Loạn dưỡng mỡ: Tình trạng mất chọn lọc mô mỡ trong cơ thể
Khoa nội tiết chuyển hóa và nghiên cứu
Đồng chủ biên
Thomas J.Braranski, MD, PhD
Janet B.McGill, MD, MA, FACE
Julie M.Silverstein, MD
Và các tác giả khác tham gia biên soạn
1 NGUYÊN LÝ CHUNG
Mô mỡ giúp bảo vệ cơ thể khỏi tình trạng ngộ độc mỡ và ngộ độc glucose.
Các tế bào mỡ dự trữ năng lượng dưới dạng lipid và khi bị thiếu hụt hoặc không có tế bào mỡ, lipid sẽ tích tụ lại trong cơ, gan và các vùng khác trong cơ thê.
Các hormon chuyển hóa (insulin và các steroids) làm tăng biệt hóa các tiền tế bào mỡ thành các tế bào mỡ trưởng thành.
Các giọt nhỏ lipid (các bào quan nhỏ dự trữ triglyceride) được tạo thành bên trong các tế bào mỡ từ các acid béo và triglycerid lưu hành trong máu.
Các tế bào mỡ có thể tăng kích thước (phì đại) và số lượng (tăng sản). Yếu tố tăng sinh peroxisome hoạt hóa thụ thể y (PPARy) là yếu tố điều hoà chính quá trình tạo mỡ.
1.1 Định nghĩa
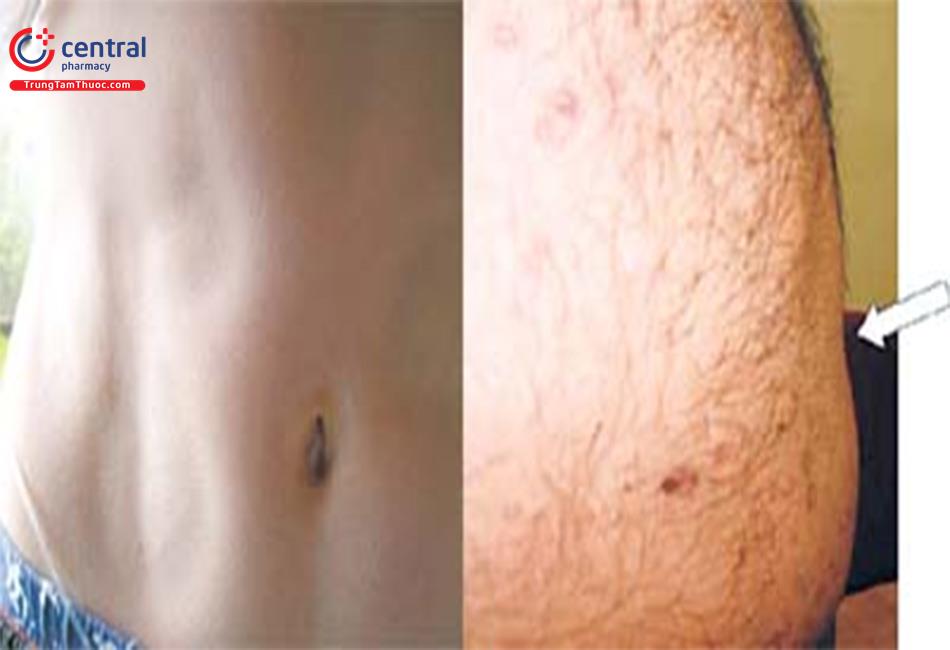
Loạn dưỡng mỡ là các rối loạn không đồng nhất được đặc trưng bằng tình trạng mất chọn lọc mỡ trong cơ thể.
- Dạng toàn thể: Tác động tới hầu hết các cơ quan trong cơ thể.
- Một phần: Chỉ tác động tới một số vùng nhất định trong cơ thể.
- Khu trú: Tác động tới các vùng dưới da tại chỗ, nhỏ hơn.
Mức độ rộng của tình trạng mất mỡ sẽ quyết định mức độ nặng của các biến chứng được gặp.
Thiếu hụt mỡ → thiếu hụt leptin → ăn vô độ và tích trữ mỡ lạc chỗ → đề kháng Insulin [2].
1.2 Phân loại
Loạn dưỡng mỡ toàn thể bẩm sinh (CGL).
Loạn dưỡng mỡ một phần có tính gia đình (FPLD).
Loạn dưỡng mỡ toàn thể mắc phải (AGL).
Loạn dưỡng mỡ một phần mắc phải (APL).
1.3 Dịch tễ học
Đã có hơn 1.000 ca được ghi nhận.
Tần suất hiện mắc <1:1,000,000 (mặc dù nhiều khả năng là số lượng báo cáo thấp hơn tần suất mắc thực sự) [2].
Các đối tượng nữ bị tác động dễ được phát hiện hơn.
1.4 Bệnh căn
Do di truyền (các kiểu phụ di truyền tính lặn và tính trội qua nhiễm sắc thể thường), mắc phải (theo cơ chế tự miễn) hoặc do thuốc.
