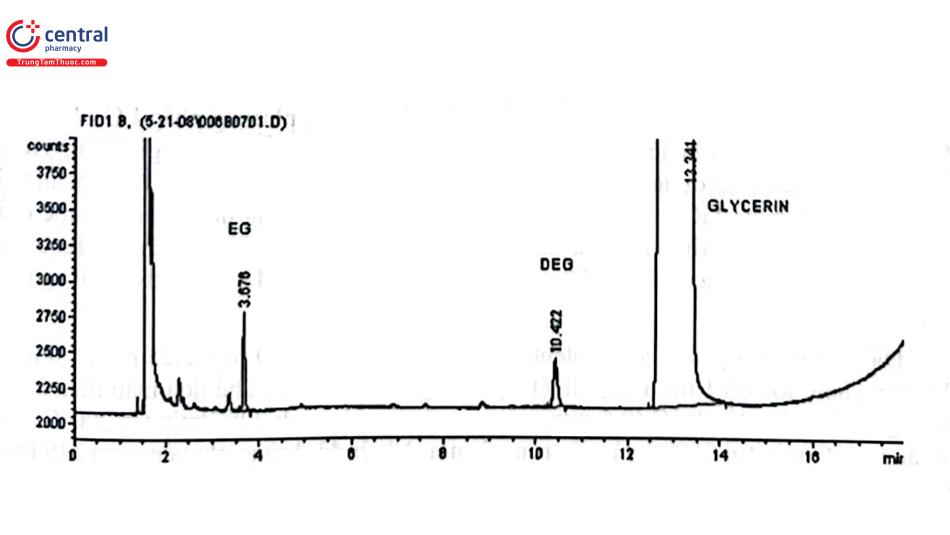
Hình 4.9 trình bày sắc ký đồ của phép thử xác định tạp chất liên quan theo phương pháp chuẩn hóa diện tích của nguyên liệu Glycerin theo tiêu chuẩn USP. Dung dịch thử là dung dịch chế phẩm trong nước nồng độ 50 mg/ml. Mẫu thử được tách bằng cột polycyanopropylphenyl siloxan 6% và polydimethylsiloxan 94% (30 m × 0,53 mm; 3,0 um). Khí mang: Heli, tốc độ 38 cm/s. Thể tích tiêm mẫu: 0,5 ul, tỷ lệ chia dòng: 1 : 10. Nhiệt độ buồng tiêm: 220 °C. Detector: FID đặt ở 250 °C. Nhiệt độ cột: tăng tuyến tính từ 100 đến 220°C, tốc độ 7,5°C/ min và giữ ở 220°C trong 4 phút.
Giới hạn cho phép:
Mỗi tạp kể cả ethylen glycol (EG) và diethylen glycol (DEG):<0,1%.
Tổng tạp: ≤ 1,0%.
1.3.2 Tạp chất nitrosamin
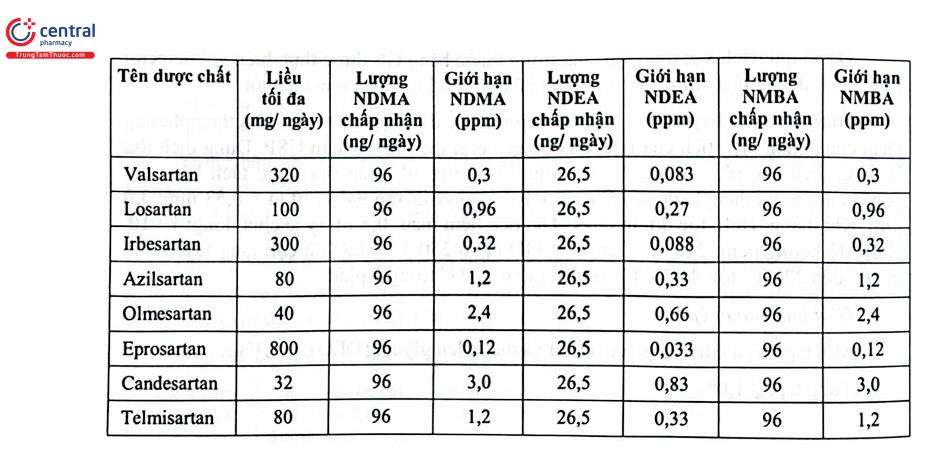
Bắt đầu từ tháng 7 năm 2018, FDA thông báo thu hồi Valsartan do có tạp N - nitrosodimetylamin (NDMA). Điều tra thêm dược chất làm thuốc và thuốc thành phẩm trong nhóm dược chất chẹn thụ thể angiotensin từ tất cả các nhà sản xuất dẫn đến quyết định thu hồi thêm Irbesartan và losartan. Các sản phẩm này phát hiện có chứa NDMA và N - nitrosodiethylamine (NDEA), cả hai chất có khả năng gây ung thư ở người.
Sau đó, các tạp chất khác, N nitrosoethylisopropylamin (NEIPA), N nitrosodiisopropylamin (NDIPA), N nitrosodibutylamin (NDBA) và acid N nitrosomel - 4 - amino - butyric (NMBA) được cảnh báo. Trong khoảng 3 năm (2018 - 2020), hơn 1100 lô thuốc sartan (valsartan, Losartan và irbesartan) đã được thu hồi do chúng chứa những tạp chất này vượt giới hạn. Giới hạn tạm thời được EMA và US FDA qui định cụ thể đối với các thuốc nhóm sartan được trình bày ở Bảng 4.11.
Xác định nitrosamin trong dược phẩm có thể sử dụng phương pháp LC - MS/MS, GC - FID và GC - MS hoặc GC - MS/MS kết nối bộ tiêm mẫu không gian hơi. Trong đó, sắc ký khí được sử dụng chủ yếu, đặc biệt là GC - MS/MS. Một qui trình phân tích năm nitrosamin bằng GC - MS/MS của hãng Agilent ứng dụng đối với các dược chất valsartan, olmesartan, irbesartan và losartan đã được công bố. Đường chuẩn được xây dựng trong khoảng nồng độ 2,5 - 100 ng/mL với chuẩn nội NDMA C13 - d6 (~ 50 ng/mL) trong methylen clorid.
Điều kiện sắc ký: với cột Agilent J&W VF - WAXms (30 m × 0,25 mm, 1,0 um); Khí mang heli, tốc độ 1 mL/min; Nhiệt độ buồng tiêm: 250°C; Thể tích mẫu tiêm 2 HL; chương trình nhiệt độ: 40°C (0,5 min), sau đó tăng tuyến tính 20°C/min tới 200°C, tiếp tục tăng 60°C/min tới 250°C và giữ trong 3 min. Nhiệt độ đường chuyển sang MS (MS Transfer Line Temperature): 250°C.