1.5 Các bệnh lý liên quan
Đái tháo đường, tăng triglycerid máu, viêm tụy cấp, bệnh lý gan mỡ không do rượu, xơ gan, buồng trứng đa nang, gai đen, xơ vữa động mạch đến sớm, bệnh lý cơ tim, các rối loạn nhịp.
2 CHẨN ĐOÁN
Tiến hành thăm khám trên các bệnh nhân có vẻ “gầy” và có các biến chứng chuyển hóa.
- Đánh giá tình trạng mất mỡ và các cơ nổi lên.
- Đặc biệt cần để ý tại vùng eo và các chi.
Tiến hành thu thập kỹ lưỡng và thấu đáo tiền sử gia đình để nắm bắt được kiểu di truyền của rối loạn này.
Xét nghiệm di truyền có thể không cần thiết trong chẩn đoán nhưng có thể giúp nhận diện được các thành viên trong gia đình có nguy cơ.
Nếu xét nghiệm di truyền âm tính, người bệnh vẫn có thể bị loạn dưỡng mỡ do hiện tại không phải tất cả các đột biến đều đã biết.
Loạn dưỡng mỡ chủ yếu vẫn là một chẩn đoán lâm sàng.
2.1 Chẩn đoán phân biệt
Chán ăn tâm thần, rối loạn suy kiệt, đói ăn, hội chứng não trung gian, hội chứng Cushing, béo phì trung tâm, u mỡ lành tính và các hội chứng lão hóa sớm hiếm gặp [3].
2.2 Chẩn đoán cận lâm sàng
Đo độ dày lớp nếp gấp da.
2.2.1 Cận lâm sàng
Các xét nghiệm có thể hỗ trợ thêm chẩn đoán
- Toàn thể: nồng độ adipocytokine rất thấp (leptin và adiponectin) [4].
- Một phần: nồng độ leptin và adiponectin rất thay đổi.
Nghiệm pháp đánh giá dung nạp Glucose, tăng lipid máu, test chức năng gan và tăng acid uric máu.
Đo nồng độ leptin giúp đánh giá đáp ứng với điều trị bằng metreleptin [3].
2.2.2 Hình ảnh học
Kiểu mất mỡ có thể được đánh giá tốt hơn với hình ảnh MRI toàn thân trên ảnh T1 và chụp Xquang hấp thụ năng lượng kép (DXA) [3].
2.2.3 Các thủ thuật chẩn đoán
Sinh thiết da có thể giúp khẳng định có tình trạng viêm mô mỡ dưới da (panniculitis) hay bệnh da tăng bạch cầu đa nhân trung tính.
Nếu nghi ngờ bệnh cơ tim hay bệnh mạch vành: làm điện tâm đồ, siêu âm tim, nghiệm pháp gắng sức, theo dõi Holter.
3 CÁC RỐI LOẠN
3.1 Loạn dưỡng mỡ toàn thể bẩm sinh
Được biết đến dưới tên gọi hội chứng Berardinelli - Seip.
Di truyền tính lặn theo nhiễm sắc thể thường.
Có 4 dưới nhóm tách biệt, xem Bảng 36.1 [2].
Sinh lý bệnh [1]:
- CGL typ 1 (CGL1): đột biến di truyền gen 1-acylglycerol-3. phosphate-O-acyltransferase 2 (AGPAT2). AGPAT2 xúc tác tổng hợp phosphatidic acid, có vai trò không thể thiếu trong sự phát triển các tế bào mỡ non.
- CGL2: đột biến di truyền gen gây loạn dưỡng mỡ bẩm sinh Berardinelli - Seip congential lipodystrophy 2 (BSCL2). BSCL2 mã hóa seipin thúc đẩy quá trình sinh tổng hợp mỡ ở giai đoạn dữ trự năng lượng dư thừa. Nếu biến đổi, sự trưởng thành các tế bào mỡ sẽ bị ảnh hưởng nghiêm trọng.
- CGL3: đột biến di truyền caveolin 1 (CAV1). CAV1: gắn và vận chuyển acid béo tới các giọt lipid.
- CGL4: đột biến di truyền của yếu tố polymerase-I và-transcriptrelease (PTRF). PTRF tạo ra caveolae (tham gia tới quá trình nạp và vận chuyển tín hiệu) và điều hòa biểu lộ caveolins 1 và 3.
Chẩn đoán
- Dễ phát hiện sau sinh và trong năm đầu sơ sinh.
- Thời kỳ sơ sinh: gan lách to, rốn lồi hay thoát vị rốn.
- Thời thơ ấu: thèm ăn vô độ, tăng trưởng nhanh, tuổi xương già hơn tuổi thực, gai đen.
- Vị thành niên: có thể bị đái tháo đường, tăng triglycerid máu, HDL thấp đi kèm với các biểu hiện lâm sàng đặc trưng.
- Khai thác tiền sử - bệnh sử:
- Có thể bị viêm tụy cấp (do tăng triglycerid), gan nhiễm mỡ không do rượu dẫn tới xơ gan.
- Nữ giới có thể bị rậm lông, âm vật to, rối loạn kinh nguyệt, buồng trứng đa nang, vô sinh [4].
- Khám thực thể
- Mất gần toàn bộ lượng mỡ trong cơ thể.
- Các cơ và tĩnh mạch nông nổi rõ.
Chẩn đoán cận lâm sàng:
- Xét nghiệm: Leptin rất thấp, tăng hemoglobin Alc (HbAl) và triglycerid, HDL thấp.
- Hình ảnh học: thăm dò xương có thể thấy các tổn thương dạng tiêu xương.
| Bảng 36. Rối loạn toàn thể bẩm sinh (hội chứng Berardinelli Seip) | ||
| Biến thể | Đột biến gen | Các đặc điểm lâm sàng |
| Loạn dưỡng mỡ toàn thể bẩm sinh 1 (CGL1) [2] | GPAT2 | Là thể dưới nhóm hay gặp nhất. Không có mỡ trong tủy xương. Tăng tiết mồ hôi. Kiểu hình giống to đầu chi. Tổn thương dạng tiêu xương ở các các xương ngoài xương trục. Đái tháo đường (tuổi trung vị: 12,5 tuổi). |
| Loạn dưỡng mỡ toàn thể bẩm sinh 2 (CGL2) [2] | BSCL2 | Là kiểu dưới nhóm hay gặp thứ 2. Thể nặng nhất. Thiếu hụt các mô mỡ cơ học (quanh nhãn cầu, bàn tay, bàn chân và các vùng quanh khớp). Thiếu mô mỡ tủy xương. Đái tháo đường (tuổi trung vị: 10 tuổi). 50% có chậm phát triển tâm thần nhẹ. |
| Loạn dưỡng mỡ toàn thể bẩm sinh 3 | CAV1 | Mới chỉ có báo cáo 1 ca bệnh. Vẫn còn mỡ cơ học và trong tủy xương. Kiểu hình thấp lùn. (CGL3) [2] Hạ Canxi máu. Kháng Vitamin D |
| Loạn dưỡng mỡ toàn thể bẩm sinh 4 CGL4 [2] | PTRF | Đã báo cáo 30 ca bệnh. Mỡ của mô mỡ nâng đỡ cơ học và trong tủy xương vẫn được bảo tồn. Tiến triển thành loạn dưỡng mỡ toàn thể ngay thời kỳ sơ sinh. Bệnh cơ bẩm sinh, hẹp môn vị, mất vững khớp đội-trục, biến dạng hành xương phía đầu xa, các rối loạn nhịp tim có thể gây đột tử. |
| Bảng 36. 2. Loạn dưỡng mỡ một phần có tính chất gia đình | ||
| Biến thể | Đột biến gen | Các đặc điểm lâm sàng |
| Loạn dưỡng mỡ toàn thể có tính chất gia đình 1 (FPLD): Koberling [2] | Chưa biết | Kiểu hình Cushing. Gỡ mỡ nổi rõ phía trên vùng mông, phần trên đùi và phần trên cánh. Chỉ gặp ở nữ. |
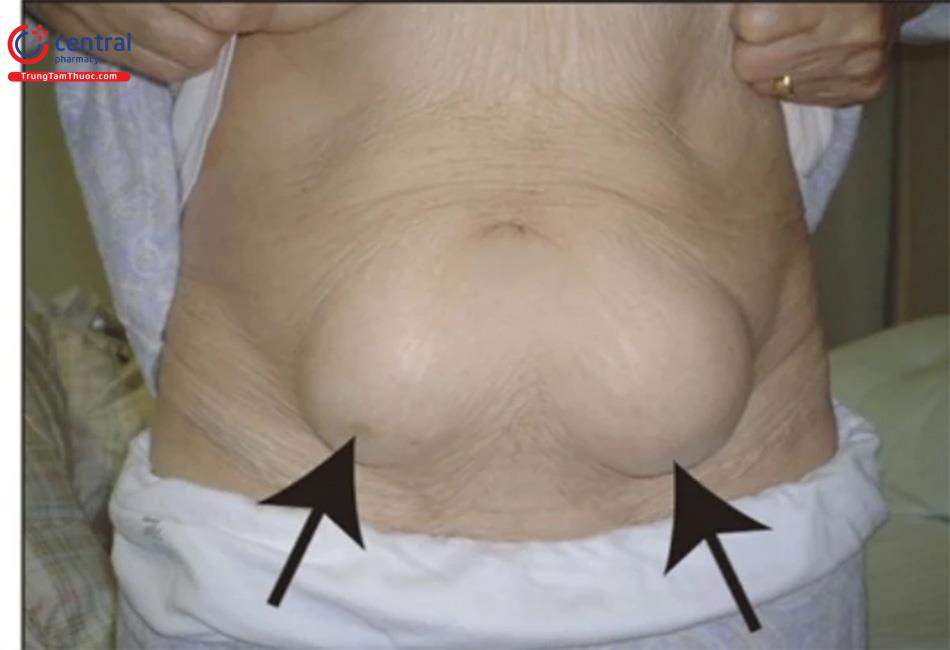
| Loạn dưỡng mỡ toàn thể có tính chất gia đình 2 (FPLD2): Dunnigan [2] | LMNA | Mất lớp mỡ dưới da ở các chi. Tăng mỡ vùng mặt, cổ, tạng bụng và môi lớn. Gai đen, phì đại cơ, các tĩnh mạch phì đại, u vàng thể phát ban, u mỡ dưới da, rậm lông. Tăng huyết áp, bệnh mạch vành, đái tháo đường, tăng triglycerid, viêm tụy, gan nhiễm mỡ. Nữ: hội chứng buồng trứng đa nang (PCSOS), thiểu sản vú, tiền sản giật, xảy thai. Nam: không có biểu hiện đặc trưng và các rối loạn chuyển hóa nhẹ hơn. |
| Loạn dưỡng mỡ toàn thể có tính chất gia đình 3 (FPLD3) [2] | PPARG | Không gia tăng mỡ ở mặt, cổ. Các biến chứng chuyển hóa điển hình. |