Điều kiện MS: Ion hóa kiểu va chạm ion mức năng lượng 40 eV, nhiệt độ nguồn ion 250°C; nhiệt độ Q1 và Q2 là 150°C; tốc độ khí va chạm (nitơ): 1,5 mL/min; tốc độ khí làm nguội (He): 4 mL/min. Thông số MS như sau:

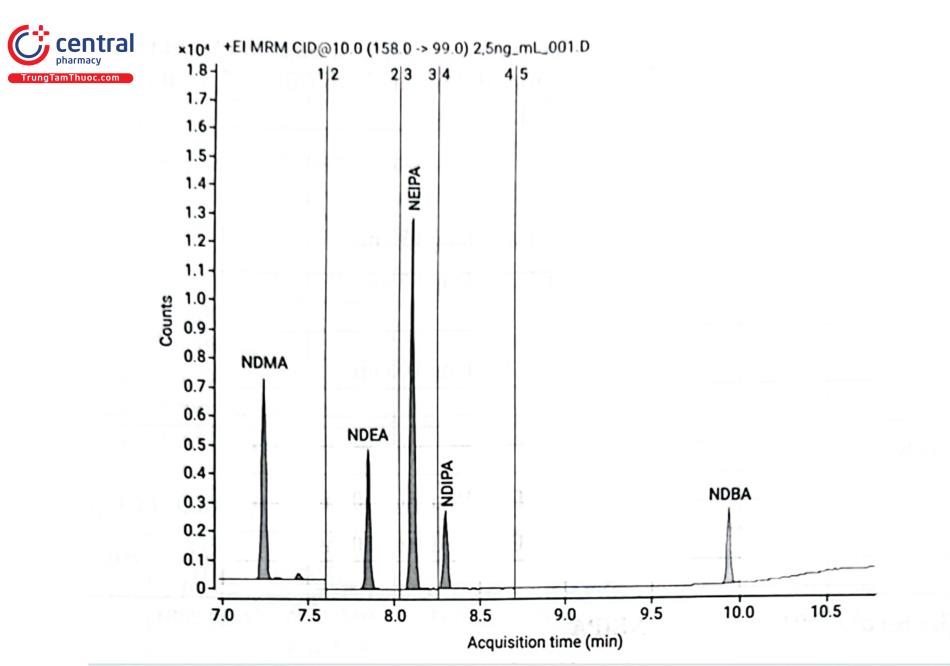
LOQ của các chất rất thấp từ 0,0025 ppm (NDBA); đến 0,005 ppm (NDEA, NEIPA, NDIPA) và cao nhất là 0,008 ppm (NDMA) đáp ứng tốt yêu cầu phương pháp phân tích để xác định dư lượng 5 nitrosamin này trong dược phẩm.
1.3.3 Xác định dung môi tồn dư
Dung môi tồn dư trong dược phẩm là các chất hữu cơ bay hơi, được sử dụng hoặc sinh ra trong quá trình sản xuất các dược chất, tá dược hoặc quá trình bào chế các dược phẩm. Các dung môi này không loại bỏ được hoàn toàn trong quá trình sản xuất. Sử dụng dung môi thích hợp có thể nâng cao sản lượng hoặc quyết định các đặc tính như dạng tinh thể, độ tinh khiết, tính hoà tan của sản phẩm. Vì vậy, đôi khi dung môi là yếu tố quyết định trong qui trình tổng hợp. Về chất lượng, chỉ yêu cầu kiểm tra đối với các dung môi đã được sử dụng hay được sinh ra trong quá trình sản xuất hoặc tinh chế các dược chất, tá dược hoặc thuốc thành phẩm.
Trong hướng dẫn của ICH, các dụng môi tồn dư được phân nhóm theo độc tính của chúng. Các dung môi nhóm 1 là chất gây ung thư có nguy cơ gây hại cho cả người dùng và môi trường. Cần tránh sử dụng những dung môi này nhưng nếu chúng được sử dụng thì cần phải kiểm soát chặt chẽ để đảm bảo chỉ có tạp chất ở mức độ vi lượng trong sản phẩm cuối cùng. Các dung môi nhóm 2 là chất không gây ung thư cho động vật và nồng độ của chúng cần được giới hạn trong các chế phẩm và nguyên liệu làm thuốc. Các dung môi nhóm 3 có tiềm năng độc hại thấp và nồng độ có thể cho phép lên đến 0,5%. Do đó, các dung môi nhóm 3 có thể được thử nghiệm bằng các kỹ thuật không đặc hiệu, chẳng hạn như mất khối lượng khi làm khô.
Vì các dung môi tồn dư không có tác dụng điều trị, các dung môi này phải được loại bỏ đến mức tối đa để đạt được các yêu cầu kỹ thuật của sản phẩm, việc thực hành tốt sản xuất (GMP) hoặc các yêu cầu chất lượng khác. Dược phẩm phải chứa một mức dung môi tồn dư không được cao hơn các dữ liệu an toàn. Phải tránh dùng một số dung môi có độc tính không thể chấp nhận được (dung môi Nhóm 1), trừ khi lợi ích của việc sử dụng chúng được xác định chắc chắn. Một số dung môi có độc tính ít nguy hiểm hơn (dung môi Nhóm 2) cũng cần phải dùng hạn chế, để bảo vệ người bệnh khỏi tác dụng độc hại. Tốt nhất là dùng các dung môi ít độc (dung môi Nhóm 3).
Trong Dược điển Việt Nam và các Dược điển tham chiếu như USP, BP, EP đều có hướng dẫn điều kiện phân tích đối với 3 nhóm dung môi. Tuy nhiên, phương pháp phân tích phải được thẩm định để chứng minh điều kiện phân tích là phù hợp các yêu cầu về độ chọn lọc, độ tuyến tính, độ đúng, độ chính xác, đặc biệt là giới hạn phát hiện và giới hạn định lượng phù hợp để phát hiện và định lượng dung môi cần phân tích.