FPLD4 FPLD5 FPLD6 Liên kết AKT [2] | PLIN1 CIDEC LIPE AKT2 | Khởi phát lúc dậy thì. Mất lớp mỡ ở mông, các chi và tăng lớp mỡ ở mặt, cổ, bụng. Gai đen, tĩnh mạch phì đại, u vàng thể phát ban, rậm lông. Các biến chứng chuyển hóa và tim mạch. |
3.2 Loạn dưỡng mỡ một phần có tính chất gia đình
Hầu hết là di truyền tính trội qua nhiễm sắc thể thường nhưng cũng có thể mang tính lặn qua nhiễm sắc thể thường và liên kết với nhiễm sắc thể giới tính X.
Có một số thể khác nhau (xem Bảng 36.2) [2].
- Dunnigan là biến thể hay gặp nhất (tần suất 1 trong 15 triệu người) [1].
- Mất mỡ dưới da ở các chi trên và chi dưới nhưng mất mỡ ở vùng thân có thể thay đổi [4].
Sinh lý bệnh [1]
- FPLD typ 2 (FPLD2): do gen LMNA. Đứt gãy lamins A và C làm thay đổi cấu trúc nhân tế bào mỡ, gây chết tế bào xảy ra sớm.
- FPLD3: liên quan tới gen PPARy. Đột biến PPARy gây ức chế biệt hóa tế bào mỡ.
- FPLD4: liên quan tới tương đồng 2 gen gây ung thư tuyến ức chuột v-AKT (v-AKT murine thymoma oncogene homolog 2 [AKT2]). Đột biến AKT2 gây ức chế biệt hóa tế bào mỡ.
- FPLD5: liên quan tới perilipin 1 (PLIN1). Đột biến PLIN1 làm cho các tế bào mỡ nhỏ dễ bị tấn công bởi các đại thực bào, làm biến đổi xơ hóa.
Chẩn đoán
- Trẻ sơ sinh và trẻ nhỏ: phân bố mỡ cơ thể bình thường.
- Dậy thì: tái phân bố mỡ
- Trưởng thành: mất mỡ đặc trưng ở mặt, các biến chứng chuyển hóa [2].
- Nữ giới giảm khả năng sinh sản, kinh nguyệt không đều và rậm lông.
- Tăng triglycerid máu thường gặp và nặng: nguy cơ bị viêm tụy cấp.
- Khám thực thể:
- Mất mỡ dưới da vùng mông và các chi.
- Mô mỡ vùng mặt, cổ và trong ổ bụng vẫn được bảo tồn. Lớp mỡ quanh cổ dày lên làm cho bệnh nhân có kiểu hình giống Cushing. " Hay gặp dấu hiệu gai đen.
- Phì đại cơ.
- Xét nghiệm: tăng HbA1c và triglycerid.
3.3 Loạn dưỡng mỡ mắc phải
Có một số thể khác nhau, bệnh căn thay đổi (xem Bảng 36.3) [2,3].
Loạn dưỡng mỡ toàn thể mắc phải (AGL), hội chứng Lawrence: tỷ lệ nữ/nam là 3/1
Loạn dưỡng mỡ một phần mắc phải (APL), hội chứng Barraquer- Simons: tỷ lệ nữ/nam là 4/1.
Tại Mỹ, hơn 100.000 các bệnh nhân nhiễm HIV có loạn dưỡng mỡ (LD-HIV) [3].
- Tỷ lệ hiện mắc cao nhất trong các bệnh lý loạn dưỡng mỡ được biết tới [1].
- Thường gặp sau điều trị ức chế Protease (PI) hay thuốc ức chế nucleoside reverse transcriptase (NRTIs) kéo dài quá 2 năm.
- Mất mỡ diễn biến nặng thêm khi điều trị HIV bằng phác đồ kháng retrovirus hiệu lực cao (HAART [highly active antiretroviral therapy]).
- Không trở lại được tình trạng cũ khi ngừng dùng thuốc ức chế protease.
- Dễ tiến triển sang bệnh lý mạch vành.
Loạn dưỡng mỡ khu trú
- Không gây rối loạn chuyển hóa.
Sinh lý bệnh [1].
- Loạn dưỡng mỡ toàn thể mắc phải (AGL)
- 25% trường hợp được kích hoạt bởi tình trạng viêm mô mỡ dưới da.
- 25% có nguồn gốc tự miễn (bắt nguồn từ các bệnh lý như viêm đa khớp dạng thấp, Lupus ban đỏ hệ thống, hội chứng Sjögren và viêm đa cơ ở người trẻ).
- 50% vô căn .
- Loạn dưỡng mỡ một phần mắc phải (APL)
- Sinh bệnh học chính xác chưa được biết.
- Có thể do biến đổi các con đường hoạt hóa bổ thể hay một biến thể của gene LMNB2.
- Loạn dưỡng mỡ do HIV (LD- HIV)
Thuốc ức chế protease: điều hoà giảm PPARy và C/EBPa gây gián đoạn quá trình sinh tổng hợp lipid và biệt hóa tế bào mỡ, có thể ức chế đồng thời metalloprotease Kẽm có liên quan tới quá trình chuyển prelamin A thành lamin A trưởng thành [4].
Thuốc ức chế enzym sao chép ngược nucleoside (NRTIs): gây rối loạn chức năng ty thể.
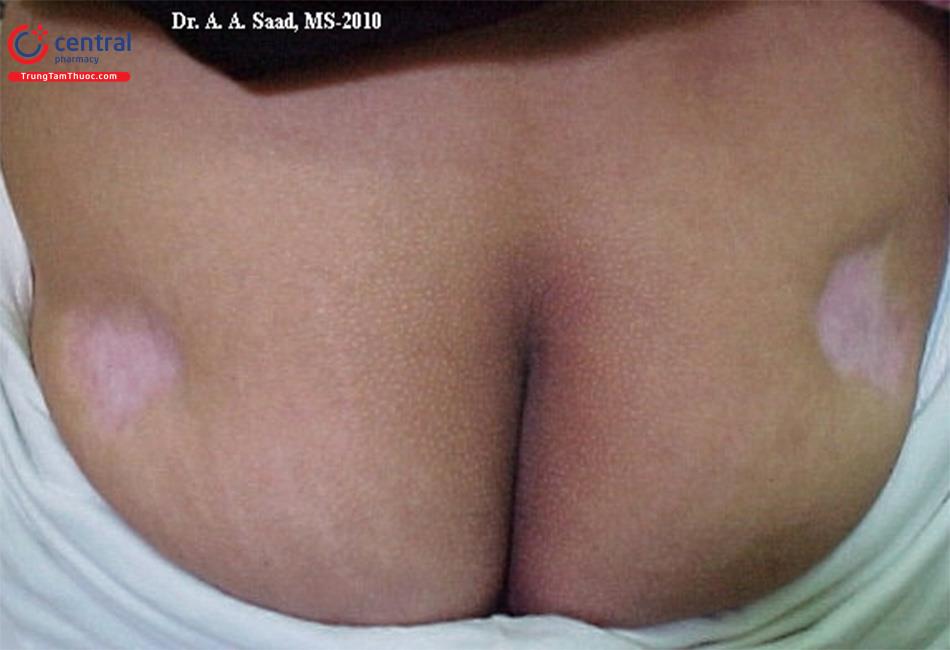
- Loạn dưỡng mỡ khu trú: thường xảy ra do viêm mô mỡ dưới da, không tương ứng với áp lực trực tiếp, tiêm các thuốc gây tổn thương mô mỡ.
| Bảng 36.5. Các bệnh lý đoạn dưỡng mỡ mắc phải | |
| Các thể | Các đặc điểm lâm sàng |
| AGL: hội chứng Lawrence [3] | Sinh ra với lượng mỡ bình thường. Thơ ấu: mất mỡ tiến triển, thường bị toàn thân. Một số vẫn duy trì mỡ trong tủy xương và ổ bụng Gan nhiễm mỡ và xơ hóa, đái tháo đường, tăng triglycerid. Xấp xỉ 25% có viêm lớp mỡ dưới da trước khi tiến triển bệnh. Xấp xỉ 25% có bệnh tự miễn. Nhiều trường hợp bị giảm yếu tố bổ thể [4]. |
| APL: hội chứng Barraquer - Simon [2,3] | Mất lớp mỡ dọc từ trên xuống khởi phát từ thời thơ ấu hoặc vị thành niên. Có thể tích trữ mỡ ở phần dưới cơ thể (hông, mông, đùi). 20% gặp viêm cầu thận màng tiến triển. 80% có giảm yếu tố 3 bổ thể. Hầu hết có yếu tố 3 bổ thể gây viêm thận (tự kháng thể lưu hành). Hiếm gặp các bệnh nhân có mảng kết tụ vàng ở võng mạc khi soi đáy mắt. Các biến chứng chuyển hóa thường không gặp. |
| Loạn dưỡng mỡ ở bệnh nhân nhiễm HIV [3] | Mất mỡ cục bộ từ các vùng nhỏ của cơ thể. Thường gây nên một hoặc nhiều điểm lõm ở da. Không xuất hiện các bất thường chuyển hóa. |
| Khu trú [3] | Mất mỡ từ từ ở cánh tay, đùi và mặt. Có thể tích tụ quá mức mỡ và gây biểu hiện hình dạng như cổ trâu, cắm đôi và tăng chu vi vòng eo. Có thể xuất hiện tình trạng tăng triglycerid máu nhưng chỉ có một vài trường hợp xuất hiện bệnh đái tháo đường. |
3.4 Các rối loạn dưỡng mỡ di truyền hiếm gặp
Có 3 rối loạn riêng biệt nhưng các rối loạn viêm tự miễn di truyền theo tính lặn có thể chồng chéo nhau kết hợp với loạn dưỡng mỡ (xem Bảng 36.4) [2].