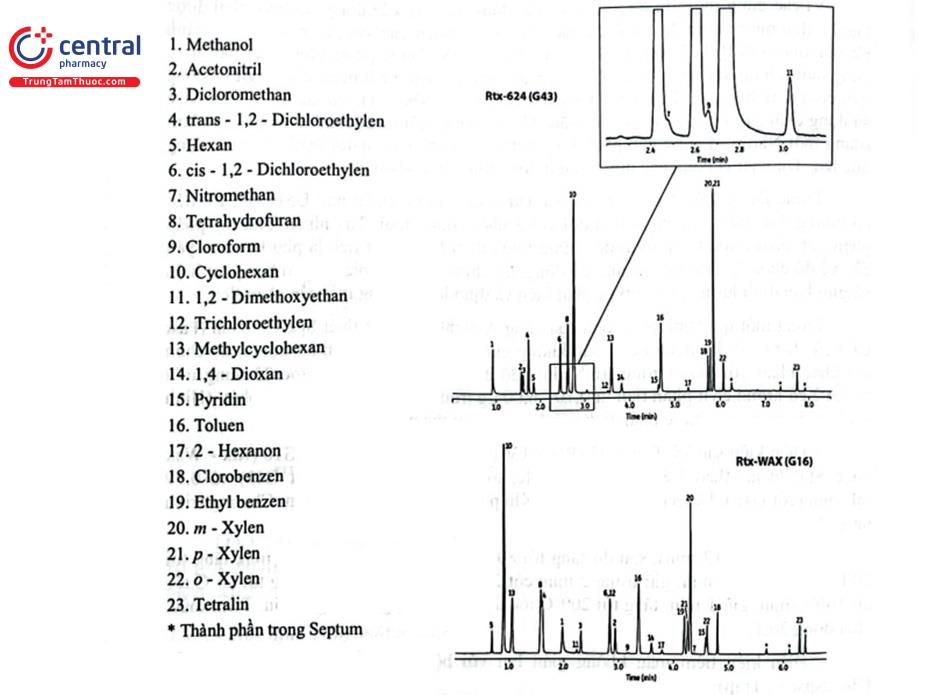
Trong một qui trình phân tích của hãng Agilent, sử dụng thiết bị GC nhanh (Fast GC) của hãng với kỹ thuật tiêm mẫu không gian hơi, phân tích trên hai cột có độ chọn lọc khác nhau với hai chương trình nhiệt độ đã tách và xác định được 23 dung môi nhóm 2 có lượng chất phân tích từ 0,10 - 6,00 ug trong thời gian khoảng 10 phút. (Hình 4.11). Các dung môi được phân tích với điều kiện cụ thể như sau:
- Điều kiện sắc ký: Cột G43 (Rtx - 1301 hoặc Rtx - 624) và cột G16 (Rtx - Wax hoặc Stabilwax) theo USP. Khí mang He, tốc độ 0,85 mL/min (cột G43) và 0,99 mL/min (cột G16). Detector: FID 250°C. Khí phụ trợ: tốc độ 45 mL/min. Chương trình nhiệt độ:
Cột 1: 50°C (2 min), sau đó tăng tới 80°C tốc độ 20°C/min, giữ 1 min, tăng tới 200°C tốc độ 40°C/min, giữ trong 2 min; cột 2: 35°C (2 min), sau đó tăng tới 60°C tốc độ 100°C/min, giữ 1 min, tăng tới 200°C tốc độ 40°C/min, giữ trong 2 min. Tiêm mẫu: chia dòng 20:1
Điều kiện tiêm mẫu không gian hơi với bộ tiêm mẫu không gian hơi bẫy (Headspace - Trap):
Nhiệt độ buồng tiêm (Inj, Temp,): 220°C Chuyển Nhiệt độ đường chuyển: 220°C Nhiệt độ lò van (Valve Oven Temp,): 220°C Tốc độ nghỉ (Standby flow rate): 50 mL/min Bẫy Nhiệt độ nghỉ (Standby Temp); 40°C Nhiệt độ quét của bẫy (Trap Sweep Temp): 40°C Preheat Mixer: On Làm nóng trước (Preheat) Thời gian trộn: 2,0 min | Bộ phận trộn làm nóng (Preheat Mixer) Thời gian ổn định: 0,5 min Nhiệt độ mẫu thử: 80°C Tốc độ quét (Sweep Flow Rate): 75 mL/min Thời gian quét (Sweep Flow Time): 5,0 min Làm khô (Dry Purge): 10,0 min, tốc độ 100 mL/min ở 25°C Phản hấp phụ Nhiệt độ làm nóng trước: 245°C Phản hấp phụ: 1,0 min ở 250°C Nhiệt độ bẫy (Trap Bake Temp): 260°C Thời gian bẫy: 6,0 min Tốc độ bẫy: 450 mL/min |
Kiểm soát dung môi tồn dư không những yêu cầu trong kiểm nghiệm nguyên liệu mà còn phải thực hiện đối với thuốc thành phẩm. Công nghệ bào chế hiện nay với mục tiêu sử dụng phương pháp bào chế hiện đại, kết hợp tối ưu hóa công thức để tăng cường Sinh khả dụng như tăng độ ổn định của dược chất trong chế phẩm được ứng dụng rộng rãi. Một trong số đó là thành phần dung môi hữu cơ được cho vào trong quá trình bào chế. Ví dụ chế phẩm viên nén Clopidogrel 75 mg thành phần bào chế viên nhân ngoài dược chất Clopidogrel bisulfat, các tá dược trong đó isopropanol được sử dụng với vai trò tá dược dính chiếm tỷ lệ khoảng 18%. Thành phần màng bao có isopropanol và dicloromethan chiếm tỷ lệ khoảng 90%. Trong quá trình bào chễ, phần lớn dung môi bay hơi, nhưng có thể vẫn còn tồn dư dung môi trong chế phẩm ảnh hưởng tới sức khỏe người dùng. Isopropanol là dung môi nhóm 3, còn dicloromethan theo phân loại là dung mỗi nhóm 2. Theo qui định, yêu cầu chất lượng của sản phẩm đối với dung môi isopropanol và dicloromethan phải không lớn hơn 5000 ppm và 600 ppm. Tồn dư các dung môi này trong chế phẩm được xác định bằng GC - FID với chế độ tiêm mẫu không gian hơi. Mẫu thử và chuẩn được chuẩn bị trong dung môi N,N - dimethylformamid (DMF), cụ thể như sau:
- Dung dịch chuẩn: Cân chính xác khoảng 500 mg isopropanol và 60 mg dicloromethan vào bình định mức 100 ml, thêm 30 ml DMF, trộn đều và thêm DMF đến vạch, lắc đều. Pha loãng chính xác 10 ml dung dịch này thành 100 ml bằng DMF. Lắc kỹ. Chuyển 10 ml dung dịch này vào lọ headspace 20 ml và đậy chặt nắp ngay.
- Dung dịch thử: Nghiền 20 viên và cân chính xác một lượng tương đương khoảng 1 viên vào lọ headspace 20 ml, thêm 10 ml DMF và đậy chặt nắp ngay.
Tiến hành phân tích ở điều kiện qui định.
Đánh giá độ phù hợp của hệ thống: Dựa vào kết quả tiêm lặp lại 6 lần dung dịch chuẩn. Phép thử có giá trị khi diện tích píc 6 lần tiêm lặp của isopropanol và dicloromethan có RSD ≤ 15,0%.
Lượng isopropanol dicloromethan trong mẫu thử tính theo ppm:
AT/AS x WS/100 x 10/100 x P/100 x10/WT x 1000000
Trong đó:
- AT và AS là diện píc isopropanol/ dicloromethan thu được của dung dịch thử và dung dịch chuẩn
- WT và Ws là khối lượng mẫu thử và isopropanol/ dicloromethan chuẩn
- P là độ tinh khiết của isopropanol/ dicloromethan chuẩn.
Cũng có thể lấy chính xác thể tích dung môi chuẩn thay cho thao tác cân khi chuẩn bị dung dịch chuẩn. Khi đó, khối lượng chuẩn sẽ tính dựa vào thể tích và tỷ trọng của dung môi chuẩn sử dụng, tuy nhiên phải chú ý nhiệt độ khi thực hiện.
Dung môi tồn dư còn được qui định phải kiểm soát trong đồ bao gói và dụng cụ y tế. Trong Dược điển Việt Nam 5, phụ lục 174 hướng dẫn cách xác định tồn dư ethylen dioxyd trong bộ dây truyền dịch tiệt khuẩn bằng ethylen dioxyd, phụ lục 17.6 cũng trình bày phương pháp xác định tồn dư ethylen oxyd trong bơm tiêm vô khuẩn bằng chất dẻo sử dụng một lần tiệt khuẩn bằng ethylen dioxyd bằng GC - FID, Giới hạn cho phép tồn dư ethylen dioxyd trong bộ dây truyền dịch và bơm tiêm đều không lớn hơn 10 ppm.
2 Ứng dụng sắc ký khí trong phân tích hóa chất bảo vệ thực vật
Có nhiều loại detector của GC có thể ứng dụng để phân tích hóa chất bảo vệ thực vật (HCBVTV), bao gồm NPD, ECD, FPD, MS,... tùy thuộc vào cấu trúc và nồng độ của chất cần phân tích.
2.1 Phân tích hóa chất bảo vệ thực vật bằng các detector thông thường
ECD là detector được ứng dụng để phân tích HCBVTV nhóm clo hữu cơ và các HCBVTV nhóm lân hữu cơ hay cúc tổng hợp mà trong phân tử có chứa clo. Nhiều qui trình đã được đánh giá và ban hành thành các qui trình chuẩn, như AOAC 985.22 ứng dụng phân tích HCBVTV nhóm lần và clo hữu cơ; AOAC 998.01 ứng dụng để phân tích HCBVTV nhóm cúc tổng hợp, EPA Method 8081B ứng dụng để phân tích HCBVTV nhóm clo hữu cơ. Ngoài ra, rất nhiều phương pháp GC - ECD đã được công bố để xác định HCBVTV trong nhiều đối tượng khác nhau. Ở Việt Nam, tác giả Trần Việt Hùng (Luận án tiến sĩ dược học, Trường Đại học Dược Hà Nội năm 2005) đã xác định đồng thời 18 hoạt chất thuốc trừ sâu nhóm clo hữu cơ, phospho hữu cơ và pyrethroid trong 66 mẫu dược liệu bằng phương pháp sắc ký khí sử dụng chiết lạnh hoặc chiết Soxhlet kết hợp phân tích bằng detector ECD, MS và NPD. Phương pháp có giới hạn phát hiện thấp ở mức nanogam hoặc dưới nanogam. Dược Điển Việt Nam 5 trình bày phương pháp phân tích 23 HCBVTV nhóm này bằng GC - ECD với cột tách silica nung chảy (30 m x 0,32 mm) phủ poly(dimethyl)(diphenyl) siloxan dày 0,25 um, sử dụng chuẩn nội carbophenothion.
NPD cũng là một detector được sử dụng do có nhiều loại HCBVTV có N và P trong phân tử, tiêu biểu là HCBVTV nhóm lân hữu cơ. Trong Dược điển Việt Nam 5 có trình bày điều kiện phân tích 13 HCBVTV nhóm này với cột tách silica nung chảy (30 m x 0,32 mm) phủ poly(dimethyl)siloxan dày 0,25 um, sử dụng chuẩn nội carbophenothion.
FPD cũng được sử dụng để phân tích HCBVTV nhóm lân hữu cơ có chứa P và S. Các tác giả sử dụng GC - FPD cùng kỹ thuật tiêm mẫu PTV để phân tích 5 HCBVTV nhóm lân hữu cơ trong dầu ô liu. Giới hạn phát hiện thấp nhất đạt được là 3 ng/kg (Giacomo Dugo, Giuseppa Di Bella, Giovanna Loredana La Torre, Marcello Saitta. Food Control, June 2005, 16(5), 435 - 438).
Tiêu chuẩn Việt Nam TCVN 8319: 2010, trình bày phương pháp xác định 21 HCBVTV nhóm lân hữu cơ, clo hữu cơ và cúc tổng hợp trong rau quả bằng GC với detector ECD và detector quang ngọn lửa, kính lọc phospho (FPD/P), sử dụng cột mao quản DB - 5 (30 m × 0,32 mm; 0,25 m).
Việc sử dụng các detector như ECD, NPD, FPD chỉ ứng dụng để phân tích một nhóm HCBVTV có tính chất hóa học tương tự. Khi phân tích các HCBVTV khác thường không đáp ứng được độ nhạy.
2.2 Phân tích hóa chất bảo vệ thực vật bằng detector khối phổ
Trong phân tích HCBVTV, CG - MS đóng vai trò rất quan trọng, đặc biệt GC - MS/MS đã đáp ứng được các yêu cầu về phân tích hàm lượng rất nhỏ với độ chính xác cao. Khi phân tích HCBVTV bằng GC - MS thường sử dụng nguồn ion hóa EI. Các kỹ thuật GC - MS có thể xác định đến hàng trăm HCBVTV trong cùng một lần phân tích. Bảng 4.12 tóm tắt một số nghiên cứu phân tích HCBVTV bằng GC - MS.
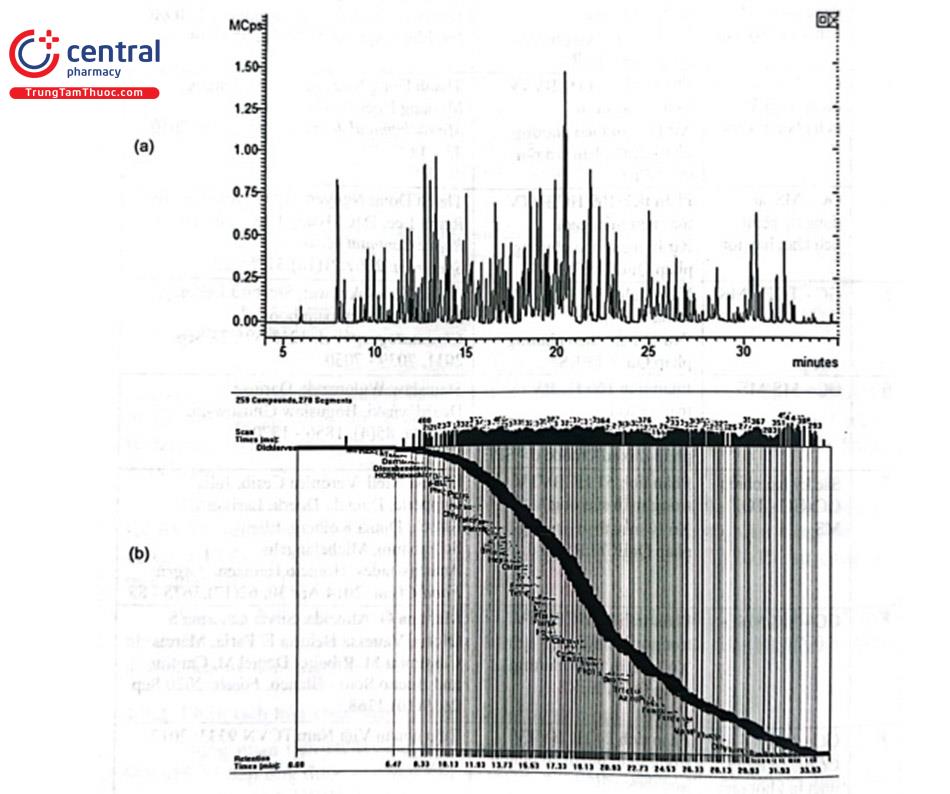
Phát huy tính năng “tuyệt vời” trong phát hiện bằng thiết bị khối phổ tứ cực chập ba, bẫy ion, thời gian bay, các nhà sản xuất không ngừng cải tiến nâng cao độ phân giải cũng như độ nhạy của thiết bị, đồng thời cột tách chuyên dùng phân tích thuốc trừ sâu cũng được phát triển để tăng cường số lượng HCBVTV có thể phân tích được đồng thời trong một lần phân tích. Hơn nữa, thư viện phổ với hàng trăm nghìn HCBVTV cho phép phân tích sàng lọc ban đầu mà có thể không cần chất chuẩn. Hình 4.12. trình bày sắc ký đồ và cửa sổ thời gian lưu phân tích 258 HCBVTV (và chuẩn nội) bằng thiết bị GC - MS/MS Bruker Scion TQ sử dụng cột tách Bruker BR - 5ms (30 m × 0,25 mm i.d.; 0,25 um).