Loạn dưỡng mỡ cũng là biểu hiện của các rối loạn gây lão hóa sớm cũng như các bệnh lý loạn dưỡng mỡ hiếm gặp (Bảng 36.5) [1,2,5-16].
Sinh lý bệnh
- Thiểu sản xương hàm dưới, điếc, hội chứng lão hoá sớm (MDP): đột biến POLD1. POLD1 gây trì hoãn tổng hợp chuỗi DNA trong quá trình phiên mã và sao chép DNA [10].
- Hội chứng lão hoá bẩm sinh Marfan: do đột biến của FBNI. FBN1 mã hóa fibrillin-1, là một thành phần cấu tạo của vi sợi (microfibril) là trụ đỡ của Elastin [11].
- Hội chứng với kiểu hình thấp lùn, tăng duỗi cơ quá mức, lõm 2 nhãn cầu, bất thường Rieger và chậm mọc răng (hội chứng SHORT [Short stature, Hyperextensibility, Ocular depression, Rieger anomaly, and Teething delay] syndrome): do đột biến PIK3R1. Truyền tín hiệu PIK3R1 tham gia vào chức năng nhiều tế bào cũng như có vai trò trong truyền tín hiệu của insulin [16].
- Hội chứng Werner: do các đột biến protein WRN. WRN mã hóa RecQ DNA helicase/exonuclease; và các đột biến gây ảnh hưởng tới sao chép DNA, dẫn tới sự mất ổn định của ADN và chết tế bào [1].
| Bảng 36.4. Các bệnh lý rối loạn tự miễn | ||
| Các biến thể | Đột biến gen | Kiểu hình |
| Hội chứng JMP [2] | PSMB8 | Co rút các khớp, loạn dưỡng cơ, thiếu máu nhược sắc. Loạn dưỡng mỡ xuất hiện từ tuổi thơ ấu. loạn dưỡng cơ, co rút các khớp, tổn thương da, thiếu máu hồng cầu nhỏ, gan lách to, tăng gammaglobulin máu. |
| Hội chứng Nakajo-Nishimura [2] | PSMB8 | Trẻ sơ sinh: nổi ban, sốt theo chu kỳ, các ban đỏ có các dạng những nốt nhỏ trên da, viêm cơ. Teo tiến triển cơ và lớp mỡ dưới da. Co cứng các khớp, gan lách to, tăng gammaglobulin máu, canxi hóa các nhân nền. |
| Hội chứng CANDLE [2] | PSMB8 | Năm đầu: sốt tái phát nhiều, tổn thương da kiểu mảng tím hình vòng, sưng đau các khớp, thiếu máu và tăng các yếu tố phản ứng pha cấp. Trẻ sơ sinh: mất lớp mỡ dưới da vùng mặt. Tuổi thơ ấu: mất dần lớp mỡ ở hai tay. |
4 ĐIỀU TRỊ
Việc điều trị rất khó khăn vì không thể đảo ngược tình trạng mất lớp mỡ [3].
Tập trung vào điều trị các biến chứng chuyển hóa và vấn đề thẩm mỹ [4].
4.1 Các thuốc điều trị [4]
Điều trị tình trạng tăng triglycerid máu nặng bằng fibrate và Dầu Cá.
Điều trị đái tháo đường bằng Metformin, có thể phải dùng cả insulin.
Đổi từ thuốc điều trị ức chế protease (PI) sang thuốc kháng retrovirus khác ở bệnh nhân bị loạn dưỡng mỡ do HIV.
Metreleptin [17].
- Chất tương đồng tổng hợp của hormon leptin.
- Điều trị tiêm dưới da hàng ngày.
- Điều chỉnh liều sau mỗi 3 - 6 tháng dựa theo các thông số chuyển hóa và thay đổi cân nặng [4].
- Loạn dưỡng mỡ toàn thể:
- Giảm rõ rệt tình trạng tăng triglycerid máu và cải thiện các thông số đường máu.
- Cải thiện tình trạng dự trữ mỡ lạc chỗ, tăng xâm nhập, protein niệu và viêm gan nhiễm mỡ.
- Được FDA phê chuẩn để điều trị loạn dưỡng mỡ toàn thể bẩm sinh (CGL) vào năm 2014.
- Loạn dưỡng mỡ một phần:
- Cải thiện tình trạng tăng triglycerid máu nhưng có tác dụng xung đột lên tình trạng đường máu.