GC đã được ứng dụng rộng rãi để xác định đồng thời nhiều HCBVTV, tuy nhiên có một số loại HCBVTV không thể phân tích được bằng GC do GC chỉ phù hợp để phân tích các HCBVTV không phân cực đến phân cực trung bình, các chất bay hơi mà không thể phân tích các hợp chất phân cực, chất không bay hơi hay chất bị phân hủy bởi nhiệt do đó, một tỷ lệ các HCBVTV vẫn phải thực hiện phân tích bằng HPLC.
3 Ứng dụng sắc ký khí trong phân tích các chất gây nghiện
Phân tích chất gây nghiện có thể ở trong nguyên liệu, chế phẩm hoặc mẫu thử sinh học. Chất gây nghiện được phân loại thành các nhóm sau:
- Các chất ma túy nhóm opiat;
- Các chất ma túy nhóm cần sa;
- Các chất ma túy nhóm ATS (Chất kích thích loại amphetamin: Amphetamin Type Stimulants);
- Cocain;
- LSD (Lysergic acid diethylamid);
- Acid y - Hydroxybutyric (GHB) và các analog;
- Dẫn chất benzodiazepin;
- Khat;
- Nấm thuộc chi Psilocybe.
Các chất có đặc tính gây nghiện, có nguy cơ bị lạm dụng nghiêm trọng, bị cấm và yêu cầu quản lý rất chặt chẽ như cần sa và các dẫn xuất của nó, cocain, heroin, methadon, Morphin, thuốc phiện. Một số chất thường được sử dụng điều trị, có nguy cơ lạm dụng thấp hơn như codein, dihydrocodein, Ephedrin được sản xuất thành thuốc thành phẩm và chịu sự quản lý theo qui định đặc biệt của Bộ Y tế.
3.1 Phân tích các chất gây nghiện trong mẫu nguyên liệu, chế phẩm
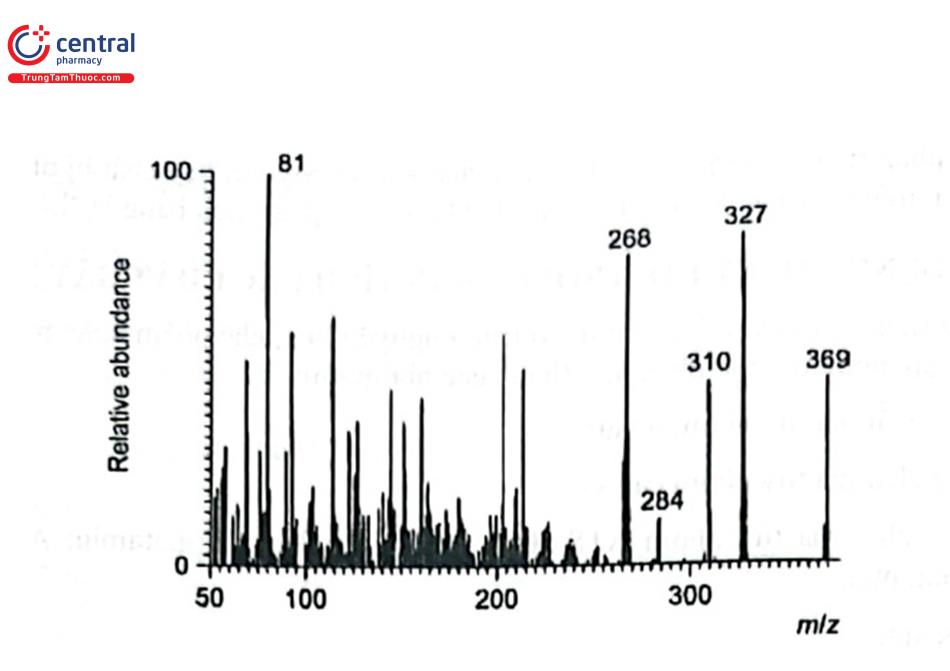
Các chất gây nghiện (ma túy) thường ở bốn dạng chính: bột; viên nén và viên nang; thực vật tươi hoặc khô và chất lỏng (dung dịch nước, dung dịch cồn). GC - MS có thể phân tách và nhận dạng hầu hết các chất ma túy đang lưu hành. Với độ nhạy cao, phương pháp này được sử dụng nhiều nhất hiện nay để xác định các chất ma túy sau khi sàng lọc sơ bộ. Phương pháp GC - MS rất quan trọng trong phân tích chất gây nghiện do các chất bất hợp pháp mới tiếp tục xâm nhập thị trường và danh sách hợp chất cần phân tích có thể lên đến hàng trăm chất. GC/MS ở chế độ quét toàn phổ (TIC) với chế độ ion hóa va chạm điện tử được áp dụng phân tích trực tiếp (không dẫn chất) để sàng lọc các chất cần kiểm soát. Các chất trong mẫu rắn thường được hòa tan trong dung môi như methanol, hexan hoặc toluen, còn các mẫu thực vật, mẫu lỏng được chiết vào dung môi hữu cơ. Mẫu thử được pha loãng tới nồng độ thích hợp để tránh quá tải cột GC hoặc detector MS. Việc định tính các thành phần khác nhau của mẫu có thể sử dụng thư viện phổ thương mại trong máy, tuy nhiên vẫn cần so sánh với mẫu chuẩn của chất ma túy cần phân tích. Hình 4.13 trình bày phổ GC - MS của heroin phân tích với chế độ ion hóa va chạm ion.
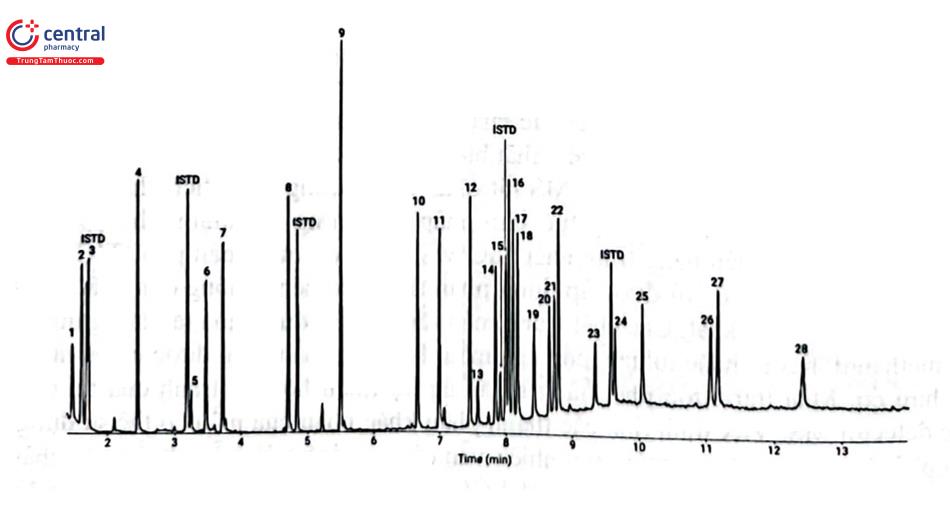
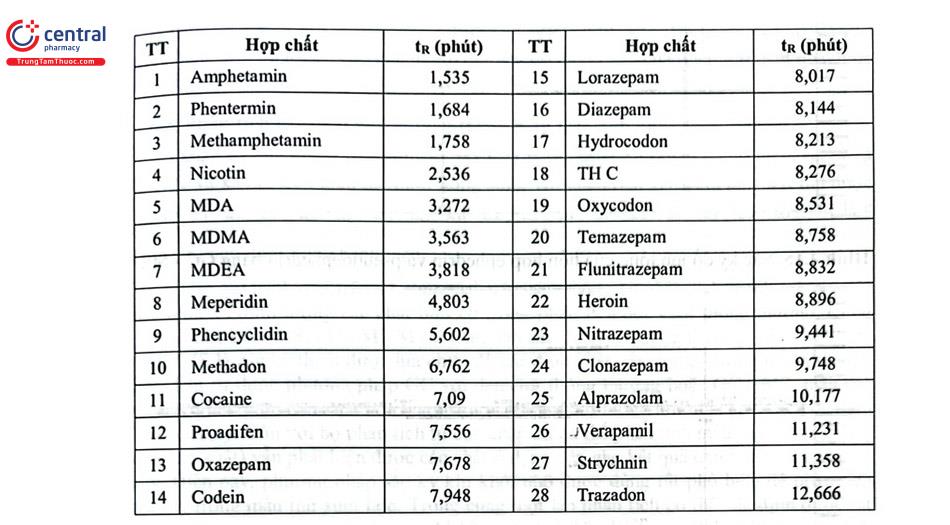
Nhiều qui trình phân tích bằng GC - MS sàng lọc các chất gây nghiện được phát triển và công bố, có thể là qui trình phân tích đồng thời từng nhóm chất hoặc một số nhóm chất. Hãng Agilent đã phát triển thành công qui trình phát hiện 28 hợp chất của các nhóm chất gây nghiện khác nhau như amphetamin, opiat, benzodiazepin, cần sa trên thiết bị GC Agilent 7890 GC kết nối MS Agilent 5977B Inert Plus GC/MSD with inert EI source. Hỗn hợp 28 chất (Bảng 4.13) có nồng độ mỗi chất 5 kg/ mL và 6 chất chuẩn nội đồng vị nồng độ mỗi chất 2 ug/ mL được tách bằng cột Agilent DB - 5ms Ultra Inert (20 m × 0,18 mm × 0,18 pm). Thể tích mẫu tiêm: 1 uL, chế độ tiêm chia dòng 20 : 1. Nhiệt độ buồng tiêm 250°C. Chương trình nhiệt độ cột: ban đầu 110°C, tăng tuyến tính 20°C/ phút tới 300°C, giữ 4,5 phút. Khí mang: Heli, tốc độ 1,5 mL/phút. Nhiệt độ đường chuyển 280°C. Nhiệt độ nguồn ion: 250°C. Nhiệt độ tứ cực: 150°C. Chế độ phổ quét với khoảng quét m/z 40 - 500. (Hình 4.14).
Tuy nhiên, GC/MS có những hạn chế trong định tính các chất gây nghiện do số lượng mảnh khối thu được khi ion hóa bằng chế độ va chạm điện tử hạn chế. Mặt khác, khó có thể tách được một số đồng phân (diastereomers and positional isomers) và dạng muối của các chất bằng GC - MS. Như vậy, sẽ không đáp ứng yêu cầu phải xác định rõ ràng chất cần kiểm soát có trong tang vật ma túy. Trong trường hợp này, GC - FTIR là một kỹ thuật thay thế được lựa chọn cho GC - MS để xác định các hợp chất hữu cơ. Tế bào đo mẫu trong GC - FTIR được bổ sung thêm bộ phận làm mát bằng nitơ lỏng cho phép phân tích trực tiếp chất phân tích kết tinh dưới dạng rắn rửa giải từ GC. Kỹ thuật này vượt trội hơn tế bào đo ống ánh sáng IR (IR light pipe) của GC - FTIR thông thường về độ nhạy, cũng như chất lượng phổ IR và cho phép so sánh trực tiếp phổ thu thập được với cơ sở dữ liệu IR hiện có.
Một nghiên cứu được tài trợ bởi Quỹ của Bộ Tư pháp Mỹ tiến hành so sánh khả năng xác định ephedrin và pseudoephedrin bằng GC - MS với GC - FTIR và đã chứng minh khả năng vượt trội của GC - FTIR. Hình 4.15 và 4.16 là sắc ký đồ và phổ đồ phân tích hai chất bằng GC - MS. Kết quả cho thấy hai chất chưa tách được và cũng không xác định được từng chất dựa vào các mảnh phổ (m/z). Hình 4,17 là phổ đồ phân tích h chất nghiên cứu bằng GC - FTIR, phổ của hai hợp chất này có sự khác biệt trong vùng vân tay hồng ngoại cho phép xác định các đồng phân không đối quang này.
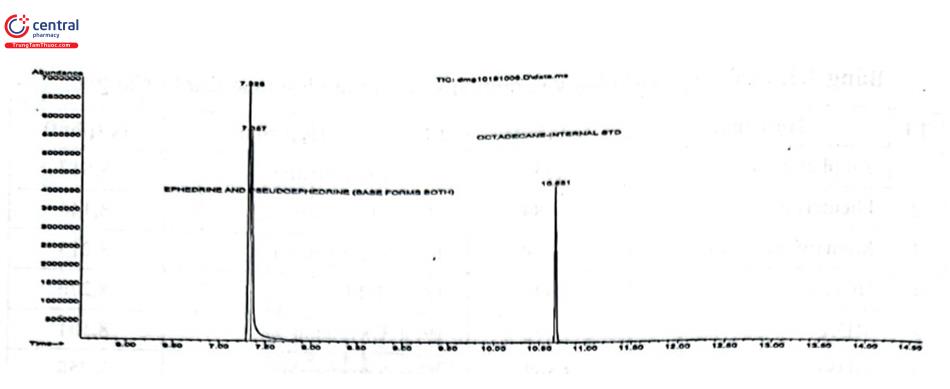


Thiết bị GC: Varian 3900 với cột VF – 5 ms (30 m × 0,25 mm, 0,25 um); khí mang Heli, tốc độ 1,1 mL/phút; chương trình nhiệt độ cột: 60C trong 1 phút, tăng 15°C/phút tới 300°C, giữ trong 3 phút; Tiêm mẫu 2 uL, nhiệt độ buồng tiêm 250°C.