- Metreleptin không được phê chuẩn để sử dụng điều trị cho các đối tượng này.
- Chỉ sử dụng theo Chương trình Đánh giá nguy cơ Myalept và Chiến lược làm giảm nhẹ (REMS).
- Cần thận trọng do đã có 3 ca bệnh bị lymphoma tế bào T và 4 ca bệnh xuất hiện tự kháng thể trung hòa leptin khi điều trị kéo dài trong các nghiên cứu không có đối chứng [17].
- Các tác dụng phụ thường gặp: hạ đường máu và phản ứng tại chỗ tiêm.
4.2 Các điều trị không dùng thuốc khác
Tham khảo ý kiến chuên gia sức khỏe tâm thần để xử trí các rối loạn cảm xúc của bệnh nhân [4].
4.3 Điều trị phẫu thuật [4]
Khuôn mặt của bệnh nhân có thể được cải thiện bằng cách ghép mô mỡ tự thân hay cấy filler da.
Hút mỡ dư thừa tại các vị trí không mong muốn.
4.4 Điều chỉnh lối sống/nguy cơ
4.4.1 Chế độ ăn
50 - 60% carbohydrate, 20 - 30% chất béo, 10 - 20% protein là phù hợp cho hầu hết các trường hợp [4].
Ăn uống quá độ có thể làm bệnh đái tháo đường và tình trạng tăng lipid máu diễn biến nặng lên cũng như thúc đẩy nhanh tình trạng thoái hóa mỡ gan.
Để hạn chế tình trạng tăng triglycerid máu nặng, ở trẻ sơ sinh cần cho trẻ ăn chế độ sữa có chứa triglycerid chuỗi trung bình (medium- chain trglyceride-based formulas) và chế độ ăn chứa ít mỡ (low-fat diets) ở các trẻ lớn hơn [4].
4.4.2 Hoạt động thể chất
Tập thể dục có thể cải thiện đề kháng insulin nhưng tránh tập quá sức nếu người bệnh có bệnh cơ tim.
Các bệnh nhân bị loạn dưỡng mỡ bẩm sinh typ 4 (CGL4), loạn dưỡng mỡ một phần có tính gia đình typ 2 (FPLD2) và hội chứng lão hóa sớm nên được sàng lọc bệnh lý tim mạch [4].
4.5 Các hướng đi trong tương lai
Các thuốc làm giảm nồng độ apoC-III:
- ApoC-III là 1 cấu phần của VLDL và HDL, là chất ức chế không cạnh tranh với lipoprotein Lipase (thủy phân triglycerid thành các acid béo tự do để hấp thu tại cơ và mô mỡ) và chuyển lipoprotein giàu triglycerid thành các chất tàn dư để được gan loại bỏ [18].
- Tăng apoC-III đi kèm với tình trạng đề kháng insulin và tăng triglycerid máu.
- Giảm apoC-III đã cho thấy làm giảm triglycerid máu.
- Các bệnh nhân loạn dưỡng mỡ có nồng độ apoC-III tăng có ý nghĩa [18].
- Nghiên cứu BROADEN là nghiên cứu ngẫu nhiên có đối chứng placebo mù đôi, pha 2/3 đối với volanesorsen (một oligonucleotide antisense với apoC-III) trong điều trị FPLD hiện vẫn đang được tiến hành.
| Bảng 36.5 các rối loạn dưỡng mỡ hiếm gặp khác | ||
| Rối loạn | Đột biến gen | Các đặc điểm lâm sàng |
| Loạn sån Mandibuloacral (MAD) typ A [5] | LMNA | Các bất thường xương: thiểu sản xương hàm dưới, xương đòn và tiêu xương đầu chi. Chậm liền các thóp sọ, răng sít, cứng khớp. Loạn dưỡng mỡ một phần. Đề kháng insulin. |
| Loạn sản Mandibuloacral (MAD) type B [6] | ZMPSTE24 | Thiếu sản xương hàm dưới và xương đòn, tiêu xương đầu chi, loạn dưỡng biểu bì. Kiểu hình lão hóa sớm. Loạn dưỡng mỡ toàn thể. |
| Lão hóa sớm không điển hình [7,8] | LMNA | Mất lớp mỡ 1 phần hoặc toàn thể. Kiểu hình lão hoá sớm. |
| Hội chứng lão hóa sớm Hutchinson - Gilford [2,9] | LMNA | Mất lớp mỡ ở hầu hết cơ thể trừ lớp mỡ nội tạng bụng. Kiểu hình lão hoá sớm. Các biến chứng tim mạch thường dẫn tới tử vong. |
| Loạn dưỡng mỡ đi kèm thiểu sản xương hàm dưới, điếc, lão hóa sớm (MDP) [10] | POLD1 | Rất ít mô mỡ dưới da. Tăng mô mỡ nội tạng ổ bụng. Thiểu sản xương hàm dưới. Điếc. Các đặc điểm kiểu hình lão hóa sớm. Tình trạng kháng insulin nặng. Nam giới: suy sinh dục và tinh hoàn lạc chỗ. |
| Hội chứng lão hóa Marfan bẩm sinh [11,12] | Fibrillin 1 | Loạn dưỡng mỡ toàn thể. Kiểu hình lão hoá sớm. Kiểu hình giống hội chứng Marfan. |
| Hội chứng lão hóa sớm bẩm sinh [13]. | CAV1 | Mất toàn bộ lớp mỡ dưới da và lớp cơ. Kiểu hình lão hóa sớm ngay khi sinh. |
| Hội chứng Werner [1,14,15] | WRN | Loạn dưỡng mỡ tứ chi. Tích mỡ ở thân và bụng. Kiểu hình lão hóa sớm xảy ra sau tuổi thiếu niên. Đề kháng insulin. Suy sinh dục. Xu hướng phát triển thành u ác tính. |
| Hội chứng với kiểu hình thấp lùn, tăng duỗi các khớp, nhãn cầu lõm, bất thường Rieger và chậm mọc răng hay hội chứng SHORT [16] | PIK3R1 | Rất ít lớp mỡ. Kiểu hình thấp lùn, duỗi các khớp quá mức, dị dạng vùng mặt, chậm mọc răng, các bất thường ổ mắt thay đổi. Tình trạng kháng insulin và/hoặc đái tháo đường thường xuất hiện ở tuổi thiếu niên. |
5 TÀI LIỆU THAM KHẢO
1. Nolis T. Exploring the pathophysiol- ogy behind the more common genet- ic and acquired lipodystrophies. J Hum Genet 2014;59(1): 16–23.