Thiết bị IR: Spectra Analysis DiscovIR - GC, FTIR unit. Độ phân giải 4 cm-1.
Sau khi đã định danh được chất phân tích (chất gây nghiện) bằng phân tích sàng lọc, thực hiện định lượng bằng GC - FID hoặc GC - MS. Khi tiến hành định lượng cần thực hiện theo phương pháp chuẩn nội để tăng độ chính xác của phép thử. Kết quả được tính dựa vào đường chuẩn thực hiện song song.
3.2 Phân tích các chất gây nghiện trong mẫu thử sinh học
Việc sử dụng chất gây nghiện, đặc biệt là ma túy gây ra hệ quả nghiêm trọng về kinh tế và xã hội trên toàn thế giới. Song song với phân tích xác định tang vật thu được ở dạng nguyên liệu và thành phẩm thì công tác phân tích độc chất với nền mẫu sinh học cũng được phát triển mạnh mẽ nhằm cung cấp bằng chứng pháp lý và phục vụ lâm sàng (cấp cứu ngộ độc) trong bệnh viện.
Các qui trình dựa trên sự phân tách sắc ký là công cụ phân tích mạnh mẽ nhất để xác định và định lượng các chất cơ trong phân tích độc chất thông thường. Cùng với HPLC, LC - MS, LC - MS/MS thì GC với các detector thông thường, GC - MS, GC - MS/MS là các kỹ thuật được lựa chọn để xác định chất gây nghiện trong mẫu thử sinh học. Khi sử dụng phương pháp GC với detector thông thường hoặc GC - MS, mẫu thử thường phải được dẫn xuất hóa hoặc làm giàu mẫu với lượng lớn mẫu (> 5 mL) để đảm bảo độ nhạy. Còn với bộ phân tích tứ cực chập ba có thể phân tích mẫu trực tiếp (không qua dẫn xuất) vẫn phát hiện được các chất ở dạng vết, cho kết quả chính xác và đáng tin cậy. Hiện nay, phương pháp sắc ký khí khối phổ được dùng rất phổ biến để phân tích ma túy trong mẫu thử sinh học. Trong cùng một lần phân tích có thể xác định được rất nhiều chất ma túy trong thời gian chỉ 10 - 30 phút. Ưu điểm của GC - MS là quá trình tách sắc ký không quá quan trọng. Mỗi chất phân tích đặc trưng bởi các giá trị m/z nhất định, do đó nếu GC không tách được các chất hoàn toàn vẫn có thể định lượng được nhờ vai trò của MS. Một số nghiên cứu được tóm tắt ở Bảng 4.14 cho thấy phương pháp GC - MS có thể phân tích được nhiều chất ma túy cùng lúc trên nền mẫu thử sinh học.

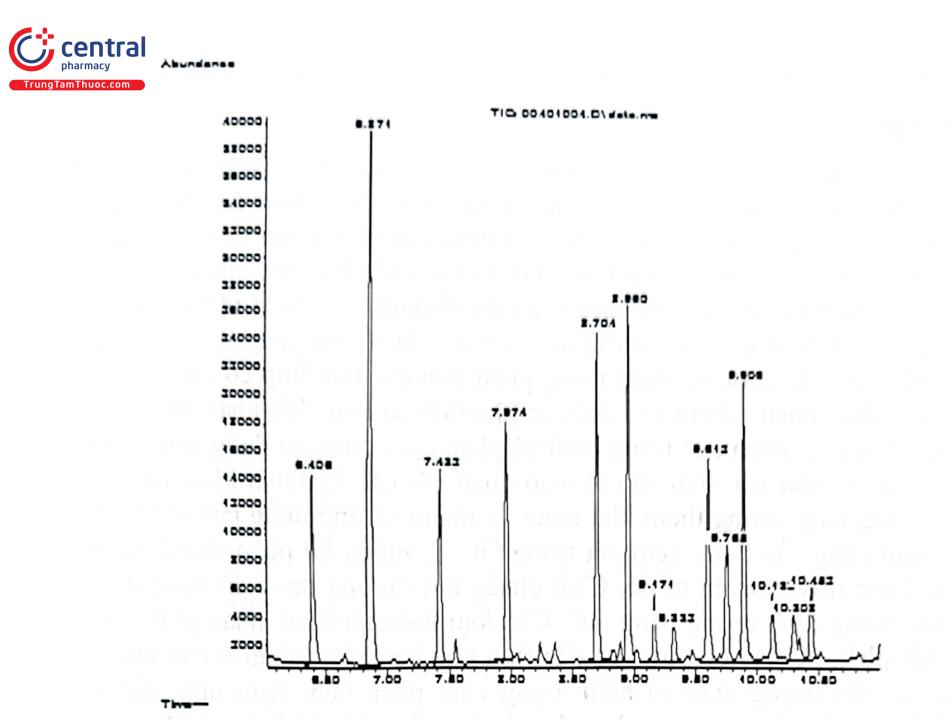
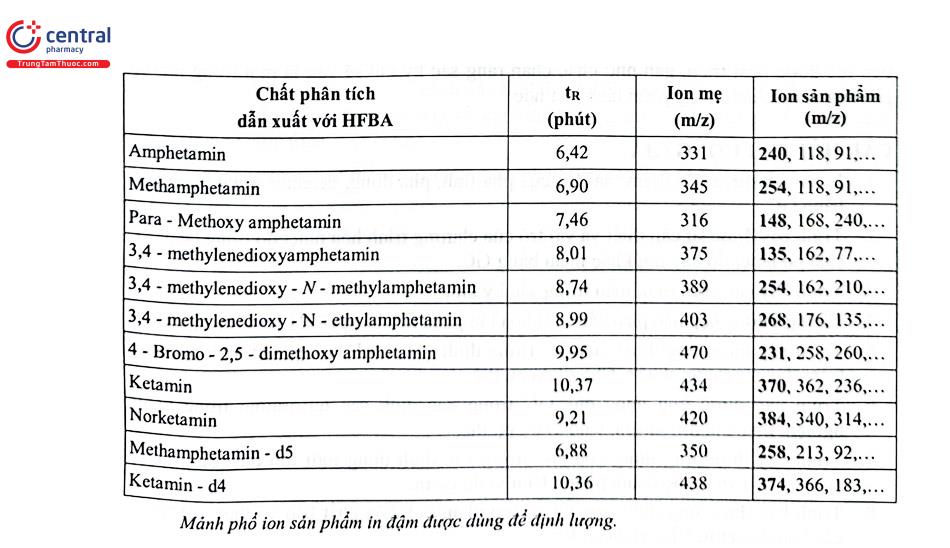
Việt Nam là một điểm nóng về ma túy do đó phương pháp phân tích ma túy cũng được phát triển mạnh. Các nhà khoa học ở Viện Khoa học hình sự - Bộ Công an, Viện Pháp y trung ương, Viện Pháp y quân đội đã có nhiều nghiên cứu về lĩnh vực này. Phương pháp phân tích sử dụng là GC - FID, LC - MS/MS, điện di mao quản, nhưng chủ yếu là GC - MS với đối tượng phân tích là máu, nước tiểu, móng tay, móng chân, tóc. Chất phân tích được chiết vào dung môi hữu cơ sau đó dẫn xuất hóa với thuốc thử thích hợp để tăng cường độ nhạy của phép phân tích. Trong một nghiên cứu gần đây, nhóm tác giả ở Viện Pháp y trung ương đã xây dựng thành công phương pháp GC - MS phân tích đồng thời 9 chất kích thích nhóm amphetamin và ketamin cũng 2 chất chuẩn nội đồng vị trong tóc và nước tiểu. Các chất được chiết bằng dung môi hữu cơ ở pH thích hợp, sau đó dẫn xuất hóa với thuốc thử heptafluorobutyric anhydrid. Các chất được tách bằng cột HP5 - MS (30 m x 250 km; 0,25 um); khí mang heli, tốc độ dòng 1 ml/phút; chương trình nhiệt độ cột; Ban đầu 80°C giữ trong 3 phút, tăng lên 40°C/min đến 200°C và giữ trong 1 phút, tiếp tục tăng 10°C/min đến 290°C và giữ trong 6 phút; thể tích tiêm mẫu 1 ul, chế độ không chia dòng; nhiệt độ buồng tiêm mẫu: 250°C. Chế độ phát hiện bằng detector khối phổ sử dụng bộ phân tích khối tử cực với điều kiện: Chế độ ion hóa dương với nguồn ion hóa va chạm điện tử, năng lượng ion hóa 70 eV, nhiệt độ nguồn ion hóa: 230°C; chế độ chọn lọc ion (SIM). Hình 4.18 là sắc ký đồ và Bảng 4.15 trình bày các mảnh ion dùng trong định tính và định lượng các chất nghiên cứu. Phương pháp có LOD nằm trong khoảng 0,02 - 0,10 ng/mg (mẫu tóc) và 0,77 - 4,29 ng/ ml (mẫu nước tiểu).
Như vậy, GC - MS là một kỹ thuật đóng vai trò rất quan trọng trong phân tích các chất ma túy. Sự phối hợp giữa dẫn xuất hóa và phân tích bằng GC - MS đem lại một phương pháp hoàn hảo để ứng dụng phân tích các chất ma túy trong mẫu thử sinh học.
4 Kết luận
Sắc ký khí đã được sử dụng để phân tích nhiều loại hợp chất trong nhiều thập kỷ vì nó ổn định và đáng tin cậy. Sự lưu giữ và tách các chất phân tích bằng sắc ký khí được chi phối bởi các tương tác liên phân tử giữa các chất phân tích và pha tĩnh. Hai yếu tố chính kiểm soát sự phân tách trong GC là bản chất hóa học của pha tĩnh và nhiệt độ cột tách. Trong thực tế, việc thay đổi nhiệt độ dễ dàng hơn so với thay đổi cột, vì vậy nhiệt độ thường là biến đầu tiên được điều chỉnh khi tối ưu hóa điều kiện phân tích. Chương trình nhiệt độ được sử dụng trong phân tích các hỗn hợp có chứa các hợp chất có độ bay hơi khác nhau. Thêm vào đó, các pha tĩnh có bản chất khác nhau về mặt hóa học (không phân cực, phân cực trung bình và phân cực) được sử dụng trong chế tạo các loại cột khác nhau như cột nhồi và cột mao quản với các kỹ thuật khác nhau như bao, WCOT, ... càng tăng cường thêm khả năng và phạm vi ứng dụng của sắc ký khí. Quá trình tiêm mẫu cũng cần được xem xét trong GC, vì nhiều lỗi phân tích hoặc hiệu suất kém có thể được truy nguyên từ nó. Chất chuẩn nội thường được sử dụng để cải thiện độ chính xác trong định lượng bằng GC. Các loại detector khác nhau phù hợp để phát hiện các chất phân tích có cấu trúc và đặc tính hóa học khác nhau ở các mức nồng độ khác nhau tùy đối tượng mẫu và hàm lượng chất phân tích. Hơn nữa, dẫn xuất hóa những chất không bay hơi tạo có thể phân tích được bằng GC đã mở rộng hơn nữa phạm vi ứng dụng của phương pháp. Một điểm cộng cho sắc ký khí đó là “phương pháp xanh” thân thiện môi trường do giảm thiểu việc sử dụng các dung môi hữu cơ trong qui trình phân tích. Trong khi các phương pháp sắc ký và phân tách mới tiếp tục được phát triển, gần như chắc chắn rằng sắc ký khí sẽ vẫn là một trong những phương pháp chính trong phân tách hóa học.
5 Tài liệu tham khảo
- Nguyễn Thị Kiều Anh, Phạm Thị Thanh Hà, Tạ Mạnh Hùng (2022), "Ứng dụng sắc ký khí trong kiểm nghiệm thuốc”, Một Số Phương Pháp Sắc Ký Dùng Trong Phân Tích Thuốc. Nhà xuất bản Y học, trang 160 - 180. Tải bản PDF tại đây.
- Trần Tử An (2007). Hóa Phân tích tập 2. Nhà xuất bản Y học.
- Bộ Y tế (2017). Dược điển Việt Nam V. Nhà xuất bản Y học.
- Bộ Y tế (2018), Thông tư 32/2018/TT - BYT: Qui định việc đăng ký lưu hành thuốc, nguyên liệu làm thuốc.
- Anthony C Moffat, M David Osselton, Brian Widdop (2011), Clarke's Analysis of Drugs and Poisons. Pharmaceutical Press.
- Mark F. Vitha (2017), Chromatography Principles and Instrumentation. John Wiley & Sons, Inc., Publication.
- United States Pharmacopeia 42, 43, 44.
- Stavros Kromidas (2016), The HPLC Expert: Possibilities and Limitations of Modern High Performance Liquid Chromatography. Wiley-VCH Verlag GmbH & Co. KGaA
- Colinf. Poole (2015), Instrumental thin layer chromatography. Elsevier Inc.
- Peter E. Wall (2005), Thin-layer Chromatography A Modern Practical Approach. The Royal Society of Chemistry.
- Guidance for industry - Bioanalytical method validation. FDA 2018.
- Québec Ministère de l'Environnement et de la Lutte contre les changements climatiques (2021), Protocole pour la validation d'une mesthode d'analyse en chimie, 4e édition.
- ICH (1996): Q2B Validation of Analytical Procedures: Methodology
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Michael W. Dong (2019), HPLC and UHPLC for practicing scientists. 2nd Edition, John Wiley & Sons, Inc.
- Danilo Corradini (2011), Handbook of HPLC. 2nd Edition, CRC Press
- Angelika Gratzfeld Hüsgen and Rainer Schuster (2011), HPLC for Food Analysis. Agilent Technologies Company.
- Camag®, Basic tool for Thin - layer Chromatography. https://www.camag.com/
- Piet de Coning John Swinley (2019), A practical guide to gas analysis by gas chromatography. Elsevier Inc.