2. Lightbourne M, Brown RJ. Genetics of lipodystrophy. En- docrinol Metab Clin North Am 2017;46(2):539-554.
3. Garg A. Clinical review#: Lipo- dystrophies: Genetic and acquired body fat disorders. J Clin Endocri- nol Metab 2011;96(11):3313-3325.
4. Hussain I, Garg A. Lipodystrophy syndromes. Endocrinol Metab Clin North Am 2016;45(4):783-797.
5. Novelli G, Muchir A, Sangiuolo F, et al. Mandibuloacral dysplasia is caused by a mutation in LMNA-en- coding lamin A/C. Am J Hum Genet 2002;71(2):426–431.
6. Agarwal AK, Fryns JP, Auchus RJ, Garg A. Zinc metalloproteinase, ZMPSTE24, is mutated in man- dibuloacral dysplasia. Hum Mol Genet 2003:12016):1995-2001.
7. Chen L, Lee L, Kudlow BA, et al. LMNA mutations in atyp- ical Werner's syndrome. Lan- cet 2003;362(9382):440–445.
8. Garg A, Subramanyam L, Agarwal AK, et al. Atypical progeroid syndrome due to het- erozygous missense LMNA mutations. J Clin Endocrinol Me- tab 2009;94(12):4971-4983.
9. Merideth MA, Gordon LB, Clauss S, et al. Phenotype and course of Hutchinson-Gilford progeria syndrome. N Engl J Med 2008;358(6):592–604.
10. Weedon MN, Ellard S, Prindle MJ, et al. An in-frame deletion at the polymerase active site of POLD1 causes a multisystem disorder with lipodystrophy. Nat Gen- et 2013;45(8):947-950.
11. Garg A, Xing C. De novo hetero- zygous FBN1 mutations in the extreme C-terminal region cause progeroid fibrillinopathy. Am J Med Genet A 2014;164A(5):1341-1345.
12. Graul-Neumann LM, Kienitz T, Robinson PN, et al. Marfan syndrome with neonatal progeroid syndrome-like lipodystrophy associated with a novel frameshift mutation at the 3' terminus of the FBN1-gene. Am J Med Genet A 2010;152A(11):2749-2755.
13. Garg A, Kircher M, Del Campo M, Amato RS, Agarwal AK; Universi- ty of Washington Center for Men- delian Genomics. Whole exome sequencing identifies de novo het- erozygous CAV1 mutations asso- ciated with a novel neonatal onset lipodystrophy syndrome. Am J Med Genet A 2015;167A(8):1796–1806.
14. Donadille B, D'Anella P, Auclair M, et al. Partial lipodystrophy with severe insulin resistance and adult progeria Werner syndrome. Or- phanet J Rare Dis 2013;8:106.
15. Navarro CL, Cau P, Lévy N. Mo- lecular bases of progeroid syn- dromes. Hum Mol Genet 2006;15 (Spec No 2):R151-R161.
16. Thauvin-Robinet C, Auclair M, Du- plomb L, et al. PIK3R1 mutations cause syndromic insulin resistance with lipoatrophy. Am J Hum Gen- et 2013;93(1):141-149.
17. Diker-Cohen T, Cochran E, Gorden P, Brown RJ. Partial and general- ized lipodystrophy: Comparison of baseline characteristics and response to metreleptin. J Clin En- docrinol Metab 2015;100(5): 1802- 1810.
18. Kassai A, Muniyappa R, Levenson AE, et al. Effect of leptin admin- istration on circulating apolipo- protein CIII levels in patients with lipodystrophy. J Clin Endocrinol Metab 2016;101(4):1790-1797.
